Back to Journals » The Application of Clinical Genetics » Volume 10
Genotype and phenotype correlation in intracranial hemorrhage in neonatal factor VII deficiency among Thai children
Authors Traivaree C ![]() , Monsereenusorn C
, Monsereenusorn C ![]() , Meekaewkunchorn A, Laoyookhong P, Suwansingh S, Boonyawat B
, Meekaewkunchorn A, Laoyookhong P, Suwansingh S, Boonyawat B ![]()
Received 17 April 2017
Accepted for publication 9 May 2017
Published 21 June 2017 Volume 2017:10 Pages 37—41
DOI https://doi.org/10.2147/TACG.S139788
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Chanchai Traivaree,1 Chalinee Monsereenusorn,1 Arunotai Meekaewkunchorn,2 Premsak Laoyookhong,3 Saranya Suwansingh,4 Boonchai Boonyawat5
1Division of Hematology/Oncology, Department of Pediatrics, Phramongkutklao Hospital and College of Medicine, 2Division of Hematology/Oncology, Department of Pediatrics, 3Division of Neonatology, Department of Pediatrics, Queen Sirikit National Institute of Child Health, Bangkok, 4Division of Hematology/Oncology, Department of Pediatrics, Chiangrai Prachanukroh Hospital, Chiang Rai, 5Division of Genetics, Department of Pediatrics, Phramongkutklao Hospital and College of Medicine, Bangkok, Thailand
Abstract: Congenital factor VII (FVII) deficiency is a rare inherited coagulopathy. The clinical manifestations and clinical findings vary widely, ranging from asymptomatic to life-threatening bleeding, including intracranial hemorrhage (ICH), with prolonged prothrombin time, normal partial thromboplastin time and normal platelet counts, which are confirmed by the low level of FVII assay. Treatment consists of fresh frozen plasma (FFP), prothrombin complex concentrates (PCCs), and recombinant activated FVII to treat bleeding and prophylactic therapy. Here, we report four patients with FVII levels <5% (severe type) who presented ICH during the neonatal period. The IVS6+1G>T was the most common (50%) mutation identified in our study, followed by the K376X nonsense mutation (37.5%). In our study, we found that genetic information affected the severity of congenital FVII deficiency with ICH.
Keywords: mutation analysis, factor VII deficiency, Thai children
Introduction
Factor VII (FVII) is a vitamin K-dependent coagulation factor synthesized in the liver and has a short circulating half-life of 2–3 hours. Congenital FVII deficiency is a rare autosomal recessive bleeding disorder that has an estimated prevalence of 1/500,000 among the general population.1 According to the natural course of the disease, bleeding manifestations and clinical findings vary widely ranging from asymptomatic to life-threatening bleeding, including intracranial hemorrhage (ICH), an important cause of mortality and morbidity among patients with congenital FVII deficiency. The severe cases are usually manifested with life-threatening bleedings with FVII levels usually <5%.2 Cases with moderate-to-mild forms may have only mild bleeding symptoms or may never bleed abnormally and usually have FVII level ~5–10%. The F7 gene (OMIM: 613878) is located on chromosome 13q34 and contains nine exons (exons 1a, 1b, and 2–8) spanning 12 kb.3,4 This gene encodes a mature 406 amino acid FVII (F7) protein composed of two chains: the light chain containing the N-terminal gamma-carboxyglutamic acid (Gla) domain and two epidermal growth factor-like (EGF) domains and the heavy chain containing the C-terminal serine protease catalytic domain. To date, >200 pathogenic mutations have been reported.5–8 Mutations of the F7 gene are very heterogeneous and have been identified throughout the gene. Missense mutations are the most common (70–80%), followed by splice-site mutations, nonsense mutations, and small deletions and insertions.
We report four patients with FVII levels <5% (severe type) who presented ICH during the neonatal period. Mutations in the F7 gene are also identified and affect the severity of congenital FVII deficiency with ICH.
Patients and methods
Patient 1
A 15-day-old girl was referred with lethargy and anemia for 2 days, suggestive of an ICH. She was born by vaginal delivery without any perinatal problems. Her birth weight was 2,530 g, and she had no family history of any bleeding disorders. Her parents originated from northeast Thailand with no history of consanguineous marriage. The investigations revealed normal platelet counts with abnormal prothrombin time (PT) of 37 s (normal 10–20 s) with international normalized ratio (INR) of 2.96. Activated partial thromboplastin time (aPTT) and thrombin time (TT) were within normal limits. Further investigations revealed that FVII assay was 2%. Brain computed tomography (CT) demonstrated an intraventricular hemorrhage (IVH) in both lateral ventricles and right frontal and occipital lobes with hydrocephalus (IVH grades III and IV). The patient required repeated transfusion of fresh frozen plasma (FFP) to maintain normal INR. A follow-up CT of the brain at 2 months showed a resolving ICH, decreased IVH and slightly improved hydrocephalus. The patient continued with FFP transfusion every 2 weeks and was followed up in the outpatient clinic regularly without any bleeding episodes since then.
Patient 2
A 1-day-old boy was referred with conditions suggestive of an ICH, who had no family history of any bleeding disorders. He was born through vaginal delivery. His birth weight was 3,170 g. His parents originated from central Thailand without a history of consanguineous marriage. At presentation, the complete blood count revealed hemoglobin (Hb) 11.2 g/L, hematocrit (Hct) 34%, white blood cell (WBC) 11,000/mL, and platelets 270,000/mL, while the coagulation profiles revealed abnormal PT 105.8 s with an INR of 10. aPTT and TT were within normal limits, and the fibrinogen level was 272 mg/dL. Further investigation revealed that FVII assay was 0.5%. Brain CT demonstrated IVH in both lateral ventricles and right temporal and left occipital areas (IVH grades III and IV). The patient required repeated transfusion of FFP to maintain normal INR every week.
Patient 3
A 4-day-old girl was brought to the emergency department with the onset of seizure for 1 day. Her parents originated from central Thailand with a history of consanguineous marriage. She was born through vaginal delivery without any perinatal problems. Her birth weight was 3,030 g. Physical examination revealed pallor and stuporous child, bulging of anterior fontanel with generalized tonic and clonic seizures. At presentation, complete blood count revealed Hb 8 g/L, Hct 24%, WBC 14,450/mL, and platelets 303,000/mL. Coagulation profiles revealed PT 54.9 s with an INR 6.40; aPTT and TT were within normal limits. Further investigation revealed a fibrinogen level of 287 mg/dL and FVII activity of 4%. CT of brain revealed extensive IVH in both lateral and third ventricles with obstructive hydrocephalus, intracerebral hemorrhage at the right temporal and occipital lobes and cerebellar vermis (IVH grades III and IV), and subarachnoid hemorrhage over the right temporal lobe with diffused brain edema. This patient expired on the sixth day due to severe ICH.
Patient 4
A 14-day-old girl was referred to the emergency department with the onset of seizure for 1 day without a family history of any bleeding disorders. Her parents originated from northern Thailand with no history of consanguineous marriage. The investigations revealed normal platelet counts and abnormal PT of 37 s with an INR of 2.96. The aPTT and TT were within normal limits. Further investigations revealed that FVII assay was 1%. Brain CT demonstrated an IVH in both lateral ventricles with hydrocephalus (IVH grade III). The patient continued regular transfusions of FFP every week.
Method
Written informed consent was provided by the parents of the patients for both participating in the study and for the publication of their case details. The study protocol was approved by the institutional review board of Phramongkutklao Hospital, Phramongkutklao College of Medicine, Thailand. The genomic DNA was extracted from peripheral blood lymphocytes using AxyPrep TM blood genomic DNA miniprep kit (Corning Incorporated, Corning, NY, USA)according to manufacturer’s protocol. Nine coding exons and exon–intron boundaries of the F7 gene were amplified by polymerase chain reaction (PCR) using nine pairs of primers as described previously.9,10 All PCR products were purified and directly sequenced in both forward and reverse directions. The mutation nomenclature conformed to HGVS (www.hgvs.org/varnomen), and the reference sequences were NM_000131.4 and NP_000122.1 for F7 cDNA and FVII position, respectively. (The numbering starts with nucleotide +1 for the A of the ATG-translation initiation codon.)
Results
Mutation analysis of the F7 gene analyzed
Patients 1 and 4
Direct DNA sequencing in patients 1 and 4 identified a homozygous c.681+1G>T (IVS6+1G>T) mutation in intron 6 of the F7 gene (Figure 1A). Heterozygosity for the same mutation was identified in both parents of patient 1 only, according to unavailable genomic DNA from the parents of Patient 4.
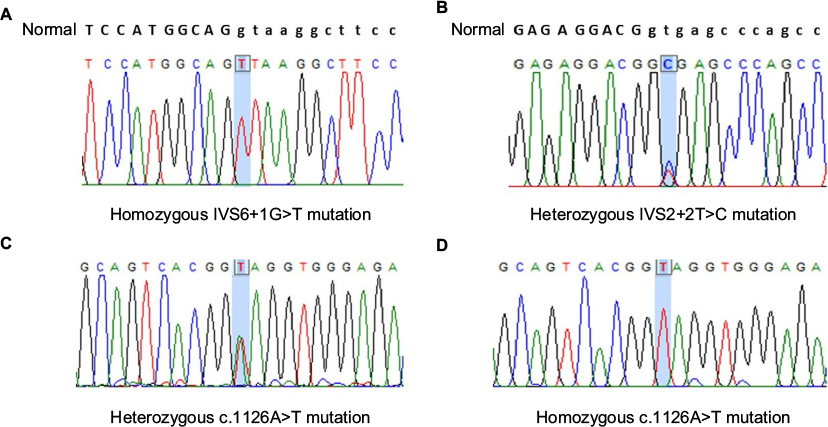 | Figure 1 Direct DNA sequencing of the F7 gene in our study revealed homozygous C.681+1G>T (IVS6+1G>T) mutation in Patients 1 and 4 (A), compound heterozygous c.291+2T>C (IVS2+2T>C) (B), and c.1126A>T (p.K376X) (C) mutations in Patient 2, and homozygous c.1126A>T (p.K376X) (D) mutation in Patient 3. |
Patient 2
Compound heterozygosity of one nonsense mutation, c.1126A>T (p.K376X), in exon 8 and one novel splice-site mutation, c.291+2T>C (IVS2+2T>C), in intron 2 of the F7 gene was identified in patient 2 (Figure 1B and C). This splice-site mutation was not identified in 100 healthy control alleles. Unfortunately, genomic DNA from both parents was unavailable.
Patient 3
In patient 3, homozygous nonsense mutation, c.1126A>T (p.K376X), in exon 8 of the F7 gene was identified (Figure 1D). Heterozygosity for the same mutation was identified in both parents.
Discussion
Congenital FVII deficiency is a rare autosomal recessive hemorrhagic disorder. The frequency is higher in countries where consanguineous marriage is more common. The disease is caused by deficient or defective blood coagulation FVII and is characterized by isolated prolonged PT and confirmed by the low level of FVII assay. FVII is a vitamin K-dependent clotting factor that is part of the extrinsic pathway of blood coagulation. The complex is formed between the naturally occurring procoagulant serine protease, activated FVII (FVIIa), and the integral membrane protein tissue factor (TF). The FVIIa/TF complex activates FX to FXa and generates some amounts of thrombin.11 This thrombin is responsible for platelet activation and prothrombin complex.
Depending on the concentration of FVII in the blood, the disorder is divided into severe (FVII<5%) and mild-to-moderate (FVII>5%) forms. The patients with mild-to-moderate form may have only mild bleeding symptoms. Our study reports patients with severe FVII deficiency who presented with ICH. The clinical signs of ICH among neonates are sometimes varied, with anemia as the most frequent symptom, followed by neurological symptoms, such as seizures, apnea, and lethargy. In survivors of ICH, the risk of sequel is considerable in many recent series.12,13 No evidence-based guidelines exist for the management of newborns with congenital FVII deficiency with replacement therapy. Treatment of congenital FVII deficiency consists of replacement therapy with FFP, prothrombin complex concentrates (PCCs), and recombinant FVIIa to treat bleeding and prophylactic therapy.14 Levels of >10% are usually hemostatic, even higher levels may be advisable. Maintaining FVII levels of at least 15% provided adequate hemostasis levels for most surgical procedures. The prophylactic therapy in congenital FVII deficiency has been a debated issue, especially because of its short half-life (3–4 hours). Karimi and Shafieian15 reported that prophylactic treatment with recombinant FVIIa among patients with FVII deficiency with a dosage of 20–30 mg/kg once or twice weekly may be beneficial in preventing hemorrhagic complications. However, this remains a controversial issue.
Because FFP is available in every hospital in our country, they can choose to receive FFP prophylaxis in the most convenient hospital. However, the data for frequency of treatment are controversial. The limitation of FFP for congenital FVII deficiency includes the volume required to provide adequate FVII replacement, life-long treatment, risk of inhibitor development, and risk of blood-borne infection.
Mutations of the F7 gene are highly variable and have been identified throughout the gene, which suggests the importance of almost every domain in FVII function. A majority of the mutations are single-nucleotide substitutions, including missense, splice-site, and nonsense mutations. Small deletions and insertions are rare.5–7,16 After direct DNA sequencing of the F7 gene was performed in our four unrelated patients with severe FVII deficiency, one novel and two previously reported mutations were identified. These mutations included one nonsense mutation, c.1126A>T (p.K376X), and two splice-site mutations, c.681+1G>T (IVS6+1G>T) and c.291+2T>C (IVS2+2T>C), of which the latter intron 2 mutation has never been reported in the literature.
The IVS6+1G>T mutation was first identified in a Taiwanese patient with severe FVII deficiency10,17 and has been occasionally reported in Latin America, Europe,5 and Asia.10,17 In Thailand, this mutation has been previously reported in three unrelated Thai patients with severe FVII deficiency.18 Two of them were homozygous for the mutation and presented life-threatening gastrointestinal and intracranial bleeding with undetected FVII levels. This mutation was found in compound heterozygosity with W424X nonsense mutation in the remaining patient, who also experienced intracranial bleeding. In our study, this intronic mutation was identified in homozygous conditions in two patients who had ICH and FVII level <5%. Even though, this mutation activates a cryptic splice site in cellular models, it secretes only trace amounts of functional FVII protein.18 The K376X nonsense mutation was identified in two patients in our study and was found to be homozygous and compound heterozygous with a novel IVS2+2T>C mutation in the third and second patients, respectively. Homozygous of K376X mutation was first reported only in a Thai patient with severe FVII deficiency manifested as ICH and low FVII activity (1.2%).19 This nonsense mutation resulted in a premature termination at the codon 316 of mature protein and gave rise to FVII protein missing the C-terminus of the catalytic domain. Two mutations in the intron 2, including IVS2+1G>C and IVS2+5G>T, have been previously reported among German patients with FVII deficiency. These mutations disrupt the donor splice site of intron 2 at nucleotides +1 and +5, respectively.20 Mutation at the nucleotide +2 of intron 2 (IVS2+2T>C) was first identified in our study and was predicted to result in abnormal splicing. Unfortunately, gene expression study cannot be performed in our institution.
Interestingly, the genotype–phenotype correlation in FVII deficiency is variable.5 Both FVII:C and FVII:Ag are poorly correlated with the bleeding phenotype. However, most of the genotypes in the patients with severe FVII deficiency are always composed of either homozygous or compound heterozygous for the mutations, preventing synthesis of any amount of functional FVII protein and resulting in FVII:C levels typically <2% of normal.5,7,16 The majority of the mutations in severe FVII deficiency are splice-site, nonsense, and frameshift mutations caused by small deletions and insertions.5–7,16 In Thailand, when all genetically confirmed severe FVII deficiency patients (four patients from our study and four previously reported patients) were included, only splice-site and nonsense mutations were identified in 62.5% and 37.5% of Thai patients, respectively. The IVS6+1G>T was the most common (56.2%) mutation identified in Thai patients, followed by K376X nonsense mutation (31.2%). The former intron 6 mutation was occasionally reported in other countries, whereas the latter nonsense mutation was reported only in Thai patients, suggesting the high prevalence and may be ethnic specificity of these mutations in Thailand. All Thai patients with severe FVII deficiency were either homozygous or compound heterozygous of deleterious mutation, for which homozygosity has been identified in 75% of the patients. Although the genotype–phenotype correlation is unreliable in overall FVII deficiency, the genetic information is still important in patients with severe FVII deficiency and provides the appropriate genetic counseling to patients and their families.
Conclusion
Congenital FVII deficiency is a rare inherited bleeding disorder. The clinical heterogeneity includes ICH with isolated prolonged PT as the hallmark of this bleeding disorder. Clinical bleeding can widely vary and does not always correlate with the level of FVII coagulant activity in the plasma. Then, genetic information is an important issue among patients with severe FVII deficiency for proper management.
Acknowledgment
This study was approved and supported with funding from the Phramongkutklao College of Medicine.
Disclosure
The authors report no conflicts of interest in this work.
References
Mannucci PM, Duga S, Peyvandi F. Recessively inherited coagulation disorders. Blood. 2004;104(5):1243–1252. | ||
Roberts HRLJ. Hemostasis and Thrombosis, Basic Principles and Clinical Practice. Philadelphia, PA: Lippincott; 1994. | ||
O’Hara PJ, Grant FJ, Haldeman BA, et al. Nucleotide sequence of the gene coding for human factor VII, a vitamin K-dependent protein participating in blood coagulation. Proc Natl Acad Sci U S A. 1987; 84(15):5158–5162. | ||
Hagen FS, Gray CL, O’Hara P, et al. Characterization of a cDNA coding for human factor VII. Proc Natl Acad Sci U S A. 1986;83(8):2412–2416. | ||
Herrmann FH, Wulff K, Auerswald G, et al; Greifswald Factor FVII Deficiency Study Group. Factor VII deficiency: clinical manifestation of 717 subjects from Europe and Latin America with mutations in the factor 7 gene. Haemophilia. 2009;15(1):267–280. | ||
Mariani G, Herrmann FH, Dolce A, et al; International Factor VII Deficiency Study Group. Clinical phenotypes and factor VII genotype in congenital factor VII deficiency. Thromb Haemost. 2005; 93(3):481–487. | ||
Giansily-Blaizot M, Aguilar-Martinez P, Biron-Andreani C, et al; Study Group of Factor Seven Deficiency. Analysis of the genotypes and phenotypes of 37 unrelated patients with inherited factor VII deficiency. Eur J Hum Genet. 2001;9(2):105–112. | ||
McVey JH, Boswell E, Mumford AD, Kemball-Cook G, Tuddenham EG. Factor VII deficiency and the FVII mutation database. Hum Mutat. 2001;17(1):3–17. | ||
Jayandharan GR, Viswabandya A, Nair SC, Chandy M, Srivastava A. Molecular basis of hereditary factor VII deficiency in India: five novel mutations including a double missense mutation (Ala191Glu; Trp364Cys) in 11 unrelated patients. Haematologica. 2007;92(7):1002–1003. | ||
Shen MC, Lin JS, Lin SW, Yang WS, Lin B. Novel mutations in the factor VII gene of Taiwanese factor VII-deficient patients. Br J Haematol. 2001;112(3):566–571. | ||
Hoffman M, Monroe DM 3rd. A cell-based model of hemostasis. Thromb Haemost. 2001;85(6):958–965. | ||
Kulkarni R, Lusher JM, Henry RC, Kallen DJ. Current practices regarding newborn intracranial haemorrhage and obstetrical care and mode of delivery of pregnant haemophilia carriers: a survey of obstetricians, neonatologists and haematologists in the United States, on behalf of the National Hemophilia Foundation’s Medical and Scientific Advisory Council. Haemophilia. 1999;5(6):410–415. | ||
Kulkarni R, Lusher JM. Intracranial and extracranial hemorrhages in newborns with hemophilia: a review of the literature. J Pediatr Hematol Oncol. 1999;21(4):289–295. | ||
Mariani G, Dolce A, Marchetti G, Bernardi F. Clinical picture and management of congenital factor VII deficiency. Haemophilia. 2004; 10(suppl 4):180–183. | ||
Karimi M, Shafieian R. Prophylactic effect of recombinant factor VIIa with congenital factor VII deficiency. Haemophilia. 2008; 14(4):851–852. | ||
McGregor JC, Pollock JG, Anton HC. Ultrasonography and possible ruptured abdominal aortic aneurysms. Br Med J. 1975;3(5975):78–79. | ||
Ariffin H, Millar DS, Cooper DN, Chow T, Lin HP. Prenatal exclusion of severe factor VII deficiency. J Pediatr Hematol Oncol. 2003; 25(5):418–420. | ||
Cavallari N, Balestra D, Branchini A, et al. Activation of a cryptic splice site in a potentially lethal coagulation defect accounts for a functional protein variant. Biochim Biophys Acta. 2012;1822(7):1109–1113. | ||
Pinotti M, Rizzotto L, Pinton P, et al; International Factor VII Deficiency Study Group. Intracellular readthrough of nonsense mutations by aminoglycosides in coagulation factor VII. J Thromb Haemost. 2006;4(6):1308–1314. | ||
Wulff K, Herrmann FH. Twenty two novel mutations of the factor VII gene in factor VII deficiency. Hum Mutat. 2000;15(6):489–496. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.