")
Back to Journals » Infection and Drug Resistance » Volume 13
Genomic Epidemiology of an Outbreak of Klebsiella pneumoniae ST471 Producing Extended-Spectrum β-Lactamases in a Neonatal Intensive Care Unit
Authors Wang Y, Luo C, Du P, Hu J, Zhao X, Mo D, Du X, Xu X, Li M, Lu H, Zhou Z, Cui Z, Zhou H
Received 26 October 2019
Accepted for publication 11 January 2020
Published 15 April 2020 Volume 2020:13 Pages 1081—1090
DOI https://doi.org/10.2147/IDR.S236212
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Yuan Wang,1 Chunyu Luo,1 Pengcheng Du,2 Jinrui Hu,3 Xiaowei Zhao,1 Dianjun Mo,1 Xiaoli Du,3 Xin Xu,1 Man Li,1 Hong Lu,1 Zhiqiang Zhou,1 Zhigang Cui,3 Haijian Zhou3
1Affiliated Hospital of Chifeng University, Chifeng 024005, People’s Republic of China; 2Beijing Key Laboratory of Emerging Infectious Diseases, Institute of Infectious Diseases, Beijing Ditan Hospital, Capital Medical University, Beijing 100015, People’s Republic of China; 3State Key Laboratory for Infectious Disease Prevention and Control, National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 102206, People’s Republic of China
Correspondence: Haijian Zhou
State Key Laboratory for Infectious Disease Prevention and Control, National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, 155, Changbai Road, Changping, Beijing 102206, People’s Republic of China
Tel +86-10-58900784
Fax +86-10-58900700
Email [email protected]
Purpose: Klebsiella pneumoniae producing extended-spectrum β-lactamases (ESBLs) causes nosocomial infections worldwide. The present study aimed to determine the molecular subtyping characteristics and antibiotic resistance mechanisms of ESBL-producing K. pneumoniae strains collected during an outbreak. Moreover, we attempted to reveal the fine transmission route of the strains within this outbreak using whole-genome sequencing (WGS).
Methods: Collecting cases and strain information were carried out. Outbreak-related strains were identified using pulsed-field gel electrophoresis (PFGE). The antibiotic susceptibility, drug-resistant genes, and molecular subtype characteristics of ESBL-producing K. pneumoniae were analyzed. The fine transmission route of the strains within this outbreak was revealed using WGS and minimum core genome (MCG) sequence typing.
Results: In mid-January, 2015, five cases of neonatal pneumonia caused by ESBL-producing K. pneumoniae were observed in the neonatal intensive care unit (NICU) of the Affiliated Hospital of Chifeng University, China. Eight ESBL-producing K. pneumoniae were isolated from these five cases, and two additional strains from another two cases were identified using PFGE. All ten isolates harbored blaCTX-M-15, blaTEM-1, blaSHV-108, and blaOXA-1 genes, and belonged to the sequence type 471 (ST471) clone. A putative transmission map was constructed via comprehensive consideration of genomic and epidemiological information. WGS identified the initial case and the “superspreader”. The genomic epidemiological investigation revealed that the outbreak was caused by the introduction of the bacteria one month before the first case appeared.
Conclusion: As far as we know, this is the first report to describe the characteristics of an ST471 ESBL-producing K. pneumoniae outbreak. The data showed that epidemiological inferences could be greatly improved by interpretation in the context of WGS and that K. pneumoniae strains isolated from the same outbreak contain sufficient genomic differences to refine epidemiological linkages on the basis of genetic lineage. These findings suggested that integration of genomic and epidemiological data can help us to have a clearer understanding of when and how outbreaks occur, so as to better control nosocomial transmission.
Keywords: whole-genome sequencing, minimum core genome, superspreader, transmission map
Introduction
Klebsiella pneumoniae, one of the “ESKAPE” pathogens, has emerged as an important nosocomial pathogen that mainly causes infections in the respiratory tract, urinary tract, and blood.1,2 The prevalence of multi-drug resistant (MDR) K. pneumoniae has increased dramatically in recent years. MDR K. pneumoniae frequently exhibits resistance to extended-spectrum cephalosporins because of its production of extended-spectrum β-lactamases (ESBLs).3 ESBL-producing bacteria cause higher morbidity, mortality, and fiscal burden.4,5 This limits efficient clinical treatment tremendously, resulting in undesirable treatment outcomes.
The outbreak of multidrug-resistant bacteria is a serious public health problem, and nosocomial outbreaks caused by ESBL-producing K. pneumoniae have been reported worldwide.6–11 ESBL-producing K. pneumoniae is the most frequently implicated pathogen causing outbreaks in neonatal intensive care units (NICUs), with mortality rates up to 31% among infected infants.12 Traditional molecular typing methods, such as pulsed-field gel electrophoresis (PFGE) and multilocus sequence typing (MLST), can define the outbreak strains well, but cannot reveal the transmission routes between strains.
Over the past decade, whole-genome sequencing (WGS) technology has developed rapidly and has been used widely in epidemiological investigations of bacterial infectious diseases, to identify outbreaks,13,14 trace pathogen transmission,15,16 discover new modes or ways of transmission,17 identify new clones,18 and track nosocomial infections and outbreaks.19 In the present study we report an outbreak of K. pneumoniae producing ESBLs that took place in a NICU and attempt to reveal the fine transmission route of the strains within this outbreak.
Materials and Methods
Review of NICU Records and Collection of Patient Information
The outbreak occurred in the NICU of the Affiliated Hospital of Chifeng University, a 2000-bed general tertiary care and University-affiliated teaching hospital in Inner Mongolia, China. This hospital accepts 2200 outpatient and emergency patients each day. The NICU has a total of 25 beds.
The medical records of patients from whom ESBL-producing K. pneumoniae was isolated were reviewed, including time of stay in the NICU, bed site, clinical symptoms, diagnosis, data of the ESBL-producing K. pneumoniae isolation, and outcomes.
Bacterial Identification and Antimicrobial Susceptibility
Bacterial identification was initially performed using the VITEK-2 automated system (BioMérieux, Craponne, France) and further confirmed using matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF-MS) (Bruker, Leipzig, Germany).
Antibiotic susceptibility testing was performed using the broth dilution method. We measured the minimum inhibitory concentration (MIC) of ampicillin (AMP), ampicillin/sulbactam (AMS), amoxicillin/clavulanate (AMC), cefazolin (CFZ), cefoxitin (CFX), cefotaxime (CTX), ceftazidime (CAZ), cefepime (FEP), aztreonam (AZM), imipenem (IMI), meropenem (MEM), amikacin (AMI), kanamycin (KAN), azithromycin (AZI), tetracycline (TET), doxycycline (DOX), tigecycline (TGC), ciprofloxacin (CIP), levofloxacin (LEV), trimethoprim-sulfamethoxazole (SXT), chloramphenicol (CHL), and colistin (CO). All antibiotics, except tigecycline and colistin, were interpreted according to the approved standard of Clinical and Laboratory Standards Institute (CLSI) 2016 guidelines.20 Susceptibility to tigecycline and colistin was defined based on the criteria proposed by the European Committee on Antimicrobial Susceptibility Testing-2018 (http://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/v_8.0_Breakpoint_Tables.pdf). Escherichia coli ATCC 25922 was used as quality control strain for the antibiotic susceptibility testing.
We screened genes for resistance to beta-lactamase (blaCTX-M, blaTEM, blaSHV, blaOXA, blaPER, blaVEB) in all strains, as described previously.21 DNA sequencing was performed on both strands of the PCR amplification products. The results were compared and aligned with reference sequences using the online BLAST database.
Pulsed-Field Gel Electrophoresis (PFGE)
We used the 1-day, standardized PFGE protocol for K. pneumoniae.22 Cell suspensions were placed in polystyrene tubes (Falcon 12 × 75 mm, from Fisher Scientific, Hampton, NH, USA), and their optical density was adjusted to 3.8–4.0 using a Densimat photometer (BioMérieux). K. pneumoniae slices were digested using 50 U per slice of XbaI (Takara, Dalian, China) for 4 h at 37 °C, and electrophoresis was performed using a CHEF-DRIII system (Bio-Rad Laboratories, Hercules, CA, USA). Electrophoresis was performed using a switch time of 6 s to 36 s for 18.5 h, and images were captured using a Gel Doc 2000 system (Bio-Rad) and converted to TIFF files. The TIFF files were analyzed using BioNumerics version 5.1 software (Applied Maths, Kortrijk, Belgium).
Multilocus Sequence Typing (MLST)
MLST using seven genes (gapA, infB, mdh, pgi, phoE, rpoB, and tonB) was performed on the isolates according to the protocol described on the K. pneumoniae MLST website (http://bigsdb.pasteur.fr/). Alleles and sequence types (STs) were assigned using the MLST database (http://bigsdb.pasteur.fr/klebsiella/klebsiella.html).
Whole-Genome Sequencing (WGS), Detection of Single Nucleotide Polymorphisms (SNPs), and Minimum Core Genome (MCG) Sequence Typing
The ten outbreak strains were selected for WGS. Genomic DNA from the bacterial strains was sequenced using Illumina sequencing by constructing two paired-end (PE) libraries with average insertion lengths of 500 bp and 2000 bp, respectively. Sequences were generated using an Illumina GA IIx apparatus (Illumina Inc., San Diego, CA, USA). Raw data was processed in four steps, including removing reads with 5 bp of ambiguous bases, removing reads with 20 bp of low quality (≤ Q20) bases, removing adapter contamination, and removing duplicated reads. Finally, 100× libraries were obtained with clean PE read data. Assembly was performed using SOAPdenovo v1.05.23
The whole-genome sequence of K. pneumoniae KPR0928 (NCBI accession number: NZ_CP008831.1) was used as the reference sequence, and clean reads of the sequenced isolates were mapped to the reference genome using bowtie 2 software under the default parameters.24 SNPs were then identified using Samtools and combined together according to the reference.25 SNPs with low quality reads (read depth < 5) and those located within 5 bps on the chromosome were removed to avoid the effect of recombination, as described in previous studies.17,26 The isolates were clustered and a heatmap was generated using the heatmap package in R. The transmission route was then reconstructed based on the emergence of different SNPs in each isolate and the case information, including the time of the onset of infection and the hospitalization time.
The multilocus sequences were identified using the pMLST 2.0 database (https://cge.cbs.dtu.dk/services/MLST/).27 Antimicrobial resistance genes were identified by searching the Comprehensive Antibiotic Research Database (CARD, https://card.mcmaster.ca/).28
Nucleotide Sequence Accession Numbers
The data from the WGS project have been deposited at GenBank under the BioProject ID PRJNA578669.
Results
Overall Description of the Outbreak
In mid-January, 2015, we identified five cases (cases 1–5 in Table 1 and Figure 1) of neonatal pneumonia caused by ESBL-producing K. pneumoniae in the NICU of our hospital. This NICU consists of two units with a total of four wards and 25 beds, and has been open since 2013.
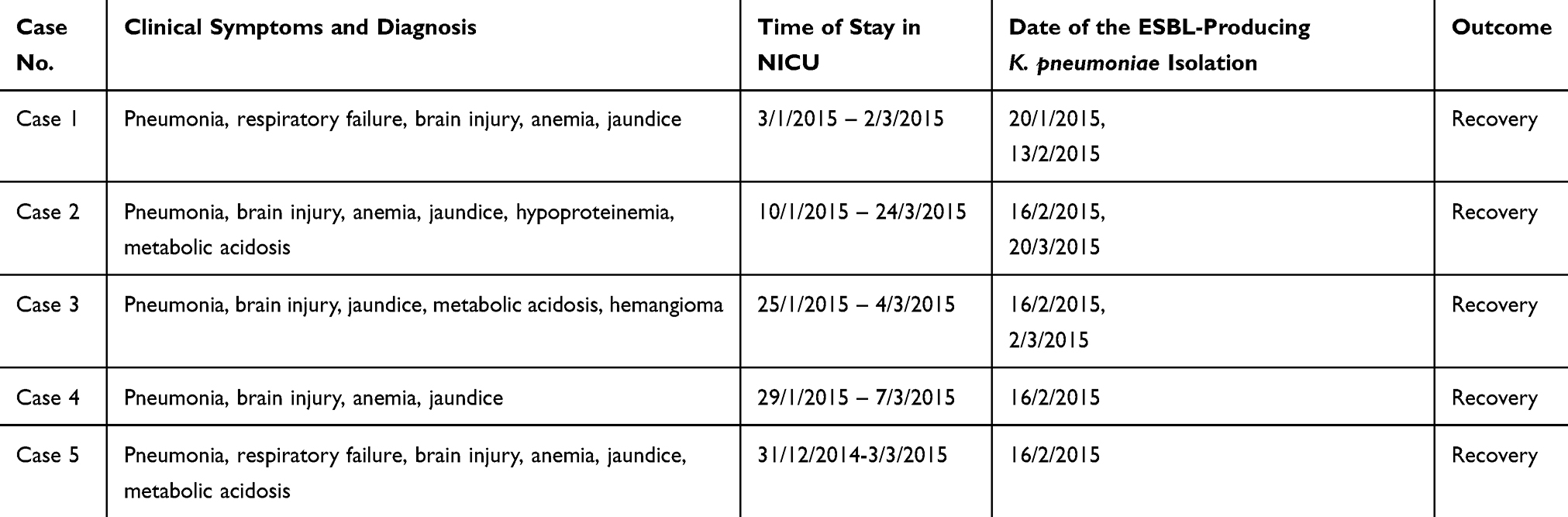 |
Table 1 Descriptions of the Five Cases Involved in the February 16th Outbreak in the NICU |
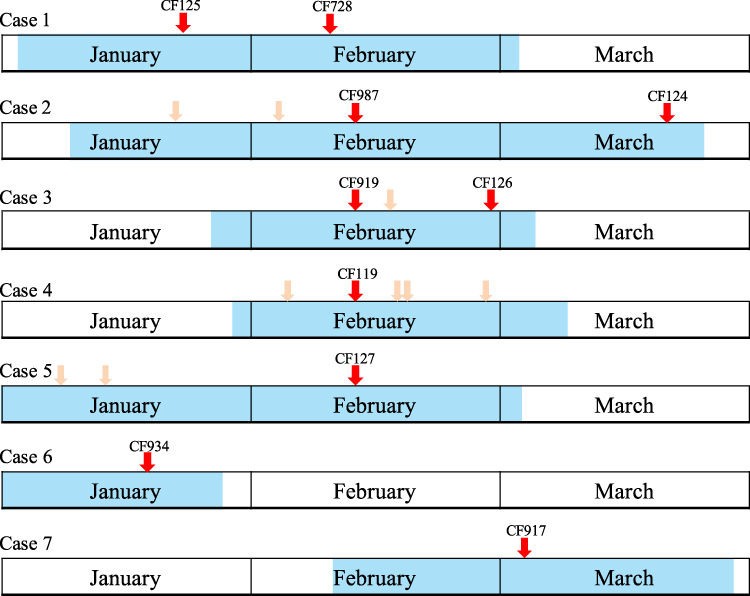 |
Figure 1 The timeline of patient admission and ESBL-producing K. pneumoniae isolation. Shadows on the timeline represent the duration of the case. Different wards are indicated in different colors. The red arrows indicate the isolation of the strains. Light red arrows represent sampling where no K. pneumoniae was isolated. |
The index case (case 1) of this outbreak was identified on February 13, 2015. On February 16, four more cases (cases 2–5) in the same unit in the NICU were observed (Table 1). The timeline of patient admission and ESBL-producing K. pneumoniae isolation is shown in Figure 1. An outbreak was declared and stringent infection control measures were implemented, including contact precautions, strengthening of hand hygiene, environmental cleaning, and enhancing antimicrobial stewardship.
Although stringent infection control measures were carried out, seven more ESBL-producing K. pneumoniae were isolated during the hospitalization of the five outbreak cases; among them, four, one, and one strains were isolated from cases 2, 3, and 5, respectively, and another one was isolated from a new case (case 7, Figure 1) in this NICU. Unfortunately, we failed to recover four of the strains (three from case 2 and one from case 5) because of poor preservation; therefore, they could not be used for subsequent experiments.
From March 27, 2015 (the discharge date of the last patients with ESBL-producing K. pneumoniae), over a period of six months, no further ESBL-producing K. pneumoniae were isolated in the NICU.
Searching for Outbreak-Related Strains Using PFGE
PFGE was used to screen outbreak-related K. pneumoniae strains among the strains isolated from the same hospital during or around the outbreak. A total of 22 K. pneumoniae strains, which were isolated between January 4 and March 17, were analyzed by PFGE. Interestingly, two strains (CF125 and CF934) were clustered together with the eight outbreak strains (Figure 2). CF125 was isolated from case 1 on January 20, 23 days before the outbreak. CF934 was isolated from the tip of a trachea cannula of a new case (case 6, Figure 1) who had been in NICU before, on January 17, 26 days before the outbreak.
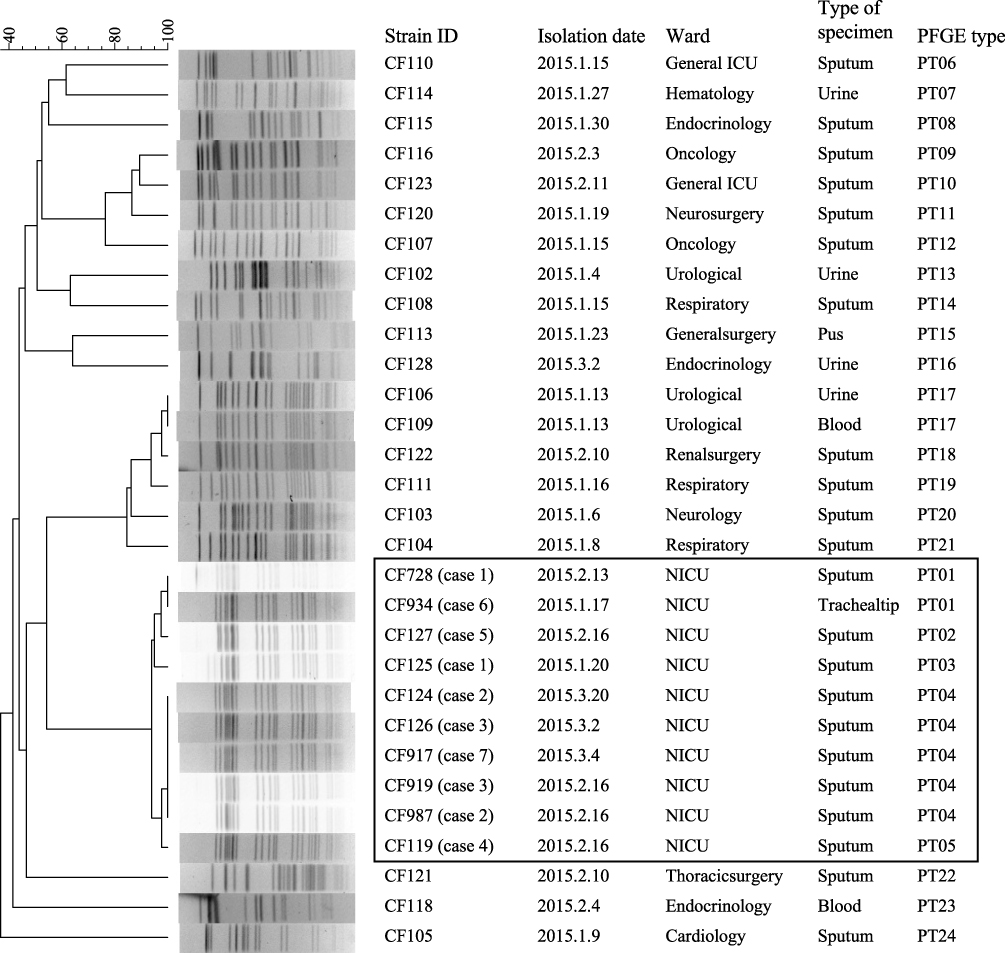 |
Figure 2 Clustering of the 30 K. pneumoniae strains based on their PFGE patterns. The outbreak and outbreak-related strains are framed. The information for strain ID, isolation date, ward, type of specimen, and PFGE type is listed to the left of the patterns. Abbreviations: NICU, neonatal intensive care unit; PFGE, pulsed-field gel electrophoresis. |
PFGE divided the ten outbreak-related strains into five different PFGE types (PT01–PT05). The dominant PFGE type (PT04) contained five isolates. The other four PFGE types showed one to three bands that were different to PT04. All ten isolates were the “same” or “closely-related” strains according to the interpretation criteria of PFGE patterns proposed by Tenover et al.29 The other 20 K. pneumoniae strains isolated from the same hospital during or around the outbreak showed more than 20 bands that were different to outbreak-related strains.
Using MLST, all ten outbreak-related strains were assigned as ST471 (allele numbers of 3, 1, 1, 68, 4, 1, and 39 for gapA, infB, mdh, pgi, phoE, rpoB, and tonB, respectively). By searching the K. pneumoniae MLST database (http://bigsdb.pasteur.fr.), we found that there was only one ST471 strain in the database, which was isolated from a human from Spain in 2012.
Antibiotic Susceptibility and Characterization of Resistance Genes
All 10 ESBL-producing K. pneumoniae strains showed same results for the antibiotic susceptibility test. All strains showed insensitivity to ampicillin, ampicillin/sulbactam, amoxicillin/clavulanate, cefazolin, cefotaxime, cefepime, trimethoprim-sulfamethoxazole, and aztreonam, and were susceptible to other 14 antibiotics (cefoxitin, ceftazidime, imipenem, meropenem, amikacin, kanamycin, azithromycin, tetracycline, doxycycline, tigecycline, ciprofloxacin, levofloxacin, chloramphenicol, and colistin). All ten K. pneumoniae strains were confirmed as positive for ESBL production using double-disk diffusion tests.
By PCR and sequence alignment of beta-lactamase genes, all 10 isolates were shown to harbor blaCTX-M-15, blaTEM-1, blaSHV-108, and blaOXA-1 genes, and tested negative for blaPER, and blaVEB. Based on the WGS results, we further characterized the antibiotic resistance genes among the 10 isolates. In addition to the four beta-lactamase genes (blaCTX-M-15, blaTEM-1, blaSHV-108, and blaOXA-1), all isolates were confirmed to possess genes for resistance to aminoglycoside (aac(3)-II, aac(6ʹ)-Ib, aph(33)-Ib and aph(6)- Id), sulfonamide (sul2), fosfomycin (fosA), trimethoprim (dfrA14, dfrA22), chloramphenicol (catB3), polymyxin (arnA), and bacitracin (bacA). Furthermore, several resistance pumps were identified, including a plasmid-mediated tetracycline resistance pump (tet_efflux).
Comparison of Outbreak Isolates Based on WGS-Based SNPs
WGS and MCG typing were performed for the 10 ESBL-producing K. pneumoniae strains, with the aim of using the SNPs found in their genomes to determine a putative transmission map of this outbreak. Genomic comparisons revealed a total of 7 MCG SNPs among the 10 strains (Figure 3). The evolutionary relationships based on MCG typing of the 10 strains are outlined and presented in Figure 4. The strains isolated from case 6 (CF934) in January showed the same SNP profile as the reference strain K. pneumoniae KPR0928. The first strain isolated from case 1 (CF125) had one SNP (T608763C) that was different to the reference strain. Another case 1 strain (CF728) had one more SNP (C154003T). The first strain (CF987) isolated from case 2 had the same SNP (T608763C) to that of the first case 1 strain (CF125). Another case 2 strain (CF124) had one more SNP (G953229A). The strains isolated from cases 4 (CF119) and 5 (CF127), both of which were isolated on February 16, had one (G2665757T) and two (C996839T, C2820669A) SNPs that were different to the case 1 and 2 strains (CF125, CF987), respectively. The first strain (CF919) of case 3, which was also isolated on February 16, had one SNP (G2550784A) that was different to the case 4 strain (CF119). The second case 3 strain (CF126), which was isolated 14 days later, showed one more SNP (G3771456A).
 |
Figure 3 Heat map of SNP distribution of ten ESBL-producing K. pneumoniae. |
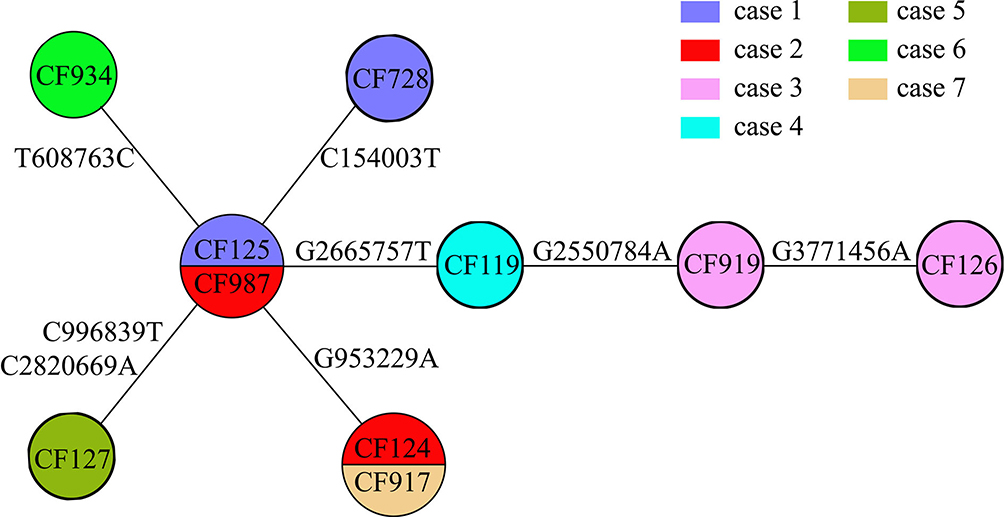 |
Figure 4 Evolutionary relationships based on MCG typing of ten ESBL-producing K. pneumoniae. Isolates from different cases are indicated in different colors. SNPs are marked on the connecting lines. |
Identification of the Origin and the Construction of a Putative Transmission Route of the Outbreak
The genomic and epidemiological information were integrated to construct the transmission route. The most likely transmission route is shown in Figure 5.
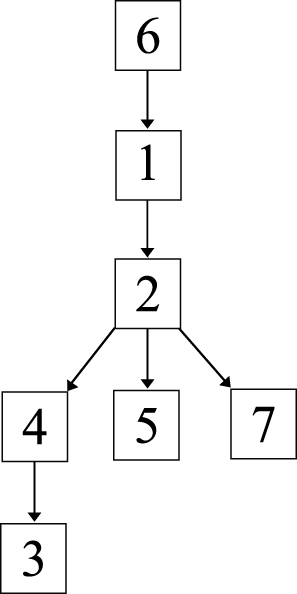 |
Figure 5 Putative map of ESBL-producing K. pneumoniae transmission during the outbreak. The transmission map was constructed using genetic and patient trace data. Nodes represent patients, and arrows indicate a transmission event directly from one patient to another. |
First, case 6 was suspected of being the source of the outbreak isolates, because the historical strain from case 6 showed the same SNP patterns to the reference strain K. pneumoniae KPR0928. This was also supported by the epidemiological information. The case 6 strain was the earliest isolated ESBL-producing K. pneumoniae strain, which was isolated 1 month before the February 16 outbreak. Thus, case 6 was the most likely source of the outbreak isolates.
Second, the first strains isolated from cases 1 and 2 showed one SNP difference to the isolates of case 6, suggesting that the strains from cases 1 and 2 were not transmitted from case 6. However, combining the epidemiological information, that the first strain of case 1 was isolated on January 20, closer to the isolation of case 6 strain; and the first strain of case 2 was isolated on February 16, suggesting that the most possible transmission route was that this ESBL-producing K. pneumoniae strain was passed from case 6 to case 1 and then to case 2. Interestingly, the second strains from both cases 1 and 2 had one SNP difference with the first strains, suggesting that there were mutations in vivo.
Third, the strains from cases 4 and 5 had one and two SNPs different to the first strains of cases 1 and 2. However, the second strain of case 1 was isolated three days before the isolation of the strains from cases 4 and 5 and showed another SNP, which suggested that the strains of cases 4 and 5 were not transmitted from case 1. Thus, case 2 was the most likely source of the strains isolated from cases 4 and 5.
Fourth, the first strain of case 3, which was also isolated on February 16, had one SNP difference with the case 4 strain. The second case 3 strain, which was isolated 15 days later, showed one more SNP. Thus, the isolates of case 3 were transmitted from case 4, which then persisted in case 3 and mutated in vivo.
Fifth, the strain from case 7 was isolated 20 days after the February 16 outbreak and showed same SNP profile as the second strain of case 2, suggesting that case 2 directly transmitted this strain to case 7.
On the whole, consistent with the genomic and epidemiological information, the integrated map identified that: (1) Case 6 was the source of the February 16 outbreak; (2) before the outbreak, the ESBL-producing K. pneumoniae was a long-term resident in case 1; (3) case 2 played a key role, as a “superspreader”, in the outbreak, probably directly causing cases 4, 5, and 7, and indirectly causing case 3.
Discussion
In this study, we described an outbreak caused by ESBL-producing K. pneumoniae in a NICU of a large university hospital in China. The ESBL-producing K. pneumoniae isolates belonged to ST471, and all harbored blaCTX-M-15, blaTEM-1, blaSHV-108, and blaOXA-1 beta-lactamase genes. ST471 is a highly uncommon ST, which has only been reported in one human case in Spain and clinical bovine mastitis in Tunisia.30 The Tunisian study pointed out the necessity to improve farm management to avoid the dissemination in this sector of ESBL-producing K. pneumoniae. It is worth noting that the outbreak reported here occurred in Chifeng City, which belongs to Inner Mongolia, the largest pastoral area in China. Therefore, a future investigation should determine whether this ST471 strain infected humans via cattle, sheep, or other livestock. Furthermore, as far as we know, this is not only the first study to report an outbreak caused by K. pneumoniae belonging ST471, but also is the first to report an outbreak caused by a K. pneumoniae strain co-harboring the blaCTX-M-15, blaTEM-1, blaSHV-108 and blaOXA-1 genes, which suggests that more attention should be paid to the K. pneumoniae isolates producing epidemic ESBLs.
CTX-M-15 is the main CTX-M-type of clinical ESBL-producing K. pneumoniae in China and other parts of the world.31–37 Furthermore, blaCTX-M-15-positive K. pneumoniae has been also widely detected in various animals, including farm cattle, pet dogs, and wild birds,38–40 indicating communication between the genetic pools of the blaCTX-M-15 gene in animals and humans. In future studies, we should investigate how these ESBLs are transmitted from animals to humans, and then implement measures to block this route to avoid such infections and outbreaks. TEM-1 is the main TEM-type of K. pneumoniae isolated from Tanzania, India, and Iran.41–43 OXA-1 and SHV-108 are rarely detected from K. pneumoniae. In a recently multicenter survey of ESBL-producing K. pneumoniae causing intra-abdominal infections from nine tertiary hospitals in China, no TEM-type and OXA-1 strain was detected, and the most common genotype for K. pneumoniae was SHV-2a.32 However, in another survey of K. pneumoniae from pediatric patients in Shanghai, China, TEM-1 and OXA-1 were detected in 68.3% and 14.6% of strains.44 Although there is no global survey of the distribution of these resistance genes at present, it can be seen from the current research reports that there are regional differences in the distribution of these beta-lactamase genes between different countries, different cities, even between different hospitals of the same region.
PFGE used to be the gold standard for molecular subtyping of outbreaks. However, although PFGE could identify outbreak-related strains, it could not reveal the evolutionary relationship and transmission route between them. Our previous research showed that WGS and MCG typing could reveal the details of transmission within a K. pneumoniae nosocomial outbreak.16 MCG typing was first used to study the population structure of Streptococcus suis and then Legionella pneumophila.17,26 In the present study, we further confirmed that WGS and MCG typing can reveal the fine transmission map and identify the “superspreader” of an outbreak. If WGS and MCG typing were applied in the early stage of an outbreak, the outbreak could be controlled. In addition, this method has the potential to be used to track other human-borne pathogens, such as Neisseria meningitidis, Bordetella pertussis, and Streptococcus pneumoniae.
Conclusion
In summary, we describe the integration of epidemiological information with high-resolution WGS and MCG typing to enhance the investigation of an outbreak of ESBL-producing K. pneumoniae infection. Our data showed that epidemiological inferences are greatly improved by interpretation in the context of WGS and that K. pneumoniae strains isolated from the same outbreak contain sufficient SNP differences to refine the epidemiological linkages on the basis of genetic lineage.
Ethics and Consent Statement
This study was approved by the scientific and ethics committees of the National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention. All clinical specimens from patients were collected for diagnostic testing in hospitals at the request of the attending doctors. All experiments on K. pneumoniae strains were reviewed and approved by the scientific and ethics committees. All experiments were performed in accordance with relevant guidelines and regulations. Furthermore, to protect patient privacy, the hospital set up a patient privacy and medical record management system according to the criminal procedure law, civil procedure law, tort liability law, and medical malpractice law in China. Excepting for the patient’s doctor, no one can enter the management system. Although no written informed consent was provided by patients or their guardians, the above measures were considered sufficient to protect the patients’ privacy.
Acknowledgments
We thank the team of curators of the Institute Pasteur MLST and whole genome MLST databases for curating the data and making them publicly available at http://bigsdb.pasteur.fr/. This work was supported by a grants from the Priority Project on Infectious Disease Control and Prevention (grant number 2017ZX10303405-002) from the Ministry of Science and Technology of the People’s Republic of China. The funding body played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Podschun R, Ullmann U. Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin Microbiol Rev. 1998;11:589–603. doi:10.1128/CMR.11.4.589
2. Boucher HW, Talbot GH, Bradley JS, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. doi:10.1086/595011
3. Falagas ME, Karageorgopoulos DE. Extended spectrum β-lactamase-producing organisms. J Hosp Infect. 2009;73:345–354. doi:10.1016/j.jhin.2009.02.021
4. Jean SS, Hsueh PR. High burden of antimicrobial resistance in Asia. Int J Antimicrob Agents. 2011;37:291–295. doi:10.1016/j.ijantimicag.2011.01.009
5. Dhillon RH, Clark J. ESBLs: a clear and present danger. Crit Care Res Pract. 2012;2012:625170.
6. Becker L, Fuchs S, Pfeifer Y, et al. Whole genome sequence analysis of CTX-M-15 producing klebsiella isolates allowed dissecting a polyclonal outbreak scenario. Front Microbiol. 2018;9:322. doi:10.3389/fmicb.2018.00322
7. Mahmoudi S, Pourakbari B, Rahbarimanesh A, et al. An outbreak of ESBL-producing Klebsiella pneumoniae in an Iranian referral hospital: epidemiology and molecular typing. Infect Disord Drug Targets. 2019;19:46–54. doi:10.2174/1871526518666180507121831
8. Peltier F, Choquet M, Decroix V, et al. Characterization of a multidrug-resistant Klebsiella pneumoniae ST607-K25 clone responsible for a nosocomial outbreak in a neonatal intensive care unit. J Med Microbiol. 2019;68:67–76. doi:10.1099/jmm.0.000884
9. Lenglet A, Faniyan O, Hopman J. A nosocomial outbreak of clinical sepsis in a Neonatal Care Unit (NCU) in Port-Au-Prince Haiti, July 2014 - September 2015. PLoS Curr. 2018;10.
10. Mshana SE, Fritzenwanker M, Falgenhauer L, et al. Molecular epidemiology and characterization of an outbreak causing Klebsiella pneumoniae clone carrying chromosomally located bla(CTX-M-15) at a German University-Hospital. BMC Microbiol. 2015;15:122. doi:10.1186/s12866-015-0460-2
11. Chen D, Hu X, Chen F, et al. Co-outbreak of multidrug resistance and a novel ST3006 Klebsiella pneumoniae in a neonatal intensive care unit: a retrospective study. Medicine (Baltimore). 2019;98:e14285. doi:10.1097/MD.0000000000014285
12. Stapleton PJ, Murphy M, McCallion N, et al. Outbreaks of extended spectrum beta-lactamase-producing Enterobacteriaceae in neonatal intensive care units: a systematic review. Arch Dis Child Fetal Neonatal Ed. 2016;101:F72–F78. doi:10.1136/archdischild-2015-308707
13. van Ingen J, Kohl TA, Kranzer K, et al. Global outbreak of severe Mycobacterium chimaera disease after cardiac surgery: a molecular epidemiological study. Lancet Infect Dis. 2017;17:1033–1041. doi:10.1016/S1473-3099(17)30324-9
14. Bloomfield SJ, Benschop J, Biggs PJ, et al. Genomic analysis of Salmonella enterica serovar Typhimurium DT160 associated with a 14-year outbreak, New Zealand, 1998–2012. Emerg Infect Dis. 2017;23:906–913. doi:10.3201/eid2306.161934
15. Gardy JL, Johnston JC, Ho Sui SJ, et al. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N Engl J Med. 2011;364:730–739. doi:10.1056/NEJMoa1003176
16. Sui W, Zhou H, Du P, et al. Whole genome sequence revealed the fine transmission map of carbapenem-resistant Klebsiella pneumonia isolates within a nosocomial outbreak. Antimicrob Resist Infect Control. 2018;7:70. doi:10.1186/s13756-018-0363-8
17. Du P, Zheng H, Zhou J, et al. Detection of multiple parallel transmission outbreak of Streptococcus suis human infection by use of genome epidemiology, China, 2005. Emerg Infect Dis. 2017;23:204–211. doi:10.3201/eid2302.160297
18. Qin T, Zhang W, Liu W, et al. Population structure and minimum core genome typing of Legionella pneumophila. Sci Rep. 2016;6:21356. doi:10.1038/srep21356
19. Onori R, Gaiarsa S, Comandatore F, et al. Tracking nosocomial Klebsiella pneumoniae infections and outbreaks by Whole-Genome analysis: small-scale italian scenario within a single hospital. J Clin Microbiol. 2015;53:2861–2868. doi:10.1128/JCM.00545-15
20. Clinical and Laboratory Standards Institute (CLSI). Performance Standards for Antimicrobial Susceptibility Testing: Twenty-First Informational Supplement. Wayne: CLSI; 2016:M100–S26.
21. Chen D, Sun C. Atlas of Clinical Microbiology Laboratory. People’s Medical Publishing House; 2016. ISBN 978-7-89456-717-8. (In Chinese).
22. Han H, Zhou H, Li H, et al. Optimization of pulse-field gel electrophoresis for subtyping of Klebsiella pneumoniae. Int J Environ Res Public Health. 2013;10:2720–2731. doi:10.3390/ijerph10072720
23. Li R, Zhu H, Ruan J, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20:265–272. doi:10.1101/gr.097261.109
24. Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357–359. doi:10.1038/nmeth.1923
25. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi:10.1093/bioinformatics/btp352
26. Chen C, Zhang W, Zheng H, et al. Minimum core genome sequence typing of bacterial pathogens: a unified approach for clinical and public health microbiology. J Clin Microbiol. 2013;51:2582–2591. doi:10.1128/JCM.00535-13
27. Larsen MV, Cosentino S, Rasmussen S, et al. Multilocus sequence typing of total genome sequenced bacteria. J Clin Microbiol. 2012;50:1355–1361. doi:10.1128/JCM.06094-11
28. Jia B, Raphenya AR, Alcock B, et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017;45(D1):D566–D573. doi:10.1093/nar/gkw1004
29. Tenover FC, Arbeit RD, Goering RV, et al. Interpreting chromosomal DNA restriction patterns produced by pulsed field gel electrophoresis: criteria for bacterial strain typing. J Clin Microbiol. 1995;33:2233–2239. doi:10.1128/JCM.33.9.2233-2239.1995
30. Saidani M, Messadi L, Soudani A, et al. Epidemiology, antimicrobial resistance, and extended-spectrum beta-lactamase-producing enterobacteriaceae in clinical bovine mastitis in Tunisia. Microb Drug Resist. 2018;24:1242–1248. doi:10.1089/mdr.2018.0049
31. An S, Chen J, Wang Z, et al. Predominant characteristics of CTX-M-producing Klebsiella pneumoniae isolates from patients with lower respiratory tract infection in multiple medical centers in China. FEMS Microbiol Lett. 2012;332:137–145. doi:10.1111/j.1574-6968.2012.02586.x
32. Liao K, Chen Y, Wang M, et al. Molecular characteristics of extended-spectrum β-lactamase-producing Escherichia coli and Klebsiella pneumoniae causing intra-abdominal infections from 9 tertiary hospitals in China. Diagn Microbiol Infect Dis. 2017;87:45–48. doi:10.1016/j.diagmicrobio.2016.10.007
33. Sader HS, Castanheira M, Flamm RK. Antimicrobial activity of ceftazidime-avibactam against gram-negative bacteria isolated from patients hospitalized with pneumonia in U.S. Medical Centers, 2011 to 2015. Antimicrob Agents Chemother. 2017;61(4):e02083–16. doi:10.1128/AAC.02083-16
34. Rocha FR, Pinto VP, Barbosa FC. The spread of CTX-M-type extended-spectrum β-Lactamases in Brazil: a systematic review. Microb Drug Resist. 2016;22:301–311. doi:10.1089/mdr.2015.0180
35. Markovska R, Stoeva T, Boyanova L, et al. Dissemination of successful international clone ST15 and clonal complex 17 among Bulgarian CTX-M-15 producing K. pneumoniae isolates. Diagn Microbiol Infect Dis. 2017;89:310–313. doi:10.1016/j.diagmicrobio.2017.08.012
36. Higashino M, Murata M, Morinaga Y, et al. Fluoroquinolone resistance in extended-spectrum β-lactamase-producing Klebsiella pneumoniae in a Japanese tertiary hospital: silent shifting to CTX-M-15-producing K. pneumoniae. J Med Microbiol. 2017;66:1476–1482. doi:10.1099/jmm.0.000577
37. Bado I, Gutiérrez C, García-Fulgueiras V, et al. CTX-M-15 in combination with aac(6ʹ)-Ib-cr is the most prevalent mechanism of resistance both in Escherichia coli and Klebsiella pneumoniae, including K. pneumoniae ST258, in an ICU in Uruguay. J Glob Antimicrob Resist. 2016;6:5–9. doi:10.1016/j.jgar.2016.02.001
38. Bandyopadhyay S, Banerjee J, Bhattacharyya D, et al. Genomic identity of fluoroquinolone-resistant blaCTX-M-15-type ESBL and pMAmpC β-Lactamase producing Klebsiella pneumoniae from Buffalo Milk, India. Microb Drug Resist. 2018;24:1345–1353. doi:10.1089/mdr.2017.0368
39. Silva MM, Fernandes MR, Sellera FP, et al. Multidrug-resistant CTX-M-15-producing Klebsiella pneumoniae ST231 associated with infection and persistent colonization of dog. Diagn Microbiol Infect Dis. 2018;92:259–261. doi:10.1016/j.diagmicrobio.2018.06.012
40. Hessman J, Atterby C, Olsen B, et al. High prevalence and temporal variation of extended spectrum β-Lactamase-producing bacteria in Urban Swedish Mallards. Microb Drug Resist. 2018;24:822–829. doi:10.1089/mdr.2017.0263
41. Sonda T, Kumburu H, van Zwetselaar M, et al. Molecular epidemiology of virulence and antimicrobial resistance determinants in Klebsiella pneumoniae from hospitalised patients in Kilimanjaro, Tanzania. Eur J Clin Microbiol Infect Dis. 2018;37:1901–1914. doi:10.1007/s10096-018-3324-5
42. Mondal AH, Siddiqui MT, Sultan I, et al. Prevalence and diversity of blaTEM, blaSHV and blaCTX-M variants among multidrug resistant Klebsiella spp. from an urban riverine environment in India. Int J Environ Health Res. 2018;6:1–13.
43. Dehshiri M, Khoramrooz SS, Zoladl M, et al. The frequency of Klebsiella pneumonia encoding genes for CTX-M, TEM-1 and SHV-1 extended-spectrum beta lactamases enzymes isolated from urinary tract infection. Ann Clin Microbiol Antimicrob. 2018;17:4. doi:10.1186/s12941-018-0256-y
44. Zhang X, Chen D, Xu G, et al. Molecular epidemiology and drug resistant mechanism in carbapenem-resistant Klebsiella pneumoniae isolated from pediatric patients in Shanghai, China. PLoS One. 2018;13:e0194000.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.