Back to Journals » Lung Cancer: Targets and Therapy » Volume 16
Genomic and Transcriptomic Profiles in Smokers and Never-Smokers Lung Squamous Cell Carcinoma Patients
Authors Canale M ![]() , Virga A, Angeli D, Fonzi E
, Virga A, Angeli D, Fonzi E ![]() , Gnetti L
, Gnetti L ![]() , Dubini A, Tedaldi G
, Dubini A, Tedaldi G ![]() , Urbini M, Bocchialini G, Petracci E, Verlicchi A
, Urbini M, Bocchialini G, Petracci E, Verlicchi A ![]() , Cravero P, Citarella F, Bennati C, Andrikou K, Delmonte A, Crinò L
, Cravero P, Citarella F, Bennati C, Andrikou K, Delmonte A, Crinò L ![]() , Carbognani P, Ulivi P
, Carbognani P, Ulivi P ![]() , Ampollini L
, Ampollini L ![]()
Received 23 January 2025
Accepted for publication 8 May 2025
Published 28 June 2025 Volume 2025:16 Pages 85—96
DOI https://doi.org/10.2147/LCTT.S517580
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Sai-Hong Ou
Matteo Canale,1 Alessandra Virga,1,* Davide Angeli,2 Eugenio Fonzi,2 Letizia Gnetti,3 Alessandra Dubini,4 Gianluca Tedaldi,1 Milena Urbini,1 Giovanni Bocchialini,5,* Elisabetta Petracci,2 Alberto Verlicchi,6 Paola Cravero,6 Fabrizio Citarella,6 Chiara Bennati,7 Kalliopi Andrikou,6 Angelo Delmonte,6 Lucio Crinò,6 Paolo Carbognani,5 Paola Ulivi,1,* Luca Ampollini5,*
1Biosciences Laboratory, IRCCS Istituto Romagnolo per Lo Studio Dei Tumori (IRST) “Dino Amadori”, Meldola, Italy; 2Unit of Biostatistics and Clinical Trials, IRCCS Istituto Romagnolo per Lo Studio Dei Tumori (IRST) “Dino Amadori”, Meldola, Italy; 3Pathology Unit, University Hospital of Parma, Parma, Italy; 4Department of Pathology, Morgagni-Pierantoni Hospital, Forlì, Italy; 5Department of Thoracic Surgery, University Hospital of Parma, Parma, Italy; 6Department of Medical Oncology, IRCCS Istituto Romagnolo per Lo Studio dei Tumori (IRST) “Dino Amadori”, Meldola, Italy; 7Department of Medical Oncology, Santa Maria Delle Croci Hospital, Ravenna, Italy
*These authors contributed equally to this work
Correspondence: Alessandra Virga, Biosciences Laboratory, IRCCS Istituto Romagnolo per lo Studio dei Tumori (IRST) “Dino Amadori”, Meldola, Italy, Email [email protected] Giovanni Bocchialini, Department of Thoracic Surgery, University Hospital of Parma, Parma, Italy, Email [email protected]
Purpose: Lung Squamous Cell Carcinoma (SCC) is a Non-Small Cell Lung Cancer (NSCLC) subtype with a strong clinical association with smoking habits and a very low incidence in never-smokers. Molecular profiling of SCC in never-smokers could unveil tumor vulnerabilities and new treatment strategies.
Patients and Methods: We considered a patient cohort of 17 former or current smokers (51.5%) and 16 never-smoker SCC patients (48.5%). TruSight Oncology® 500, investigating hotspots in 523 cancer-related genes, Tumor mutation burden (TMB) and microsatellite instability (MSI), and RNA sequencing was performed on tumor tissue. Genomic and transcriptomic profiles were compared between smokers and never-smoker patients.
Results: The most frequently altered genes were TP53 (67%), CDKN2A (20%) and PIK3CA (17%), with no substantial differences between groups, except for TP53 which was more frequently mutated in smokers (86.7% vs 46.7%, p = 0.05), who also showed a higher TMB with respect to non-smokers (median 11 mut/Mb vs 5.5 mut/Mb, p = 0.028); all patients were stable for MSI score (median 1.87 vs 1.82, p = 0.87). Activating mutations in EGFR and MET were found in one and two never-smokers, respectively. Three smoker patients had simultaneous amplifications in FGF3, FGF19 and FGF4. Enrichment analyses showed that cyclin-dependent protein Ser/Thr kinase activity and PI3K signaling pathways were affected in both groups, while cellular damage response was exclusively altered in never-smokers. Unsupervised hierarchical clustering on transcriptomes effectively identified different specific transcriptional subtypes between smokers and never-smokers. Gene set enrichment analysis highlighted that tumors from never-smokers are characterized by dysregulation in cell membrane potential and ion homeostasis across cell membrane pathways.
Conclusion: Genomic and transcriptomic profiles deeply differentiate SCC occurring in never-smokers with respect to SCC in smoker patients. Moreover, SCC could carry canonical NSCLC) activating mutations. Our data suggest that deep molecular analyses resolve tumor heterogeneity and may help with new algorithm-based treatment strategies for SCC.
Keywords: lung squamous cell carcinoma, smoking habits, next generation sequencing, transcriptomics
Introduction
Lung cancer (LC) is the most diagnosed and the most lethal malignancy worldwide, with almost 2.5 million new cases and 1.8 million deaths in 2022.1 Non-Small Cell Lung Cancer (NSCLC) is the most prevalent histology, accounting for around 85% of LC cases; based on histologic features, NSCLC is further classified into other subtypes, of which adenocarcinoma (ADC, 70% of all NSCLC cases) and Squamous Cell Carcinoma (SCC, about 30% of NSCLC cases) are the most represented forms.2 Even though they are both classified as NSCLC, SCC and ADC deeply differ in both biological and clinico-pathological features.3 In fact, ADCs are mainly characterized by acinar, papillary, solid or mucinous patterns, with thyroid transcription factor (TTF-1) and napsin-A staining to support the diagnosis; more common in younger patients, in women, and it is more addicted to oncogene-activating mutations (eg EGFR mutations, EML4-ALK fusions), which are rarely detected in SCC.3–5 Conversely, SCC histology is histo-pathologically classified into keratinizing, non-keratinizing and basaloid subtypes, with squamous cell patterns (supported by p40 and p63 staining) or keratinization aspects (supported by cytokeratin 5/6 staining).3,6–8 Moreover, ADC and SCC deeply differ by transcriptomic profiling, highlighting different biologically relevant pathways involved in carcinogenesis and differentiation of tumor histology, and also molecular factors influencing patient prognosis, eg the availability of targeted therapies only for patients with ADC, which is often diagnosed with activating mutations.9–11
Smoking is the most common etiology for lung cancer, associated with around 80% of LC cases, as LC diagnosis is associated with both number of cigarettes/day and years of tobacco consumption.12–14 Nonetheless, even though it could arise in smokers, ADC is more common in never-smoker patients, especially oncogene-addicted ADC, while SCC diagnosis is strongly associated with smoking habits.5,15,16 While clinical and molecular features of both non-smokers and smokers ADC patients have been well elucidated, molecular profiles of SCC arising in never-smoker patients remain an uninvestigated field of research. As LC histology could influence patients’ prognosis and therapy efficacy, outlining genomic and transcriptomic features of SCC in never-smoker patients could uncover tumor vulnerabilities, and specific signatures to help patient stratification.17 Moreover, it could give insights on SCC tumor biology, as in the last years many indications are suggesting that SCC could be considered as an independent tumor histology, that deeply differs from ADC, even though still classified together.3
The main aim of the present study was to investigate the tissue molecular profile of non-smoker SCC patients using a comprehensive DNA next-generation sequencing (NGS) targeted panel and whole-transcriptome analysis and to compare it with that of a similar group of SCC patients with a history of smoking.
Materials and Methods
Case Series
This was a retrospective study on SCC patients with a histologically confirmed diagnosis of lung SCC between 2004 and 2022 at the University Hospital of Parma, Santa Maria delle Croci Hospital of Ravenna and Morgagni-Pierantoni Hospital of Forlì, in Italy. Diagnostic haematoxylin-eosin slides were retrieved and reviewed by two expert pathologists to confirm SCC histology, prior to inclusion within the study. Patient demographics and clinical features, including tumor histology, age, gender, and smoking history were obtained through medical chart review. Formalin-fixed paraffin-embedded (FFPE) tumor specimens were retrieved from the Pathology Units and revised by dedicated expert pathologists to contain at least 50% of tumor cells. FFPE slides were provided for centralized analysis at Istituto Romagnolo per lo Studio dei Tumori (IRST) “Dino Amadori” for molecular analysis. The protocol was approved by C.E.ROM. Ethical Committee (study code IRST-B045). The present study was conducted in accordance with the Declaration of Helsinki 1964 and later versions, and all patients signed the informed consent.
Targeted Panel DNA Sequencing
The neoplastic area was scraped to collect the DNA. The tumor-derived DNA was isolated using the QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions and quantified with the Qubit dsDNA BR Assay kit (Thermo Fisher Scientific, Waltham, MA, USA).
The TruSight™ Oncology 500 (TSO500) target panel (Illumina, San Diego, CA, USA) was applied to detect a wide range of genetic alterations, including single nucleotide variants (SNVs) and copy number variants (CNVs) in more than 500 genes, as well as genetic signatures as tumor mutational burden (TMB) and microsatellite instability (MSI). Fragmentation of 40 ng of gDNA was performed using the ME220 ultrasonicator (Covaris, Woburn, Massachusetts, USA). Libraries were enriched for the 523 target genes in the panel following the manufacturer’s instructions. After quantity and quantity controls, 8 final libraries for each run were pooled and loaded onto the NextSeq™ 550 sequencer (Illumina).
The analysis was performed with DRAGEN™ TruSight™ Oncology 500 Analysis v2 software starting from fastq files. This tool can identify low-frequency somatic variants in coding exons and other biologically significant regions in 523 DNA biomarker-related genes. The manufacturer’s quality control criteria were used to determine whether a TSO500 result was valid, including median insert size ≥ 70 bp, median exon coverage ≥ 50 count, and percentage of exons with coverage of at least 100 count ≥80%. Variants with a variant allele frequency (VAF) greater than 5% were called. Variants frequently present in the normal population (GnomAD > 1%) were excluded (Karczewski et al, 2020). Pathogenicity of the variants was defined following ACMG guidelines (American College of Medical Genetics and Genomics).18 Variants classified as “pathogenic” or “likely pathogenic” by Varsome (VarSome Clinical 11.7.5) were considered as clearly pathogenic and not retained in the analysis together with variants of uncertain significance (VUS). Alterations classified as “benign” or “likely benign” by Varsome were excluded.19 The selected cutoff values were >20% of unstable microsatellite sites to define MSI and >10 mut/Mb to define high TMB.20,21 The minimum number of usable microsatellites to determine MSI status was 40. The OncoPrint was built with the ComplexHeatmap R package.22
RNA Sequencing
Additional FFPE slides were scraped to extract RNA from tumor cells. FFPE RNA isolation kit (Roche Diagnostics, Vienna, AUT) was used to collect RNA, following manufacturer instructions. RNA quantification was performed with the Qubit 3.0 instrument using the RNA High Sensitivity kit (Thermo Fisher Scientific).
RNA libraries were generated starting from 500 ng of purified total RNA, using the Illumina Stranded Total RNA Prep with Ribo-Zero Plus (Illumina) on an automated Hamilton STARlet platform (Reno, NV, USA). Briefly, the total RNA was depleted, fragmented and denatured. The two-strand cDNA was synthesized and ligated with anchors. Fragments were cleaned up and amplified to generate libraries. Library quantity and quality were assessed on Qubit 3.0 instrument and BioAnalyzer 2100 (Agilent, Santa Clara, CA, USA). All libraries were sequenced on a single run on a NovaSeq 6000 platform (Illumina) with a NovaSeq 6000 S2 Reagent Kit v1.5, and two steps of 101 cycles of paired-end sequencing were performed.
Transcript-level read count was performed with kallisto v0.46.2, then raw counts were collapsed to gene-level with tximport v1.12.1, then Differential Expression Analysis (DEA) was performed with DESeq v1.22.1121–123. The same software was used to produce PCA plots, while for heatmaps and hierarchical clustering Seaborn v0.12.1 was used. Gene Set Enrichment Analysis (GSEA) was performed with the package GSEApy v0.9.16,124–126. For QC analyses, DESeq2’s Variance Stabilizing Transformation (VST) was applied to raw read counts, and the 500 genes with the highest variance across the dataset were selected to perform PCA and hierarchical clustering; VST-transformed counts were also used as input for GSEA, after removing genes <1 across all samples. GSEA was analyzed by Gene Ontology (GO) Gene Function, v. 2018.
Statistical Analysis
Data were summarized by median, first (IQ) and third (IIIQ) quartiles, for continuous variables and by means of absolute frequencies and percentages for categorical ones. The association between continuous variables and smoking status was assessed by the Wilcoxon–Mann–Whitney test whereas that with binary variables with the Pearson’s χ2 test of the Fisher exact test, as appropriate. P-values for the association between pathogenic alterations and clinical data were adjusted using the Benjamini–Yekutieli method. However, since this is an exploratory study, we decided to report the unadjusted p-values in the text and complement them with the adjusted values. For this analysis, the odds ratio (OR) and corresponding 95% confidence intervals (CI) were also reported.
Results
Patients Clinico-Pathological Features
In this study, we collected RNA and DNA samples from all patients. However, we encountered some limitations: 2 DNA samples from smokers were quantitatively insufficient, and the quality of 5 RNA samples was not suitable for transcriptome analysis (2 from smokers and 3 from never-smokers). Overall, adequate samples for at least one of the molecular analyses were available for 16 never-smoker SCC patients and 17 SCC patients who were current or former smokers (ie 6 current smokers and 11 former smoker patients).
Among the 33 patients, the median age at diagnosis of SCC was 71 years (IQ-IIIQ 63–76), male patients were 55%, and most of the patients had a stage I SCC (45% stage I, 30% stage II, 18% stage III, and 6% stage IV). Patient characteristics by smoking status are summarized in Table 1. Median packs/year (P/Y) for former and current smokers was 51 [IQ-IIIQ 40–74]. Specifically, median P/Y was 50 [IQ-IIIQ 30–85] and 53 [IQ-IIIQ 40–60] for former and current smokers, respectively, while for the 11 former smokers, the median time since cessation was 5 years (IQ-IIIQ 2–20). No substantial differences were present between the patients with and without smoking habits, except for a higher prevalence of stage I tumors among never-smokers as compared to current or former smokers. For paired comparison analysis, we considered molecular features of never-smoker patients with respect to patients with a smoking history (former and current smokers), as exposed to the risk factor of smoking habit for cancer occurrence.
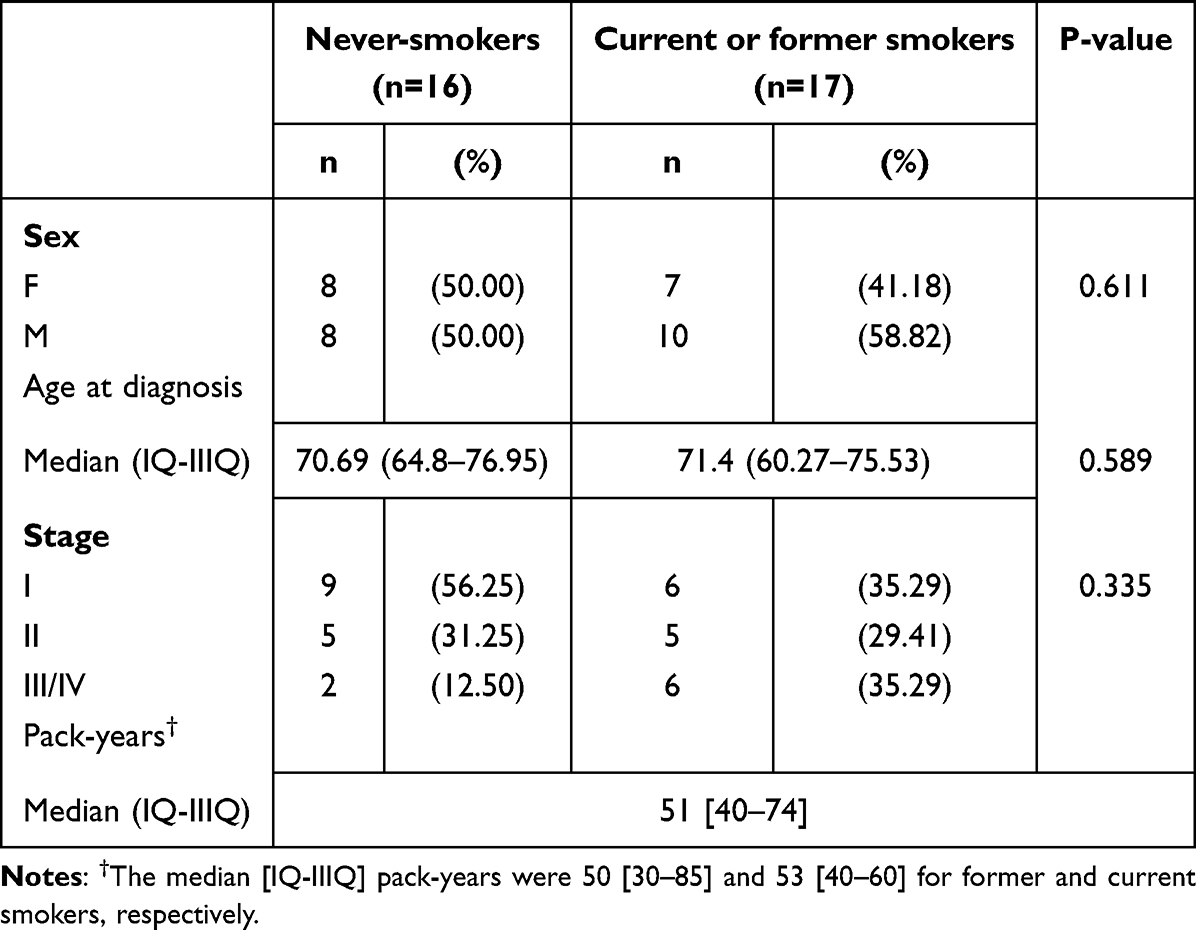 |
Table 1 Patients’ Characteristics and Clinical Associations |
DNA Targeted Sequencing
To investigate the presence of driver mutations and the molecular profile of never-smoker patients as well as to compare it with that of smokers or former smokers, a wide DNA mutation sequencing focused on 500 cancer-related genes, was performed. DNA sequencing analysis was performed on DNA with enough quantity and quality (n.15 patients with smoking habits [ie n.5 current smokers and n.10 former smokers] and n.15 never-smoker patients). By DRAGEN analysis software according to ACMG guidelines, overall 278 variants were identified, of which 82 were classified as pathogenic, and 22 CNV were found; considering only pathogenic variants, the most frequently mutated genes were TP53, 67% (87% [13/15] in current and former smokers patients, and 47% [7/15] in never-smokers), CDKN2A, 20% (27%, [4/15] in current and former smoker patients, and 13% [2/15] in never-smokers) and PIK3CA, 17% (27% [4/15] in current and former smoker patients, and 7% [1/15] in never-smokers. Interestingly, we found that one never-smoker patient had an activating insertion/deletion in exon 20 of EGFR, while further 2 never-smoker patients had a MET exon 14 skipping mutation (one exon 14 point mutation and one exon 14 splicing variant, respectively). Moreover, two patients (one smoker and one never-smoker) had an amplification of EGFR. We also found that tumors from 3 smoker patients had concomitant alterations in the FGF pathway, presenting simultaneous amplifications in FGF3, FGF19 and FGF4. The spectrum of mutations with a frequency higher than 6% is reported in Figure 1.
 |
Figure 1 Oncoprint plot for gene alterations in the patients’ cohort. Altered genes at a higher frequency than 6% are reported, ordered by number of mutations per patient. Columns represent individual samples and are divided by smoking habits. The different colors represent the different alterations, as indicated in the figure legends. The bar plots at the top and right of the oncoprint indicate the count of events found respectively in each sample and in each gene. In the lower part of the oncoprint, the smoking habit is reported for each sample. |
No associations were found between patients’ clinical features and pathogenic mutations. With regard to TP53, it was found altered in 13 out of 15 smokers or former smokers (86.7%) with respect to 7 out of 15 never-smokers (46.7%, OR: 7.43, 95% CI: 1.23–45.01), showing a borderline statistical significance (p = 0.05) that disappeared after adjusting for multiple comparisons (adjusted p = 1.00).
Overall, smoker or former smokers patients had a higher rate of pathogenic alterations with respect to never-smokers (67.1% vs 32.9%, p = 0.004), also resulting in a higher TMB for these patients (median 11 mut/Mb, IQ-IIIQ 8.8–15.3, vs 5.5 mut/Mb, IQ-IIIQ 3.2–14.4, p = 0.028) (Figure 2A and B). MSI assessment revealed that all patients were classified as microsatellite stable, with no differences between smokers or former smokers and never-smoker patients (median 1.87 IQ-IIIQ 1.0 −2.7 vs median 1.82 IQ-IIIQ 0–2.7, p = 0.87) (Figure 2C).
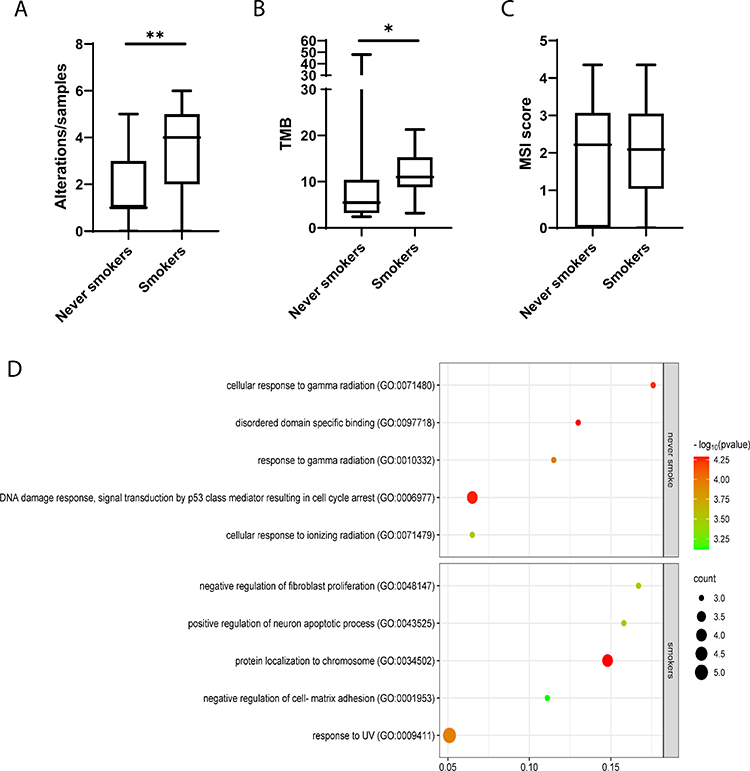 |
Figure 2 Box plots and Pathway enrichment bubble plot. The box plots comparing the rate of pathogenic alterations (A), tumor mutation burden (B) and microsatellite stability (C) between never-smokers and smokers or former smokers. Box Plots were obtained using GraphPad Prism 8.4.3 (GraphPad Software, Inc., San Diego, CA). *p-value: 0.028 **p-value: 0.004. (D) The plot showed only the differentially enriched pathways between the 2 groups of smoker and never-smoker patients. In the scatter plot are presented the top 5 enriched GO pathways. Pathway enrichment bubble was generated using SRplot.23 |
Finally, we performed a GO enrichment pathway analysis comparing gene expression between never-smokers and smokers or former smokers. In both groups of patients, replicative senescence pathways, regulation of cyclin-dependent protein serine/threonine kinase activity pathways, cell cycle and several PI3K signaling pathways were enriched with alterations. Only patients with current or past smoking habits showed alterations in the regulation of fibroblast proliferation, neuronal apoptotic process, cell-matrix adhesion, MAPK cascade, base excision repair and UV response pathways. On the other hand, never-smokers showed several pathways of radiation response, DNA damage response, and signal transduction by p53 class mediator enriched (Figure 2D and Supplementary Table 1).
Transcriptome Analysis Marks the Difference Between Smoker and Never-Smoker Patients
A whole-transcriptome analysis in RNA from patients’ tumors was performed. Total mRNA analysis was performed on tissue mRNA with at least >20 million successfully sequenced reads, ie 13 never-smoker and 15 smoker or former smoker patients. By considering only differentially expressed genes with a fold change > 2 and an adjusted p-value < 0.05, unsupervised clustering, and identified 277 genes for which mRNA expression was able to effectively discriminate never-smoker patients from smoker and former ones (Figure 3A and Supplementary Figure 1).
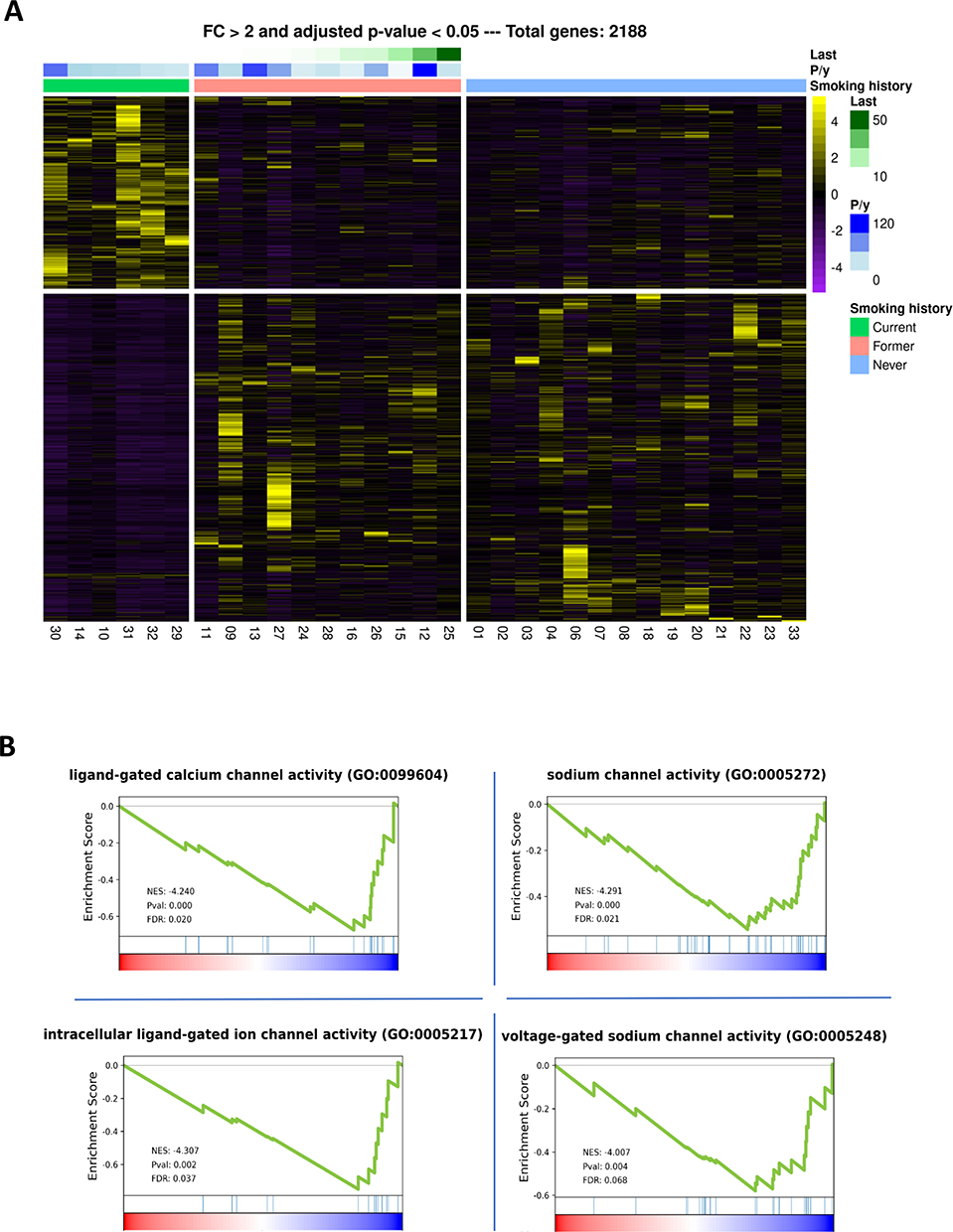 |
Figure 3 Heatmap performed by unsupervised clustering in the whole patient cohort and Gene Set Enrichment Analysis. (A) Considering a fold change > 2 and p ≤ 0.05, 2188 genes were found differentially expressed between the clinical subgroups of smokers, former smokers and never-smoker patients. Genes (rows) and patients (columns) are ordered by hierarchical clustering based on gene expression, using the Euclidean distance method and average linkage clustering for both genes and samples. (B) The analysis showed the transcriptome for pathways differentiating smokers patients versus never-smoker patients. A negative or positive Normalized Enrichment Score (NES) indicates an enriched pathway in the never-smokers group or in the smokers group, respectively. FDR: False Discovery Rate. |
By GSEA in GO Gene Function pathway enrichment analysis, we identified 3 enriched pathways in never-smoker patients with respect to smoker ones, and one pathway linked to these pathways had a trend (ie GO:0099604, GO:0005272, GO:0005217, GO:0005248), as shown in Figure 3B and Table 2. In particular, all these pathways were related to cell membrane potential and ion homeostasis across cell membranes, especially calcium and sodium channel activity, suggesting that dysregulation in membrane potential homeostasis could have a role in cancer onset and progression.
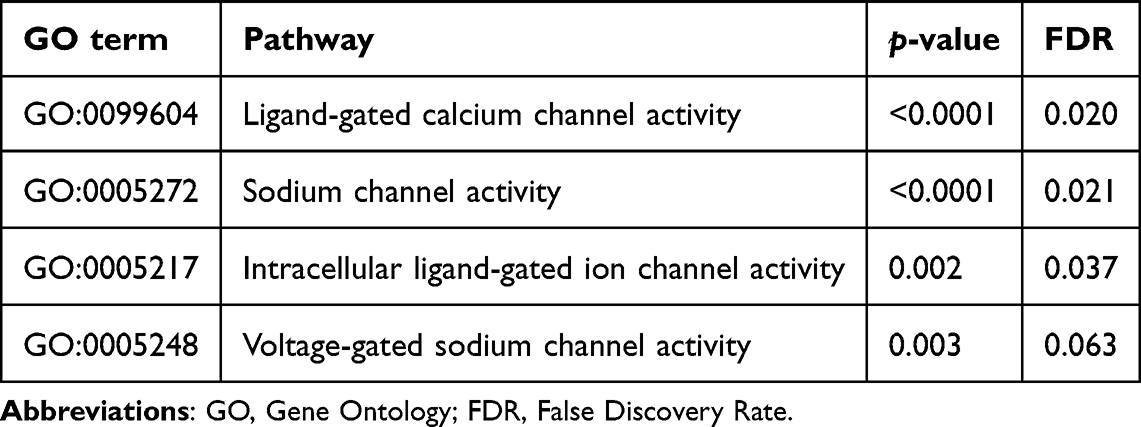 |
Table 2 Gene Set Enrichment Analysis of Transcriptome for Pathways Differentiating Smokers Patients versus Never-Smoker Patients. p-value and FDR are Specified for Each Pathway |
Discussion
Molecular profiles of SCC arising in never-smoker patients remain an unexplored area of research. In this study, we performed a wide molecular characterization including genomic and transcriptomic analyses to highlight which molecular features distinguish SCCs arising in never-smoker patients from the ones arising in patients with a history of smoking. The results of this study reveal distinct transcriptomic profiles between SCC in never-smoker and smoker patients. While the most commonly altered genes were similar between the two groups, including TP53, PIK3CA, and CDKN2A, there were notable differences observed. Based on the mutational profiles of the entire cohort, the most altered genes are comparable to the typical SCC profile.24
Molecular profiling of patients with NSCLC led to the identification of activating targetable mutations, and targeted therapy was demonstrated to confer a great benefit for patients carrying these mutations, even in the adjuvant setting.25–27 On the other hand, these mutations are exclusively present in lung ADC patients, while for lung SCC, only the SQUIRE trial demonstrated that the addition of necitumumab to gemcitabine confers a survival benefit for patients with higher EGFR expression.28 Interestingly, in our case series, we identified 3 targetable mutations in 3 different never-smoker patients (ie EGFR exon 20, MET exon 14 skipping and MET non canonical variant), highlighting that about 20% of never-smoker SCC patients could carry potential targetable mutations. On the other hand, we observed multiple FGF pathway alterations in 3 smokers, suggesting possible therapeutic implications also in this set of patients. In addition, in the genomic landscape of patients with SCC, the majority of mutated genes are TP53 and CDKN2A (90% and 70%, respectively), as reported in the review of key literature data performed by Roy S. Herbst et al.29 This finding is confirmed in our small cohort in which we were able to identify TP53 and CDKN2A as the most altered genes (67% and 20%, respectively).24 CDKN2A encodes for p16 and p14 proteins, and the loss of p16 leads to a disruption of the RB pathway, making CDKN2A a known tumor suppressor gene altered in lung SCC, whose mutations have been demonstrated to be associated with SCC patients’ prognosis when co-occurring with TP53 mutations.30,31 As expected, smokers had a significantly higher rate of TP53 mutations as they are associated with different squamous cell cancer histologies, especially in patients with smoking habits.32–34 As a consequence, smoker patients also carried a significantly higher rate of pathogenic alterations (67.1% vs 32.9, p = 0.004), resulting in a higher TMB (median 11 mut/Mb vs 5.5 mut/Mb, p = 0.028) for the accumulation of DNA damage and of gene alterations.35,36 The clinical significance of TMB in patients with SCC was highlighted as it has been demonstrated to be a strong predictor of better clinical outcomes, particularly in patients treated with single-agent immune checkpoint inhibitors.37 The landscape of mutations in driver and non-driver genes showed an urgent need for molecular evaluation of patients with SCC, allowing the improvement of their management and treatment options. By GO enrichment analysis, both groups showed enrichment of pathways related to PI3K, replicative senescence and regulation of cyclin-dependent kinases, suggesting that dysregulation of cell cycle control and escape from senescence are critical events in SCC pathogenesis, regardless of smoking status, as also demonstrated by the induction of apoptosis and senescence following the pharmacological inhibition of CDK4/6 in several types of solid tumors.38–40 Several PI3K signaling pathways were also enriched with gene alterations in both smokers and never-smokers. The PI3K/AKT/mTOR pathway is a key regulator of cell growth, proliferation, and survival of many cancers, including SCC. Targeting this pathway may represent another therapeutic opportunity for patients with SCC, and several clinical trials are nowadays evaluating PI3K target molecules (NCT: NCT02785913; NCT: 03065062). Defects in PI3K/AKT/mTOR signaling pathways were associated with enhanced cellular responsiveness to irradiation and chemotherapy, which balance the effect of mutations, which can be translated into second events, which tend to undermine the balancing mechanism against tumors.41,42
RNA analysis demonstrated that lung SCC also exhibits distinct transcriptomic characteristics based on smoking history, suggesting that both genomic and transcriptomic changes play critical roles in shaping the tumor microenvironment and influencing treatment outcomes. By unsupervised clustering analysis, we successfully discriminated between never-smoker and smoker patients based on their transcriptomic profiles, leading to the identification of 277 genes that were significantly associated with smoking status. Interestingly, the analysis revealed a specific subgroup of smoker patients whose transcriptomic features were more closely aligned with those of never-smokers; in terms of expressed proteins, this association was already seen, as reported by Jacqueline M. Vink et al.43 Interestingly, former smokers were clustered as an intermediate transcriptomic profile between current and never-smokers, suggesting that smoking cessation is directly reflected in tumor transcriptomics plasticity of gene expression. Then, this plasticity underscores the potential of gene expression profiles after quitting smoking, which could influence tumor behavior and treatment responses.44,45
On tumor RNA, we investigated the tumor-enriched pathways by GSEA, identifying key pathways related to cell membrane potential and ion homeostasis, particularly involving calcium and sodium channel activity, while a fourth pathway linked to these was reported with a strong trend (GO:0099604, GO:0005272, GO:0005217, GO:0005248). These pathways are crucial in maintaining cellular homeostasis and signaling. Ion channels are known to be involved in lung carcinoma, exhibiting different expression patterns compared to normal tissue.46 In particular, sodium channels are transmembrane proteins that regulate action potentials, neuronal excitability, and ion transport,47,48 and the targeting of these channels could lead to the identification of new potential biomarkers and therapeutic targets.49 The significant function of calcium signaling in lung cancer has also been documented. The expression levels of certain specific Ca2+ channels and calcium-binding proteins are modified in lung cancer, thereby influencing cell proliferation, metastasis, apoptosis, and tumor development.50,51 Recent research highlights the crucial role of Mitofusin-2 (MFN2) in regulating mitochondrial function and calcium homeostasis in lung adenocarcinoma, suggesting its potential as a therapeutic target.52 Additionally, the selective T-type Ca²+ channel blocker KYS05047 has demonstrated anti-proliferative effects in A549 lung cancer cells, indicating that targeting calcium signaling pathways could be an effective strategy for treating this malignancy.53 The transcriptomic profiles are an emerging effective tool to better characterize tumor at a molecular level, providing new insights for new treatment strategies, and could be included in prognostic and predictive tools to patients stratification. In the context of lung cancer, this evidence has been achieved for Small-Cell Lung Cancer molecular subtyping, and the translation of this approach to other lung cancer subtypes orphans of targetable markers could revolutionize treatment algorithms.54 In summary, ion channels, particularly sodium and calcium channels, play a crucial role in various aspects of lung cancer pathogenesis, including tumor invasion, progression, and treatment-related side effects, underscoring their potential as valuable biomarkers and therapeutic targets for LC. On the other hand, several studies have reported ion channel overexpression in SCC, but the ion channel overexpression in never-smoker SCC patients is not fully understood.46,55 Understanding these mechanisms may provide insights into targeted therapies and improved patient management strategies.
The major study limitations are related to the retrospective nature of patient enrolment, and to the number of patients enrolled, which needs to be larger in confirmation studies. On the other hand, SCC is not commonly diagnosed in never-smokers, limiting the enrolment power. Moreover, smoking habits, in terms of time from smoking cessation for former smokers and pack/years could influence molecular features, as also highlighted by our transcriptomic profiling; further studies including a higher number of patients and considering clinical variables of different smoking habits are warranted to confirm the first results of this exploratory study.
Conclusion
In this study, we showed that about 20% of never-smoker SCC patients carry a potentially targetable alteration, suggesting the importance, in this subset of patients, of a molecular characterization as for ADC patients. We also confirmed the involvement of the PI3K/AKT/mTOR pathway in SCC independently of smoking habits, reinforcing the utility of PIK3 inhibitors development as a potential treatment strategy in this disease. Moreover, we highlighted that SCCs arising in never-smokers display peculiar transcriptomics profiles, suggesting that smoking habits influence tumor phenotype.
Abbreviations
ADC, adenocarcinoma; CNVs, copy number variants; FFPE, Formalin-fixed paraffin-embedded; GO, Gene Ontology; LC, IQ, first quartiles; IIIQ, third quartiles; Lung cancer; MSI, microsatellite instability; NGS next generation sequencing; NSCLC, non small cell lung cancer; SCC, Lung Squamous Cell Carcinoma; SNVs, single nucleotide variants; TTF-1, thyroid transcription factor; TMB, Tumor mutation burden; TSO500, TruSight™ Oncology 500; VAF, variant allele frequency; VST, Variance Stabilizing Transformation; VUS, variants of uncertain significance.
Ethics Approval and Consent to Participate
All methods used in our studies involving human participants adhered strictly to the ethical guidelines of our institutional research committee, aligning with the principles outlined in the 1964 helsinki declaration and its subsequent modifications, or equivalent ethical norms. The protocol was approved by C.E.ROM. Ethical Committee (study code IRST-B045) All patients considered in the present study signed written informed consent.
Acknowledgments
This work was supported thanks to the contribution of Ricerca Corrente by the Italian Ministry of Health within the research line “Precision, gender and ethnicity-based medicine and geroscience: genetic-molecular mechanisms in the development, characterization and treatment of tumors”.
The abstract of this paper has been uploaded to ESMO Open website (https://www.esmoopen.com/) as a preprint (https://www.esmoopen.com/article/S2059-7029(24)01635-1/fulltext).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74(3):229–263. doi:10.3322/caac.21834
2. Davidson MR, Gazdar AF, Clarke BE. The pivotal role of pathology in the management of lung cancer. J Thorac Dis. 2013;5 Suppl 5(Suppl 5):S463–78. doi:10.3978/j.issn.2072-1439.2013.08.43
3. Relli V, Trerotola M, Guerra E, Alberti S. Abandoning the notion of non-small cell lung cancer. Trends Mol Med. 2019;25(7):585–594. doi:10.1016/j.molmed.2019.04.012
4. Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553(7689):446–454. doi:10.1038/nature25183
5. de Groot P, Munden RF. Lung cancer epidemiology, risk factors, and prevention. Radiol Clin North Am. 2012;50(5):863–876. doi:10.1016/j.rcl.2012.06.006
6. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 world health organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol. 2015;10(9):1243–1260. doi:10.1097/JTO.0000000000000630
7. Kadota K, Nitadori JI, Woo KM, et al. Comprehensive pathological analyses in lung squamous cell carcinoma: single cell invasion, nuclear diameter, and tumor budding are independent prognostic factors for worse outcomes. J Thorac Oncol. 2014;9(8):1126–1139. doi:10.1097/JTO.0000000000000253
8. Sánchez-Danés A, Blanpain C. Deciphering the cells of origin of squamous cell carcinomas. Nat Rev Cancer. 2018;18(9):549–561. doi:10.1038/s41568-018-0024-5
9. Girard L, Rodriguez-Canales J, Behrens C, et al. An expression signature as an aid to the histologic classification of non-small cell lung cancer. Clin Cancer Res. 2016;22(19):4880–4889. doi:10.1158/1078-0432.CCR-15-2900
10. Hou J, Aerts J, den Hamer B, et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS One. 2010;5(4):e10312. doi:10.1371/journal.pone.0010312
11. Liu J, Yang XY, Shi WJ. Identifying differentially expressed genes and pathways in two types of non-small cell lung cancer: adenocarcinoma and squamous cell carcinoma. Genet Mol Res. 2014;13(1):95–102. doi:10.4238/2014.January.8.8
12. Alberg AJ, Brock MV, Ford JG, Samet JM, Spivack SD. Epidemiology of lung cancer: diagnosis and management of lung cancer, 3rd ed: American college of chest physicians evidence-based clinical practice guidelines. Chest. 2013;143(5 Suppl):e1S–e29S. doi:10.1378/chest.12-2345
13. Alduais Y, Zhang H, Fan F, Chen J, Chen B. Non-small cell lung cancer (NSCLC): a review of risk factors, diagnosis, and treatment. Medicine. 2023;102(8):e32899. doi:10.1097/MD.0000000000032899
14. Peto R, Darby S, Deo H, Silcocks P, Whitley E, Doll R. Smoking, smoking cessation, and lung cancer in the UK since 1950: combination of national statistics with two case-control studies. BMJ. 2000;321(7257):323–329. doi:10.1136/bmj.321.7257.323
15. Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101(36):13306–13311. doi:10.1073/pnas.0405220101
16. Sun S, Schiller JH, Gazdar AF. Lung cancer in never smokers--a different disease. Nat Rev Cancer. 2007;7(10):778–790. doi:10.1038/nrc2190
17. Hirsch FR, Spreafico A, Novello S, Wood MD, Simms L, Papotti M. The prognostic and predictive role of histology in advanced non-small cell lung cancer: a literature review. J Thorac Oncol. 2008;3(12):1468–1481. doi:10.1097/JTO.0b013e318189f551
18. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
19. Kopanos C, Tsiolkas V, Kouris A, et al. VarSome: the human genomic variant search engine. Bioinformatics. 2019;35(11):1978–1980. doi:10.1093/bioinformatics/bty897
20. Jung J, Heo YJ, Park S. High tumor mutational burden predicts favorable response to anti-PD-(L)1 therapy in patients with solid tumor: a real-world pan-tumor analysis. J Immunother Cancer. 2023;11(4):e006454. doi:10.1136/jitc-2022-006454
21. Wei B, Kang J, Kibukawa M, et al. Evaluation of the TruSight oncology 500 assay for routine clinical testing of tumor mutational burden and clinical utility for predicting response to pembrolizumab. J Mol Diagn. 2022;24(6):600–608. doi:10.1016/j.jmoldx.2022.01.008
22. Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32(18):2847–2849. doi:10.1093/bioinformatics/btw313
23. Tang D, Chen M, Huang X, et al. SRplot: a free online platform for data visualization and graphing. PLoS One. 2023;18(11):e0294236. doi:10.1371/journal.pone.0294236
24. Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–525. doi:10.1038/nature11404.
25. Ramalingam SS, Yang JCH, Lee CK, et al. Osimertinib as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer. J Clin Oncol. 2018;36(9):841–849. doi:10.1200/JCO.2017.74.7576
26. Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–1703. doi:10.1056/NEJMoa1006448
27. Wu YL, Tsuboi M, He J, et al. Osimertinib in resected EGFR-mutated non-small-cell lung cancer. N Engl J Med. 2020;383(18):1711–1723. doi:10.1056/NEJMoa2027071
28. Thatcher N, Hirsch FR, Luft AV, et al. Necitumumab plus gemcitabine and cisplatin versus gemcitabine and cisplatin alone as first-line therapy in patients with stage IV squamous non-small-cell lung cancer (SQUIRE): an open-label, randomised, controlled Phase 3 trial. Lancet Oncol. 2015;16(7):763–774. doi:10.1016/S1470-2045(15)00021-2
29. Herbst RS, Gandara DR, Hirsch FR, et al. Lung master protocol (Lung-MAP)-A biomarker-driven protocol for accelerating development of therapies for squamous cell lung cancer: SWOG S1400. Clin Cancer Res. 2015;21(7):1514–1524. doi:10.1158/1078-0432.CCR-13-3473
30. Wikman H, Kettunen E. Regulation of the G1/S phase of the cell cycle and alterations in the RB pathway in human lung cancer. Expert Rev Anticancer Ther. 2006;6(4):515–530. doi:10.1586/14737140.6.4.515
31. Wang P, Wang F, He H, et al. TP53 and CDKN2A mutations in patients with early-stage lung squamous cell carcinoma: an analysis of the correlations and prognostic outcomes. Ann Transl Med. 2021;9(16):1330. doi:10.21037/atm-21-3709
32. Ahrendt SA, Chow JT, Yang SC, et al. Alcohol consumption and cigarette smoking increase the frequency of p53 mutations in non-small cell lung cancer. Cancer Res. 2000;60(12):3155–3159.
33. Brennan JA, Boyle JO, Koch WM, et al. Association between cigarette smoking and mutation of the p53 gene in squamous-cell carcinoma of the head and neck. N Engl J Med. 1995;332(11):712–717. doi:10.1056/NEJM199503163321104
34. Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. doi:10.1038/nature12634
35. Smolle E, Pichler M. Non-smoking-associated lung cancer: a distinct entity in terms of tumor biology, patient characteristics and impact of hereditary cancer predisposition. Cancers. 2019;11(2):204. doi:10.3390/cancers11020204
36. Derman BA, Mileham KF, Bonomi PD, Batus M, Fidler MJ. Treatment of advanced squamous cell carcinoma of the lung: a review. Transl Lung Cancer Res. 2015;4(5):524–532. doi:10.3978/j.issn.2218-6751.2015.06.07
37. Xu Y, Li H, Huang Z, et al. Predictive values of genomic variation, tumor mutational burden, and PD-L1 expression in advanced lung squamous cell carcinoma treated with immunotherapy. Transl Lung Cancer Res. 2020;9(6):2367–2379. doi:10.21037/tlcr-20-1130
38. Wagner V, Gil J. Senescence as a therapeutically relevant response to CDK4/6 inhibitors. Oncogene. 2020;39(29):5165–5176. doi:10.1038/s41388-020-1354-9
39. Anders L, Ke N, Hydbring P, et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011;20(5):620–634. doi:10.1016/j.ccr.2011.10.001
40. Niu Z, Jin R, Zhang Y, Li H. Signaling pathways and targeted therapies in lung squamous cell carcinoma: mechanisms and clinical trials. Signal Transduct Target Ther. 2022;7(1):353. doi:10.1038/s41392-022-01200-x
41. Brognard J, Clark AS, Ni Y, Dennis PA. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 2001;61(10):3986–3997.
42. Schuurbiers OCJ, Kaanders JHAM, van der Heijden HFM, Dekhuijzen RPN, Oyen WJG, Bussink J. The PI3-K/AKT-pathway and radiation resistance mechanisms in non-small cell lung cancer. J Thorac Oncol. 2009;4(6):761–767. doi:10.1097/JTO.0b013e3181a1084f
43. Vink JM, Jansen R, Brooks A, et al. Differential gene expression patterns between smokers and non‐smokers: cause or consequence? Addict Biol. 2017;22(2):550–560. doi:10.1111/adb.12322
44. Conte G, Pacino SA, Urso S, et al. Changes in oral health and dental esthetic in smokers switching to combustion-free nicotine alternatives: protocol for a multicenter and prospective randomized controlled trial. JMIR Res Protoc. 2024;13:e53222. doi:10.2196/53222
45. Williams SA, Kivimaki M, Langenberg C, et al. Plasma protein patterns as comprehensive indicators of health. Nat Med. 2019;25(12):1851–1857. doi:10.1038/s41591-019-0665-2
46. Ko JH, Gu W, Lim I, Bang H, Ko EA, Zhou T. Ion channel gene expression in lung adenocarcinoma: potential role in prognosis and diagnosis. PLoS One. 2014;9(1):e86569. doi:10.1371/journal.pone.0086569
47. Chen-Izu Y, Shaw RM, Pitt GS, et al. Na+ channel function, regulation, structure, trafficking and sequestration. J Physiol. 2015;593(6):1347–1360. doi:10.1113/jphysiol.2014.281428
48. Cardoso FC, Lewis RJ. Sodium channels and pain: from toxins to therapies. Br J Pharmacol. 2018;175(12):2138–2157. doi:10.1111/bph.13962
49. Roger S, Rollin J, Barascu A, et al. Voltage-gated sodium channels potentiate the invasive capacities of human non-small-cell lung cancer cell lines. Int J Biochem Cell Biol. 2007;39(4):774–786. doi:10.1016/j.biocel.2006.12.007
50. Yang H, Zhang Q, He J, Lu W. Regulation of calcium signaling in lung cancer. J Thorac Dis. 2010;2(1):52–56.
51. Cui C, Merritt R, Fu L, Pan Z. Targeting calcium signaling in cancer therapy. Acta Pharm Sin B. 2017;7(1):3–17. doi:10.1016/j.apsb.2016.11.001
52. Zhang J, Pan L, Zhang Q, et al. MFN2 deficiency affects calcium homeostasis in lung adenocarcinoma cells via downregulation of UCP4. FEBS Open Bio. 2023;13(6):1107–1124. doi:10.1002/2211-5463.13591
53. Rim HK, Lee HW, Choi IS, et al. T-type Ca2+ channel blocker, KYS05047 induces G1 phase cell cycle arrest by decreasing intracellular Ca2+ levels in human lung adenocarcinoma A549 cells. Bioorg Med Chem Lett. 2012;22(23):7123–7126. doi:10.1016/j.bmcl.2012.09.076
54. Gay CM, Stewart CA, Park EM. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell. 2021;39(3):346–360. doi:10.1016/j.ccell.2020.12.014
55. Huang Z, Sun Z, Zhang X, et al. Loss of stretch-activated channels, PIEZOs, accelerates non-small cell lung cancer progression and cell migration. Biosci Rep. 2019;39(3). doi:10.1042/BSR20181679
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.