")
Back to Journals » OncoTargets and Therapy » Volume 11
Genome-wide profiling reveals cancer-related genes with switched alternative polyadenylation sites in colorectal cancer
Authors Yang X, Wu J, Xu W, Tan S, Chen C , Wang X, Sun J, Kang Y
Received 2 February 2018
Accepted for publication 28 May 2018
Published 31 August 2018 Volume 2018:11 Pages 5349—5357
DOI https://doi.org/10.2147/OTT.S164233
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Arseniy Yuzhalin
Xiaochen Yang,1 Jun Wu,1 Wei Xu,1 Sheng Tan,2 Changyu Chen,3 Xiaoyan Wang,4 Jielin Sun,4 Yani Kang1
1School of Biomedical Engineering, Bio-ID Center, Shanghai Jiao Tong University, Shanghai, 200240, People’s Republic of China; 2The CAS Key Laboratory of Innate Immunity and Chronic Disease, School of Life Sciences, University of Science and Technology of China, Hefei, Anhui 230027, People’s Republic of China; 3Department of General Surgery, The First Affiliated Hospital of Anhui University of Traditional Chinese Medicine, Hefei, Anhui, 230031, People’s Republic of China; 4Key Laboratory of Systems Biomedicine (Ministry of Education), Shanghai Center for Systems Biomedicine, Shanghai Jiao Tong University, Shanghai, 200240, People’s Republic of China
Background: Alternative polyadenylation (APA) is an important post-transcriptional regulation in eukaryotic cells. It plays considerable roles in many biological processes and diseases, such as cell differentiation, proliferation and cancer. Colorectal cancer (CRC) is one of the most common malignancies worldwide, which is among the top five in incidence and mortality of all cancers in China. Although there have been some studies on the APA of CRC, the normal and carcinoma samples used for genome-wide profiling were not matched. The purpose of this study was to obtain genes with switched 3'-untranslated region (UTR) that may be associated with intracellular regulation of CRC by analyzing APA patterns of strict control groups from clinical patients.
Materials and methods: CRC and matched normal tissues were acquired from surgical specimens from three CRC patients. Their libraries of 3'-terminal fragments of mRNA with poly(A) tails were constructed by 3T-seq technology and sequenced by Illumina Hiseq X Ten. APA patterns of cancer and matched normal tissues were analyzed by bioinformatics analysis, and a representative gene, GPI, was verified by quantitative reverse transcription PCR.
Results: Overall, we identified 35,076 poly(A) sites in total. Compared to the matched normal tissues, we detected 350, 405 and 375 genes with significantly APA-mediated 3'-UTR alteration in cancer tissues of three patients, respectively. Forty-seven genes with switched 3'-UTR were shared in all three patients. In addition, most of these genes have shortened 3'-UTRs, some of which were associated with cancers, such as GPI.
Conclusion: Our studies found several genes with switched 3'-UTR in CRC patients, which may provide some important clues for more in-depth study of the cellular regulation in CRC from the perspective of post-transcriptional regulation. It may also help in the search for new biomarkers of CRC.
Keywords: alternative polyadenylation, CRC, 3T-seq, 3'-UTR
Introduction
Almost all eukaryotic pre-messenger RNAs (pre-mRNAs) and a portion of noncoding transcripts have poly(A) sites (PAS) and are polyadenylated.1–3 Recent discoveries have found that more than two-thirds of all genes have multiple PAS,1 which means that alternative polyadenylation (APA) can take place. APA can be typically divided into the following two categories: untranslated region (UTR)-APA and coding region (CR)-APA.4 APA is extensively used to regulate gene expression by producing transcript isoforms with diverse 3′-UTRs. Alternative 3′-UTR may affect stability, cellular localization and translation efficiency of transcript isoforms, even if the encoded proteins have the same sequences.5,6 Recent studies have shown that APA alterations are displayed in a series of biological processes, such as development, cell differentiation, proliferation, neuron activity and cancer.7
A study has shown that ~70% of protein-coding genes have conserved microRNA (miRNA) target sites within their 3′-UTRs.1 During transformation, the cell starts using the PAS most proximal to the open reading frame (ORF) to generate a short 3′-UTR, which makes the mRNA resistant to miRNA by eliminating miRNA-binding sites.8 A recent study reported a widespread preferential usage of proximal PAS in cancers, such as breast, lung, liver and colorectal cancers (CRCs),2 even if the role of APA in transformation and cancer is still not very clear.
CRC, which is one of the most common malignancies worldwide, remains among the top five in incidence and mortality of all cancers in China, even though it is not a high prevalence area in comparison with Western Europe and North America.9 Although Morris et al10 have studied APA in CRC patients, their normal and carcinoma samples in the high-throughput sequencing data used for analysis were not matched. The establishment of the control group in that study was not sufficiently rigorous, which may influence the accuracy of APA sites obtained through high-throughput sequencing.
In this study, we used a previous published method, the 3T-seq technology, to profile genome-wide APA sites in CRC patients to analyze the effects of this kind of post-transcriptional modification on CRC. Comparisons between cancer samples and the matched normal samples help to understand the role of APA in clinical patients.
Materials and methods
Collection of human tissue samples
Fresh tissue samples from three CRC patients were collected at the Anhui Medical University, First Affiliated Hospital, Anhui, China. This study plan was approved by the institutional review boards of Anhui Medical University and was carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki). All patients had signed an informed consent form. All samples were examined by one experienced pathologist, and clinical information of all individuals are listed in Table 1. After being collected, the samples were put in liquid nitrogen quickly and preserved in low temperature environment. TRIzol (Thermo Fisher Scientific, Waltham, MA, USA) was used to isolate total RNA from tissue samples following the manufacturer’s protocol. Total RNA quantity was determined with Nanodrop 2000 (Thermo Fisher Scientific), and quality was assessed by running a 1% agarose gel electrophoresis, stained with 4S Red Plus Nucleic Acid Stain (Sango, Shanghai, China).
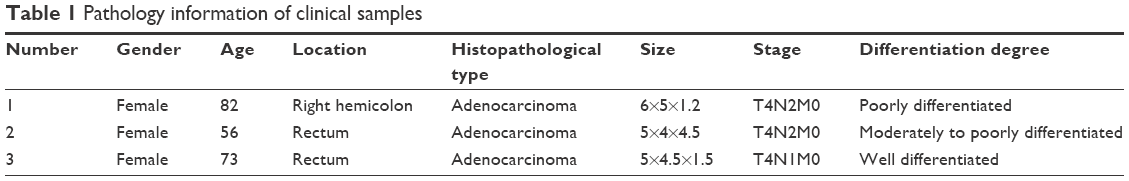 | Table 1 Pathology information of clinical samples |
3T-seq library preparation
The libraries were prepared following the previous published method.11 In general, after being extracted, ~50 μg of total RNA was incubated with Dynabeads™ M-280 streptavidin (Thermo Fisher Scientific) with biotin-modified reverse transcription GsuI-Oligo (dT)20 primers. Catalyzed by Super Script III (Thermo Fisher Scientific), the first strand was synthesized, in which 5-methylated-dCTP had replaced dCTP to prevent GsuI from cutting the new synthetic chain. When the second strand of cDNA chain was synthesized, dNTP mixture contained dUTP instead of dTTP. Then, cDNA was randomly fragmented to ~200–400 bp with Fragmentase (NEB, Ipswich, MA, USA). After that 3′-terminal fragments were released from beads by GsuI (Fermentas, Waltham, MA, USA) digestion. Next, Illumina p5/p7 adaptors were ligated to the released cDNA. Before PCR amplification, the second strand of cDNA which had dUTP was digested by USER (NEB) to achieve chain specificity sequencing. Finally, a series of 3T-seq libraries were sequenced by Illumina Hiseq X Ten. Raw sequence data have been submitted to the EMBL-EBI ArrayExpress (accession number: E-MTAB-6403).
Data analysis
The data analysis process is similar to our previous study.11 Briefly, raw reads were filtered with custom C++ scripts to obtain usable and valid reads. The selected reads were then mapped to the UCSC human reference genome (hg19) with bowtie2.12 The PAS were identified through iteratively clustering the neighbor sites. The linear trend test was employed to identify the genes with significantly switched 3′-UTR. The function annotation was performed with the online tool DAVID (https://david.ncifcrf.gov).
Quantitative reverse transcription PCR validation
One microgram of total RNA was reverse transcripted by using PrimeScript™ RT reagent kit (TAKARA, Kyoto, Japan). RT primer mixture used in reverse transcription was a mixture of oligo dT and random hexamer primers. qRT-PCR was performed with PowerUp SYBR Green Master Mix (Thermo Fisher Scientific). Reactions were run in triplicate and normalized against ACTB. Primers design and data analysis were based on those in a previous study.13 Primer sequences are shown in Table 2.
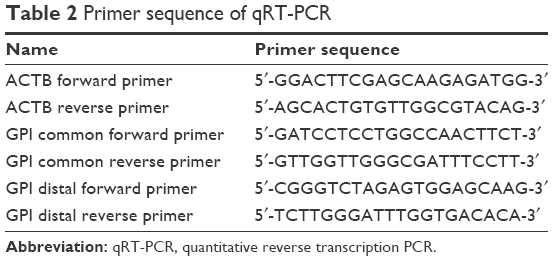 | Table 2 Primer sequence of qRT-PCR |
Results
Identification of polyadenylation sites
After total RNA was extracted, we constructed libraries for sequencing by using the 3T-seq technology.11 The 3′-terminal fragments were subjected to deep sequencing by Illumina Hiseq X Ten. Data from high-throughput sequencing were exhibited on Integrative Genomics Viewer (IGV) software (Figure 1). Totally, we generated ~107 million reads, of which ~52% are uniquely mappable (Table 3). Possible polyadenylation sites, whose downstream 20 nt bases included continuous 8 or 12 or more adenine ribonucleic acids, were identified as internal priming and filtered out.14 Approximately 43 million reads passed internal priming filter, and most of them were concentrated near the annotated transcription termination sites (TTSs) (Figure 2A). Identification method of PAS was based on that in a previous study;15 totally, we identified 34,982 PAS in patient 1, 34,187 PAS in patient 2 and 35,303 PAS in patient 3. We examined the presence of a hexamer motif such as AAUAAA within 20–30 nt upstream of the identified sites and reported that motifs could be found in this interval (Figure 2B). For example, among the 34,982 identified PAS in patient 1, 13.7% were mapped to UCSC TTS and 42.1% to 3′-UTR regions (Figure 2C). Among expressed genes detected in this study, an average of 64.9% genes had three or more PAS in three patients (Figure 2D).
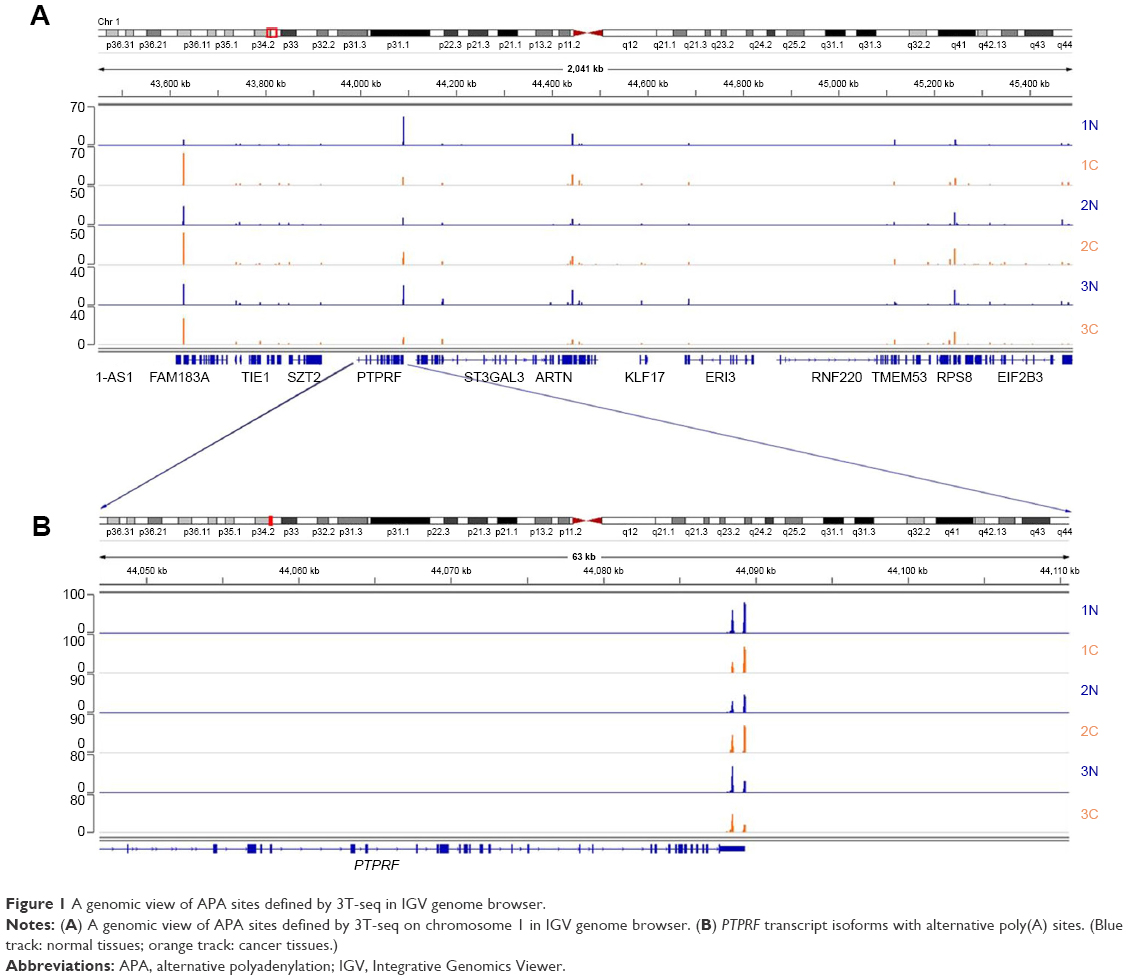 | Figure 1 A genomic view of APA sites defined by 3T-seq in IGV genome browser. |
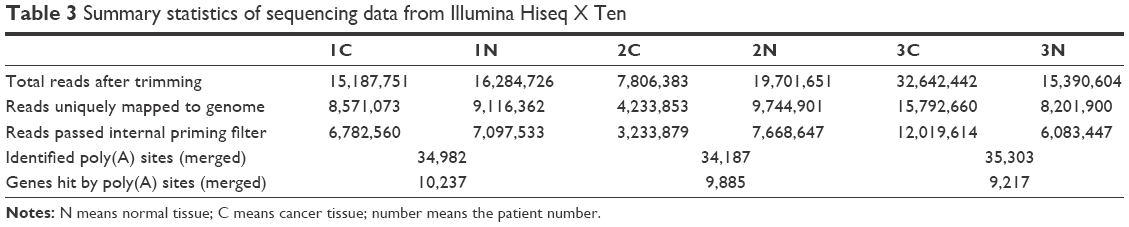 | Table 3 Summary statistics of sequencing data from Illumina Hiseq X Ten |
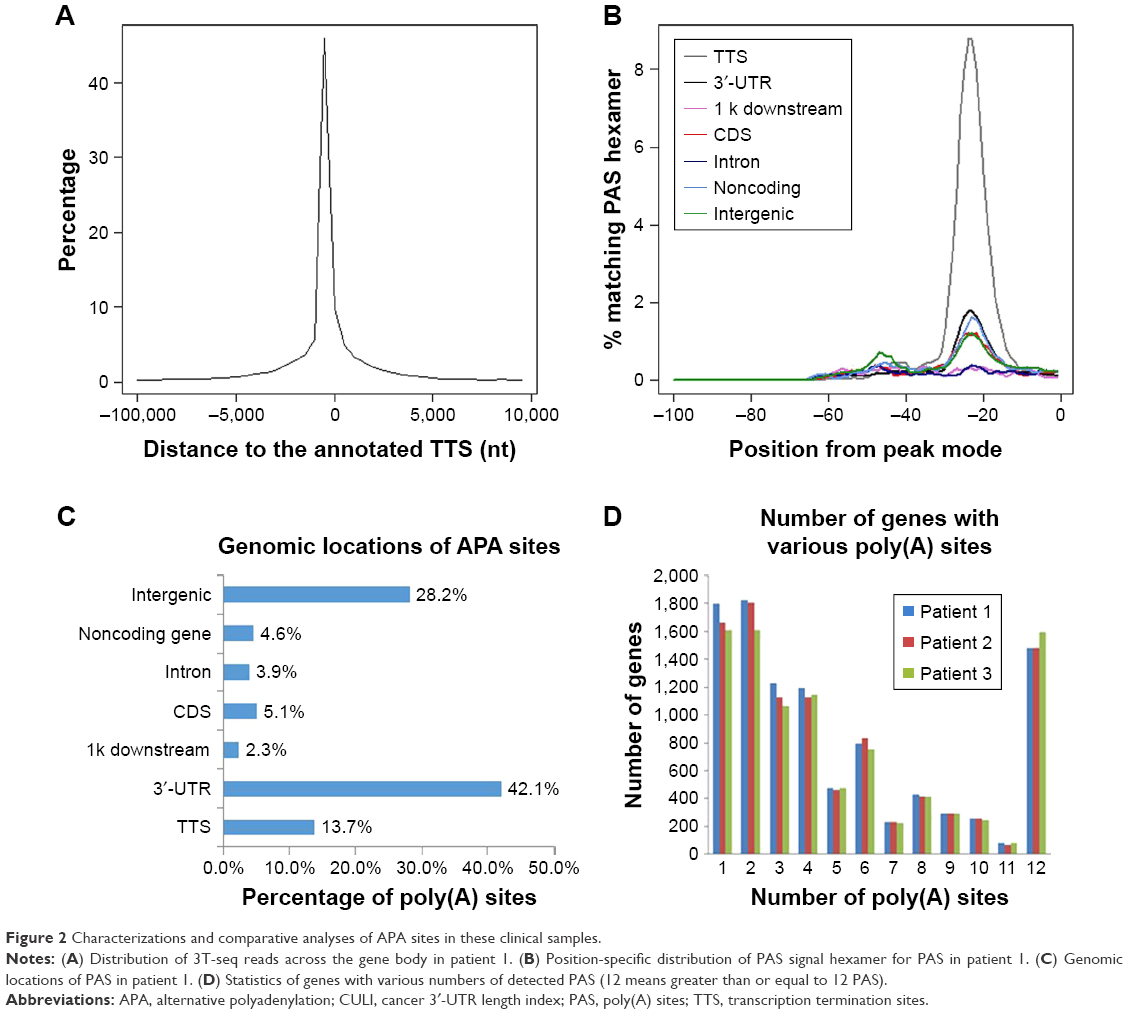 | Figure 2 Characterizations and comparative analyses of APA sites in these clinical samples. |
Variation analysis of APA between clinical samples
To facilitate comparison between samples, the cancer 3′-UTR length index (CULI) was adopted to quantitatively characterize the 3′-UTR alteration in CRC patients.15 A positive CULI means that a gene harbors lengthened 3′-UTR in cancer tissues compared with their corresponding normal tissues, and a negative CULI suggests the shortened one (Table S1). With this standard, the number of genes was identified with a significant difference in 3′-UTR length between cancer tissues and their corresponding normal tissues. In patient 1, the number was 350, and in patients 2 and 3, the numbers were 405 and 375, respectively (Figure 3A). In patient 1, 79.1% genes in cancer tissues had shortened 3′-UTR, while in other patients, the percentage became 88.1% (in patient 2) and 50.4% (in patient 3). To obtain the relationship between APA alteration and gene expression, we compared the APA change with mRNA abundance, and the result showed that there was no linear correlation on transcriptome scale (Figure 3B–D).
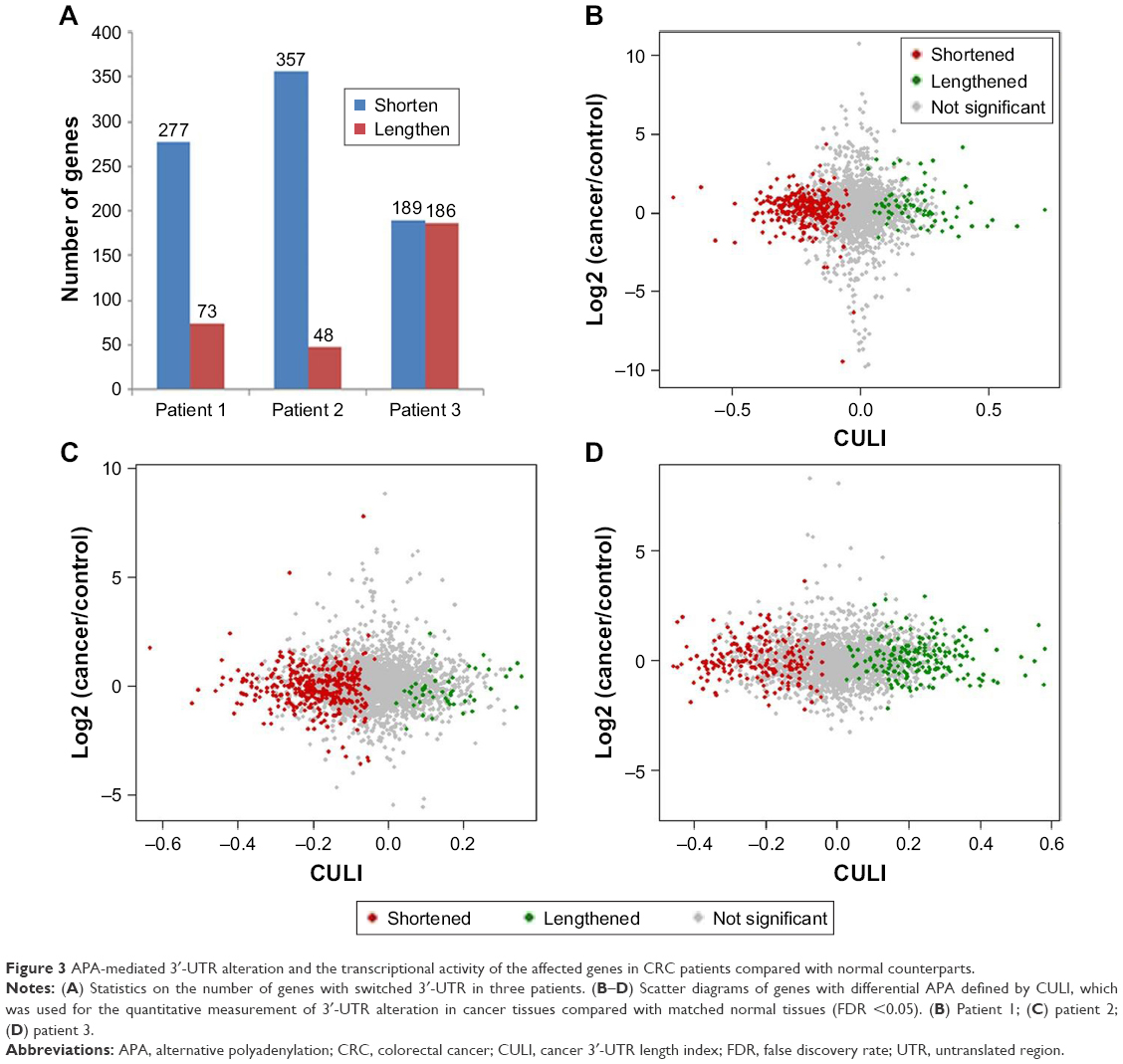 | Figure 3 APA-mediated 3′-UTR alteration and the transcriptional activity of the affected genes in CRC patients compared with normal counterparts. |
Functional enrichment of genes with switched APA sites
To understand the biological consequences of altered patterns in clinical CRC patients, genes with shortened 3′-UTR were analyzed by DAVID (https://david.cifcrf.gov). Analysis of 277 genes with 3′-UTR shortening in patient 1 yielded proliferation-related biological processes that were statistically overrepresented, including metabolic process, mRNA processing and RNA splicing (Figure 4A and Table S2). Similar results were also observed in other two patients (Figure 4B and C and Table S2).
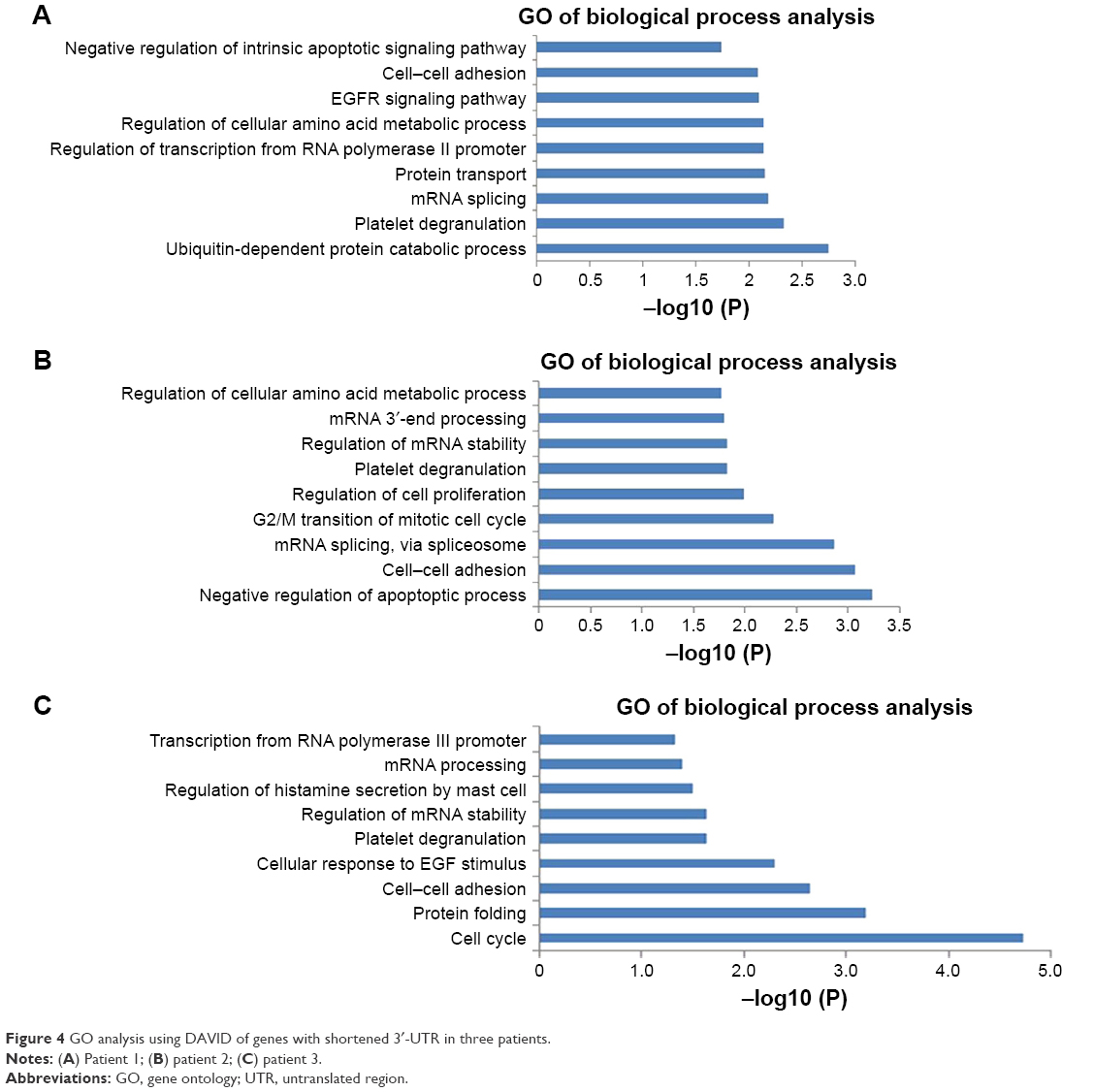 | Figure 4 GO analysis using DAVID of genes with shortened 3′-UTR in three patients. |
Genes associated with cancer preferentially use proximal APA in cancer samples
Because shortened 3′-UTR increased the stability of the mRNA and meanwhile reduced cis elements that can interact with the transcription regulation of trans function factor (such as RNA binding protein) or miRNA interaction, these genes were more likely to escape gene silencing induced by miRNA, which then leads to higher expression level.5 Based on this, genes that had switched APA patterns between cancer tissues and their corresponding normal tissues in three patients were further analyzed. Especially, some of them had a tendency to preferentially use proximal APA and higher expression level in cancer tissues.
There were 35 genes with shortened 3′-UTR present in all three patients. Gene ontology analysis showed that most of them are related to the metabolic process (Table S3). GPI, which was one of them, showed increased expression in cancer tissues in all three patients (Figure 5A). qRT-PCR was used to prove the shortening of 3′-UTR. The design of primers was based on a previous research, common primers targeted the ORF, and distal primers were located just before the distal PAS (Figure 5B).13 The relative use of the distal PAS was calculated, and genes preferentially used proximal APA in cancer tissues when the value was negative. Taking GPI as an example, qRT-PCR results showed that it tended to use proximal PAS in the cancer tissues of three patients (Figure 5C).
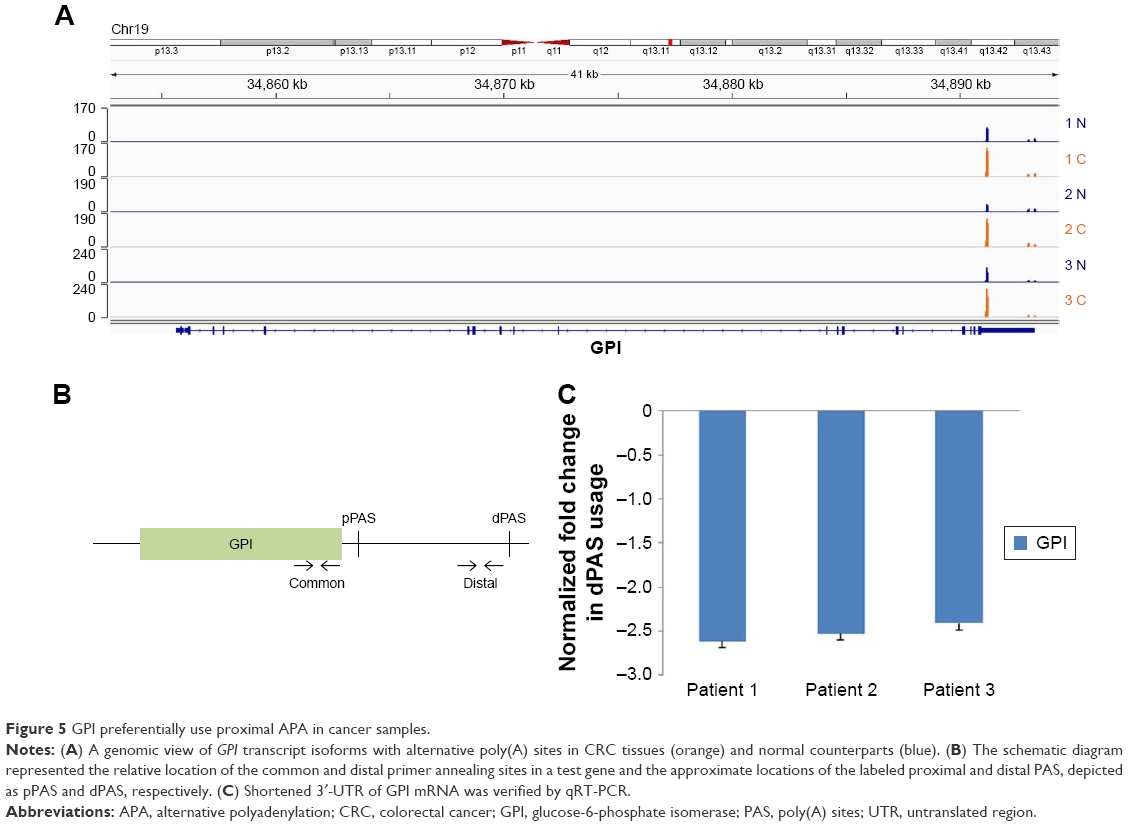 | Figure 5 GPI preferentially use proximal APA in cancer samples. |
Discussion
With an increasing number of studies reported, APA events are involved in various biological processes.7 APA can affect mRNA stability, translation and localization. The shortening of the 3′-UTR can eliminate miRNA-binding sites, which can be found in longer 3′-UTR and usually result in the escape of miRNA-regulated programmed cell death.22 The APA events in the whole genome, which may have a major impact on mechanisms of tumorigenesis and antitumor, can be investigated by means of genome-wide analyses. Through comparison of cancer tissue samples and their corresponding normal tissue samples, APA events in cancer tissue samples were found to have significant differences with APA patterns of normal tissue samples. There were 350 genes that have changed 3′-UTR in a cancer tissue sample of patient 1, and 79.1% of them had shortened 3′-UTR. The number of genes in the other two patients was 405 and 375, respectively, and 3′-UTR shortened genes accounted for 88.1% and 50.4%, respectively (Figure 2B). It was interesting to note that the proportion of genes with shortened 3′-UTR seemed to be consistent with the disease stage. However, cases were too few to draw from this conclusion.
3′-UTR shortening could increase gene expression level by eliminating miRNA-binding sites.22 However, in our data, genes with shortened 3′-UTR did not always have higher expression level. In patient 1, 180 of 277 genes had shortened 3′-UTR and higher level of expression in the cancer tissue. In patient 2, the number became 168 of 357, and it became 83 of 189 in patient 3. This suggested that the length and expression of 3′-UTR were not simply negatively correlated. In addition, similar phenomenon has been found in a previous study.15 The mechanism of the reduction of 3′-UTR to the expression level has not been fully understood.
Cancer tissues in three patients all preferentially used proximal APA of GPI. GPI, alternatively known as PGI or PHI, has been identified as the autocrine motility factor (AMF), which can regulate tumor cell growth and stimulate metastasis.16–18 Overexpression of AMF has been shown to induce epithelial-to-mesenchymal transition (EMT) in some cancers.19,20 Elevated serum GPI levels have been used as a prognostic biomarker for various cancers, including CRC.21 Nevertheless, 3′-UTR-shortened genes only partially overlapped in three patients. This may be due to the heterogeneity of individuals. When the number of samples increases, the heterogeneity may be more obvious. In recent studies, Morris et al10 also found a series of genes with changed PAS in CRC, some of which were overlapped with our results. However, 3T-seq was more focused on 3′-UTR PAS, and relative to 3′seq when paired-end sequencing was used; this method largely avoided sequencing desynchronization. More importantly, the normal control that Morris et al10 used in high-throughput sequencing was not sufficiently rigorous.
Conclusion
Briefly, in this study we used a robust approach, 3T-seq, to profile global APA sites in three patients and observed that hundreds of genes exhibit shortened 3′-UTR, and some of them have been reported to play a key role in cancer. Comparative results provide some clues for more in-depth study of the cell regulation mechanism of CRC from post-transcriptional regulation.
Acknowledgments
This work was supported by the Development Program for Basic Research of China (2014YQ09070904), National Natural Science Foundation of China (31671299), Shanghai Science and Technology Committee Program (17JC1400804), Medical Engineering Cross Fund (YG2017ZD15 and YG2015QN35) and Laboratory Innovative Research Program of Shanghai Jiao Tong University (17SJ-18).
Disclosure
The authors report no conflicts of interest in this work.
References
Derti A, Garrett-Engele P, Macisaac KD, et al. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012;22(6):1173–1183. | ||
Lin Y, Li Z, Ozsolak F, et al. An in-depth map of polyadenylation sites in cancer. Nucleic Acids Res. 2012;40(17):8460–8471. | ||
Shepard PJ, Choi EA, Lu J, Flanagan LA, Hertel KJ, Shi Y. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA. 2011;17(4):761–772. | ||
Rehfeld A, Plass M, Krogh A, Friis-Hansen L. Alterations in polyadenylation and its implications for endocrine disease. Front Endocrinol. 2013;4:53. | ||
Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science (New York, N. Y.). 2008;320(5883):1643–1647. | ||
Ji Z, Lee JY, Pan Z, Jiang B, Tian B. Progressive lengthening of 3′ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc Natl Acad Sci U S A. 2009;106(17):7028–7033. | ||
Elkon R, Ugalde AP, Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nat Rev Genet. 2013;14(7):496–506. | ||
Masamha CP, Wagner EJ. The contribution of alternative polyadenylation to the cancer phenotype. Carcinogenesis. 2017;39(1):2–10. | ||
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA: a cancer journal for clinicians. Mar. 2016;66(2):115–132. | ||
Morris AR, Bos A, Diosdado B, et al. Alternative cleavage and polyadenylation during colorectal cancer development. Clin Cancer Res. 2012;18(19):5256–5266. | ||
Lai DP, Tan S, Kang YN, et al. Genome-wide profiling of polyadenylation sites reveals a link between selective polyadenylation and cancer metastasis. Hum Mol Genet. 2015;24(12):3410–3417. | ||
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. | ||
Masamha CP, Xia Z, Yang J, et al. CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature. 2014;510(7505):412–416. | ||
Beaudoing E, Freier S, Wyatt JR, Claverie JM, Gautheret D. Patterns of variant polyadenylation signal usage in human genes. Genome Res. 2000;10(7):1001–1010. | ||
Fu Y, Sun Y, Li Y, et al. Differential genome-wide profiling of tandem 3′ UTRs among human breast cancer and normal cells by high-throughput sequencing. Genome Res. 2011;21(5):741–747. | ||
Stoker M, Gherardi E, Perryman M, Gray J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature. 1987;327(6119):239–242. | ||
Watanabe H, Takehana K, Date M, Shinozaki T, Raz A. Tumor cell autocrine motility factor is the neuroleukin/phosphohexose isomerase polypeptide. Cancer Res. 1996;56(13):2960–2963. | ||
Silletti S, Raz A. Autocrine motility factor is a growth factor. Biochem Biophys Res Commun. 1993;194(1):446–457. | ||
Tsutsumi S, Hogan V, Nabi IR, Raz A. Overexpression of the autocrine motility factor/phosphoglucose isomerase induces transformation and survival of NIH-3T3 fibroblasts. Cancer Res. 2003;63(1):242–249. | ||
Li Y, Che Q, Bian Y, et al. Autocrine motility factor promotes epithelial-mesenchymal transition in endometrial cancer via MAPK signaling pathway. Int J Oncol. 2015;47(3):1017–1024. | ||
Baumann M, Brand K, Matthias Baumann KB. Purification and characterization of phosphohexose isomerase from human gastrointestinal carcinoma and its potential relationship to neuroleukin. Cancer Res. 1988;48(24 Pt 1):7018–7021. | ||
Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138(4):673–684. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.