Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 21
Genetically Predicted Use of Common Analgesics and Risk of Chronic Obstructive Pulmonary Disease: A Bidirectional Mendelian Randomization Study
Received 8 December 2025
Accepted for publication 18 April 2026
Published 20 May 2026 Volume 2026:21 586858
DOI https://doi.org/10.2147/COPD.S586858
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Guojun Chen, Wanhui You
Department of Integrated Traditional Chinese and Western Medicine, Changde Hospital, Xiangya School of Medicine, Central South University (The First People’s Hospital of Changde City), Changde, Hunan, 415000 People’s Republic of China
Correspondence: Wanhui You, Department of Integrated Traditional Chinese and Western Medicine, Changde Hospital, Xiangya School of Medicine, Central South University (The First People’s Hospital of Changde City), Changde, Hunan, 415000, People’s Republic of China, Email [email protected]
Background: Observational studies suggest a link between common analgesic use and chronic obstructive pulmonary disease (COPD), but causality is unclear. This study aimed to investigate the bidirectional causal relationship between the genetically predicted use of paracetamol, aspirin, and ibuprofen, and COPD risk using a two-sample Mendelian randomization (MR) approach.
Methods: We performed a bidirectional MR study using summary statistics from large-scale genome-wide association studies (GWAS). Genetic instruments for paracetamol, aspirin, and ibuprofen use were from the UK Biobank (N=457,547). COPD summary statistics were from the FinnGen consortium (24,138 cases, 409,070 controls). The primary analysis used the inverse-variance weighted (IVW) method. We conducted extensive sensitivity analyses (MR-Egger, weighted median, weighted mode, MR-PRESSO) to assess pleiotropy and heterogeneity.
Results: Genetically predicted paracetamol use was significantly associated with an increased risk of COPD (IVW OR: 6.00, 95% CI: 2.43– 14.82, P < 0.001). Conversely, genetically predicted aspirin use was associated with a decreased risk of COPD (IVW OR: 0.19, 95% CI: 0.05– 0.71, P = 0.014). Genetically predicted ibuprofen use showed no significant association with COPD risk (IVW OR: 0.47, 95% CI: 0.06– 3.82, P = 0.483). In the reverse analysis, genetic liability to COPD was not causally associated with the use of any of the three analgesics (P > 0.05). Sensitivity analyses supported the robustness of these findings, showing no significant directional pleiotropy or heterogeneity for the primary results.
Conclusion: This MR study provides genetic evidence supporting a causal relationship between paracetamol use and an increased risk of COPD, and between aspirin use and a decreased risk. These findings suggest that the choice of analgesic may have important implications for COPD risk, warranting further clinical investigation. The diagram illustrates the Mendelian Randomization Approach using a DNA Helix and Genetic Instruments (SNPs). Three drugs are analyzed: Paracetamol, Aspirin and Ibuprofen. Paracetamol is linked to an increased COPD risk, shown by an arrow leading to lungs with COPD. Aspirin is associated with a decreased COPD risk, leading to healthy lungs. Ibuprofen shows no significant effect, leading to lungs without a specific condition. An arrow from lungs with COPD indicates reverse causation is not supported.A diagram showing the impact of drugs on COPD risk using Mendelian Randomization Approach.
Keywords: analgesics, chronic obstructive pulmonary disease, mendelian randomization, paracetamol, aspirin, ibuprofen, causal inference
Introduction
Chronic Obstructive Pulmonary Disease (COPD) is a major global health burden, affecting approximately 400 million people worldwide and ranking as the third leading cause of death.1 It is a progressive and largely irreversible lung condition characterized by persistent respiratory symptoms and airflow limitation, arising from abnormalities in the airways and/or alveoli, typically caused by significant exposure to noxious particles or gases.2 The pathophysiology of COPD is complex, involving chronic inflammation, oxidative stress, and an imbalance of proteases and antiproteases, leading to structural changes in the lungs such as emphysema and chronic bronchitis. Key risk factors for COPD are well-established and include tobacco smoking, exposure to biomass fuels, occupational dusts and fumes, and genetic predisposition.3,4 The high prevalence and mortality associated with COPD underscore the need for a better understanding of its etiology and modifiable risk factors.
Common analgesics, including paracetamol (acetaminophen), aspirin, and ibuprofen, are among the most widely used medications globally for pain and fever management. Given their widespread use, often as over-the-counter products, their potential long-term effects on chronic diseases have been a subject of interest. Observational studies have reported associations between the use of these analgesics and various respiratory conditions. For instance, increased use of paracetamol has been linked to a higher risk of both asthma and COPD, as well as reduced lung function, in a dose-dependent manner with an adjusted odds ratio of 1.16 (95% CI 1.09–1.24) for COPD per increasing category of intake.5 The proposed mechanism for paracetamol’s effect on the respiratory system involves the depletion of glutathione, a critical antioxidant in the lungs. This depletion can lead to increased oxidative stress, a key factor in the pathogenesis of COPD, as paracetamol use results in a dose-dependent decrease in pulmonary glutathione levels.6 For non-steroidal anti-inflammatory drugs (NSAIDs) like aspirin and ibuprofen, the mechanism of action involves the inhibition of cyclooxygenase (COX) enzymes, which can alter the balance of pro-inflammatory and anti-inflammatory eicosanoids. In susceptible individuals, this can lead to bronchospasm, and aspirin-exacerbated respiratory disease (AERD) is a well-described clinical syndrome characterized by asthma, chronic rhinosinusitis with nasal polyposis, and respiratory reactions upon exposure to COX-1 inhibitors.7
Despite these plausible biological mechanisms and observational findings, establishing a causal relationship between analgesic use and COPD risk is challenging. Observational studies are prone to limitations including confounding factors (eg., smoking, underlying health conditions), reverse causation, and exposure misclassification, which can introduce bias in estimating causal effects.8 The observational design raises concerns about residual confounding and reverse causality.9 Therefore, it remains unclear whether the observed associations are causal.
To overcome these challenges, we employed Mendelian randomization (MR), an analytical method that uses genetic variants as instrumental variables (IVs) to infer causal relationships between exposures and outcomes.10 The random assortment of genes at conception mimics a natural randomized trial, allowing MR to mitigate confounding and be less susceptible to reverse causation, thus providing more robust evidence for causality than traditional observational studies.
In this study, we employed a two-sample, bidirectional MR approach to investigate the causal relationship between the genetically predicted use of three common analgesics—paracetamol, aspirin, and ibuprofen—and the risk of COPD. We acknowledge that using genetic variants as proxies for medication use has inherent limitations, but it provides a valuable opportunity to examine long-term effects while minimizing traditional confounding from factors like smoking. We hypothesized that the use of these analgesics might causally influence COPD risk, and conversely, that a diagnosis of COPD might lead to altered patterns of analgesic use. By leveraging large-scale genome-wide association study (GWAS) summary statistics, this study aims to provide robust evidence on these potential causal links. Our findings could have significant implications for clinical practice and public health recommendations regarding pain management in individuals at risk of or with COPD.
Methods
Study Design
This study employed a two-sample, bidirectional Mendelian randomization (MR) design to investigate the causal relationships between the use of common analgesics (paracetamol, aspirin, and ibuprofen) and the risk of COPD. The MR method is predicated on three core assumptions for the genetic variants used as instrumental variables (IVs): (1) relevance – they must be strongly associated with the exposure of interest; (2) independence – they must not be associated with any confounders of the exposure-outcome relationship; and (3) exclusion restriction – they must affect the outcome only through the exposure, with no independent pathway (ie., no horizontal pleiotropy).10–12 In the forward MR analysis, we assessed the causal effect of analgesic use on COPD risk. In the reverse MR analysis, we assessed the causal effect of COPD on analgesic use. A bidirectional MR design further allows for the investigation of causality in both directions, helping to disentangle the temporal relationship between two associated traits.10 An overview of the study design is presented in Figure 1. All data used in this study were obtained from publicly available summary statistics from genome-wide association studies (GWAS), and thus, no new ethical approval was required.
 |
Figure 1 Study Design A directed acyclic graph (DAG) illustrating the bidirectional Mendelian randomization design used in this study. In the forward analysis, genetic instruments (SNPs) are used to assess the causal effect of analgesic use (paracetamol, aspirin, ibuprofen) on COPD risk. In the reverse analysis, genetic instruments are used to assess the causal effect of COPD on analgesic use. The design relies on the three core assumptions of MR: (1) the genetic variants are strongly associated with the exposure, (2) the genetic variants are not associated with confounders, and (3) the genetic variants influence the outcome only through the exposure. |
Data Sources and Selection of Genetic Instruments
Analgesic Use GWAS Data
Summary statistics for the use of paracetamol, aspirin, and ibuprofen were obtained from the UK Biobank GWAS dataset, available through the IEU OpenGWAS database.13 The specific GWAS IDs used were: ukb-b-17595 for paracetamol use, ukb-b-7137 for aspirin use, and ukb-b-8888 for ibuprofen use. These datasets are based on a European population of 457,547 individuals. The exposure phenotypes were based on self-reported use of each analgesic, treated as a binary trait.
For each analgesic, we selected single nucleotide polymorphisms (SNPs) as IVs that reached the genome-wide significance threshold (P < 5 × 10−8). To ensure the independence of the IVs, we performed clumping using a stringent linkage disequilibrium (LD) threshold of r2 < 0.001 within a 10,000 kb window, based on the European 1000 Genomes Project reference panel. We calculated the F-statistic for each SNP to assess instrument strength, using the formula F = (beta/se)2, where beta and se are the effect size and standard error of the SNP-exposure association, respectively. SNPs with an F-statistic less than 10 were considered weak instruments and were excluded from the analysis. The mean F-statistics for the selected instruments were well above 10 for all exposures, indicating sufficient strength to mitigate weak instrument bias. The proportion of variance (R2) in the exposure explained by each set of instruments was also calculated (details in Supplementary Table S1 and Table S2). Proxy SNPs were identified (LD r2 > 0.8) for any SNPs that were not present in the outcome GWAS data. Finally, we removed palindromic SNPs with intermediate allele frequencies (minor allele frequency > 0.42) to avoid ambiguity in aligning effect alleles between exposure and outcome datasets.
COPD GWAS Data
Summary-level GWAS data for COPD were obtained from the FinnGen consortium (Release 12), with the GWAS ID finngen_R12_J10_COPD.14 This dataset included 24,138 cases and 409,070 controls, totaling 433,208 participants of European ancestry. COPD cases were defined based on International Classification of Diseases (ICD) codes. The same procedures for IV selection—genome-wide significance, clumping, F-statistic calculation, and removal of weak and palindromic SNPs—were applied when COPD was treated as the exposure in the reverse MR analysis.15
Details of the GWAS summary statistics used in this study, including the number of instrumental variables for each analysis, are provided in Table 1.
 |
Table 1 Details of the GWAS Summary Statistics Used in the MR Analysis |
Statistical Analysis
The statistical analysis was performed using the “TwoSampleMR” package in R (version 4.2.1).
Primary Mendelian Randomization Analysis
The primary MR analysis was conducted using the random-effects inverse-variance weighted (IVW) method. The IVW method combines the Wald ratios (SNP-outcome effect / SNP-exposure effect) for each SNP in a meta-analysis, weighting each by the inverse of its variance. It provides the most precise causal estimate under the assumption that all IVs are valid or that any pleiotropy is balanced and not directional.16
Sensitivity Analyses
To assess the robustness of our findings and test for violations of the MR assumptions, several sensitivity analyses were performed.
MR-Egger Regression: This method can detect and adjust for directional pleiotropy. It is similar to the IVW method but allows the intercept term in the regression to be non-zero. A non-zero intercept is indicative of directional pleiotropy, and the slope of the regression provides a causal estimate adjusted for this pleiotropy. The validity of the MR-Egger estimate relies on the Instrument Strength Independent of Direct Effect (InSIDE) assumption.11
Weighted Median Estimator: This method provides a consistent causal estimate if at least 50% of the weight in the analysis comes from valid IVs. It is robust to the presence of some invalid instruments, making it less sensitive to outliers.17
Weighted Mode Estimator: This method is based on the assumption that the largest cluster of SNPs with similar causal effect estimates represents the true causal effect, making it robust to a plurality of invalid instruments.18
MR-Pleiotropy Residual Sum and Outlier (MR-PRESSO) Test: This method identifies horizontal pleiotropy by detecting outlier SNPs and provides a causal estimate after removing them. We used the MR-PRESSO global test to assess overall pleiotropy and, if significant, corrected the causal estimate by removing identified outliers.19
Heterogeneity and Pleiotropy Assessment
Cochran’s Q Test: Heterogeneity among the causal estimates from individual SNPs was assessed using Cochran’s Q statistic. Significant heterogeneity (P < 0.05) may indicate the presence of pleiotropy or other violations of MR assumptions.20
MR-Egger Intercept Test: The intercept from the MR-Egger regression was examined to test for directional pleiotropy. A P-value < 0.05 for the intercept suggests the presence of significant directional pleiotropy.
Leave-One-Out Analysis: We performed a leave-one-out sensitivity analysis by systematically removing one SNP at a time and re-estimating the causal effect. This helps to determine if the overall causal estimate is disproportionately influenced by a single SNP.
Funnel and Scatter Plots: We generated funnel plots and scatter plots to visually inspect for heterogeneity and potential pleiotropic outliers.
Multiple Testing Correction
For the primary analysis results, we applied a false discovery rate (FDR) correction to account for multiple testing across the different analgesics and analysis directions. An FDR-corrected P-value < 0.05 was considered statistically significant.
Results
Primary Analysis: Causal Effect of Analgesic Use on COPD Risk
The bidirectional MR estimates for the causal association between analgesic use and COPD risk are summarized in Table 2. The results of the primary IVW analysis indicated a statistically significant causal relationship between genetically predicted paracetamol use and an increased risk of COPD. For each standard deviation increase in the genetic predisposition to paracetamol use, the odds of developing COPD increased six-fold (OR: 6.00, 95% CI: 2.43–14.82, P < 0.001). This association remained significant after FDR correction (P-FDR < 0.001).
 |
Table 2 Bidirectional MR Estimates for the Causal Association Between Analgesic Use and COPD Risk |
In contrast, genetically predicted aspirin use was associated with a significantly reduced risk of COPD. The IVW analysis showed that genetic predisposition to aspirin use was associated with 81% lower odds of COPD (OR: 0.19, 95% CI: 0.05–0.71, P = 0.014). This finding also remained significant after FDR correction (P-FDR = 0.021).
For ibuprofen, the IVW analysis did not reveal a statistically significant causal association with COPD risk (OR: 0.47, 95% CI: 0.06–3.82, P = 0.483).
These results are visually represented in the forest plot in Figure 2.
 |
Figure 2 Forest plot of the causal effects of genetically predicted analgesic use on the risk of COPD. The plot shows the odds ratios (ORs) and 95% confidence intervals (CIs) from the inverse-variance weighted (IVW) analysis for the effect of paracetamol, aspirin, and ibuprofen use on COPD. The squares represent the OR, and the horizontal lines represent the 95% CI. |
Sensitivity Analyses for the Effect of Analgesics on COPD
The results from the sensitivity analyses (MR-Egger, weighted median, and weighted mode) were largely consistent with the primary IVW findings, although with wider confidence intervals.
For paracetamol, the weighted median (OR: 5.47, 95% CI: 1.75–17.12, P = 0.004) and weighted mode (OR: 5.88, 95% CI: 1.15–30.12, P = 0.051) methods provided estimates of similar direction and magnitude to the IVW analysis, supporting a positive causal association. The MR-Egger analysis also showed a similar direction of effect, though it did not reach statistical significance (OR: 0.69, 95% CI: 0.02–25.65, P = 0.845), likely due to the lower statistical power of this method.
For aspirin, the weighted median method showed a directionally consistent protective effect (OR: 0.14, 95% CI: 0.02–0.78, P=0.025), further supporting the primary finding. The MR-Egger and weighted mode estimates were also directionally consistent, though not statistically significant.
Heterogeneity and pleiotropy diagnostics for the analgesic-to-COPD analyses are presented in Table 3. Cochran’s Q test showed no significant heterogeneity for the paracetamol and aspirin analyses (P > 0.05), suggesting that the causal estimates across the SNPs were consistent. The MR-Egger intercept test did not indicate significant directional pleiotropy for any of the analgesics (P > 0.05 for all). Furthermore, the MR-PRESSO global test found no significant evidence of pleiotropic outliers for any of the analgesic exposures.
 |
Table 3 Sensitivity Analyses for Heterogeneity and Pleiotropy |
Scatter plots (Figure 3), leave-one-out plots (Figure 4), and funnel plots (Figure 5) provided further visual confirmation of the robustness of the primary findings. The leave-one-out analysis for paracetamol and aspirin demonstrated that the overall causal estimates were not driven by any single SNP.
 |
Figure 3 Scatter plot of the causal effects of genetically predicted (A) paracetamol (B) aspirin use on the risk of COPD. Each dot represents a single SNP, with the x-axis showing the SNP’s effect on paracetamol use and the y-axis showing the SNP’s effect on COPD risk. The lines represent the causal estimates from different MR methods (IVW, MR-Egger, Weighted Median). |
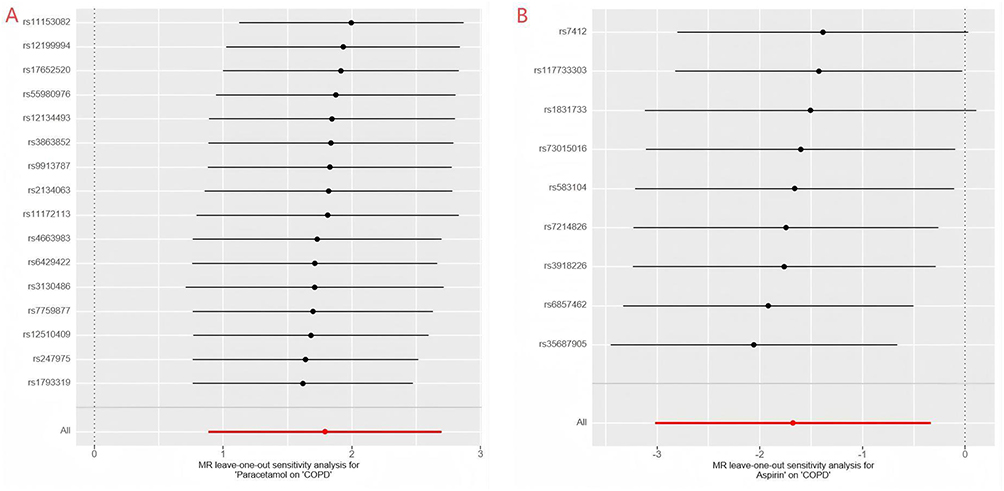 |
Figure 4 Leave-one-out plot for the causal effect of genetically predicted (A) paracetamol (B) aspirin use on the risk of COPD. The plot shows the IVW causal estimate after removing each SNP one at a time. This analysis assesses the influence of individual SNPs on the overall causal estimate. |
 |
Figure 5 Funnel plot for the causal effect of genetically predicted (A) paracetamol (B) aspirin use on the risk of COPD. The funnel plot is used to visually inspect for heterogeneity and potential pleiotropy. Each dot represents a single SNP’s causal estimate. In the absence of heterogeneity and pleiotropy, the dots should be symmetrically distributed around the overall causal estimate. |
Reverse Analysis: Causal Effect of COPD on Analgesic Use
The results of the reverse MR analysis, which investigated the causal effect of genetic liability to COPD on the use of common analgesics, are presented in Table 2. In the primary IVW analysis, there was no evidence of a causal association between genetic predisposition to COPD and the use of paracetamol (OR: 1.03, 95% CI: 0.94–1.13, P = 0.512), aspirin (OR: 0.99, 95% CI: 0.91–1.08, P = 0.824), or ibuprofen (OR: 1.01, 95% CI: 0.93–1.10, P = 0.755). These results are visually summarized in Figure 6.
 |
Figure 6 Forest plot of the causal effects of genetically predicted COPD on the use of common analgesics. The plot shows the odds ratios (ORs) and 95% confidence intervals (CIs) from the IVW analysis for the effect of COPD on the use of paracetamol, aspirin, and ibuprofen. |
Sensitivity Analyses for the Effect of COPD on Analgesics
The results from the sensitivity analyses for the reverse direction (Table 2) were consistent with the primary IVW findings, with all methods (MR-Egger, weighted median, and weighted mode) showing no significant causal effect of COPD on the use of any of the three analgesics.
Heterogeneity and pleiotropy diagnostics are shown in Table 3. Cochran’s Q test did not indicate significant heterogeneity for any of the analyses in the reverse direction (P > 0.05). Similarly, the MR-Egger intercept and MR-PRESSO global tests showed no evidence of directional pleiotropy or outliers. These findings support the conclusion that there is no robust evidence for a causal effect of COPD on the use of these common analgesics.
Discussion
In this comprehensive bidirectional Mendelian randomization study, we investigated the causal relationships between the use of three common analgesics—paracetamol, aspirin, and ibuprofen—and the risk of chronic obstructive pulmonary disease (COPD). Our findings provide novel genetic evidence that supports a differential causal role of these analgesics in the etiology of COPD. Specifically, we found that genetically predicted use of paracetamol is causally associated with an increased risk of COPD, whereas genetically predicted use of aspirin is causally associated with a decreased risk. In contrast, we did not find evidence for a causal link between ibuprofen use and COPD risk. Furthermore, our reverse MR analysis showed no evidence of a causal effect of COPD on the use of any of these analgesics, suggesting that the observed associations are not due to reverse causation.
The finding of a strong positive association between acetaminophen use and increased COPD risk is a key finding of this study. The magnitude of this effect is substantial, suggesting that acetaminophen use may be an important and potentially modifiable risk factor for COPD. This finding aligns with previous observational studies that have reported a dose-dependent association between acetaminophen use and an increased risk of COPD and reduced lung function.5 The biological plausibility of this association is supported by the “glutathione depletion” hypothesis. Acetaminophen metabolism can deplete cellular levels of glutathione, a crucial antioxidant in the lung epithelial lining fluid.21 Since oxidative stress is a central mechanism in the pathogenesis of COPD, the reduction of antioxidant defenses by acetaminophen could exacerbate lung damage from smoking or other environmental insults, thereby increasing COPD risk.5 Our study provides genetic support for this causal pathway, moving beyond the correlational evidence from observational studies.
Conversely, our finding of a protective effect of aspirin use on COPD risk is equally significant. Observational studies have reported a reduction in COPD exacerbations and mortality associated with aspirin use; for example, a retrospective cohort study found a 40% lower in-hospital mortality among aspirin users hospitalized for acute exacerbation of COPD.22 The mechanism underlying this protective effect is likely related to aspirin’s anti-inflammatory properties. As a non-steroidal anti-inflammatory drug (NSAID), aspirin inhibits cyclooxygenase (COX) enzymes, leading to reduced production of pro-inflammatory prostaglandins.23 Given that chronic inflammation is a hallmark of COPD, the anti-inflammatory action of aspirin could plausibly mitigate the inflammatory processes that drive the disease’s progression. It is important to note that our findings contrast with concerns about NSAID use in patients with certain respiratory conditions, such as aspirin-exacerbated respiratory disease (AERD). However, our study focuses on the risk of developing COPD in the general population rather than exacerbations in individuals with pre-existing conditions, suggesting that the overall effect of aspirin on COPD risk may be protective. Recent MR studies have also hinted at a protective role for aspirin in other inflammatory conditions,24,25 and our results extend this to COPD.
Our study did not find a significant causal association between ibuprofen use and COPD risk. This neutral finding is interesting, as ibuprofen is also an NSAID that inhibits COX enzymes.26 The lack of a significant effect may be due to several factors. First, the genetic instruments for ibuprofen use might have been weaker or less specific than those for aspirin, leading to lower statistical power. Indeed, the wide confidence intervals in the sensitivity analyses for ibuprofen suggest lower precision, possibly due to the smaller number of available SNPs. Alternatively, there might be genuine pharmacological differences between aspirin and ibuprofen in their effects on the respiratory system. For instance, aspirin is an irreversible inhibitor of COX, whereas ibuprofen is a reversible inhibitor,27 which might lead to different long-term effects on inflammatory pathways. It is also possible that the net effect of ibuprofen on COPD risk is null, with potential benefits from its anti-inflammatory action being counteracted by other effects.
While our findings are compelling, the large effect size observed for paracetamol (OR of 6.00) warrants a cautious interpretation. Such a strong association is unusual for a complex disease and may be influenced by several factors. Although our sensitivity analyses did not detect significant pleiotropy, we cannot entirely rule out unmeasured pleiotropic pathways where the genetic instruments for paracetamol use might influence COPD risk through mechanisms other than paracetamol metabolism itself. This unusually large estimate could also reflect specific genetic pathways that confer a particularly high susceptibility. Therefore, while the direction of the effect is consistent across analyses, the magnitude of the risk should be interpreted with caution pending replication in further studies.
The bidirectional design of our study is a major strength, as it allowed us to formally test for reverse causation. The lack of any significant causal effect of COPD on the use of paracetamol, aspirin, or ibuprofen provides strong evidence against the hypothesis that the associations observed in previous studies were due to individuals with early or undiagnosed COPD symptoms using analgesics for pain or fever. This strengthens the causal interpretation of our findings in the forward direction.
This study has several strengths, but some limitations must be acknowledged. First, the use of summary-level data prevented us from performing stratified analyses, for example, by smoking status, which is the primary risk factor for COPD. While the MR design inherently mitigates confounding from such environmental factors due to the random allocation of genes, an interaction between genetic predisposition to analgesic use and smoking cannot be ruled out. Second, the genetic instruments for analgesic use were based on self-reported, binary data from the UK Biobank, which may be subject to misclassification and does not capture information on dose, frequency, or duration of use. This is a significant limitation, as the effects of these drugs could be dose-dependent. Such misclassification is likely to be non-differential and would typically bias the results towards the null. Third, our study population was restricted to individuals of European ancestry, which may limit the generalizability of our findings to other populations. This is particularly relevant as the pharmacokinetics and metabolism of analgesics like paracetamol can vary across different ethnic groups,28,29 potentially altering their effects. Genetic polymorphisms influencing drug response can also differ between populations,30 further underscoring the need for research in non-European ancestries. Fourth, while MR is a powerful tool for causal inference, we cannot completely rule out the possibility of residual horizontal pleiotropy. However, our extensive sensitivity analyses, including the MR-Egger intercept test and MR-PRESSO, did not detect any evidence of significant directional pleiotropy, increasing confidence in our results.
Conclusion
In conclusion, this bidirectional MR study provides genetic evidence supporting a causal role for common analgesics in the risk of COPD. Our findings cautiously suggest that genetic liability to paracetamol use increases the risk of COPD, while genetic liability to aspirin use may decrease it. These results have important clinical and public health implications, highlighting that the potential risks and benefits of different analgesic choices warrant further consideration. For individuals at high risk of COPD, such as smokers, the potential long-term respiratory effects of paracetamol may be a relevant consideration. Conversely, the potential protective effect of aspirin warrants further investigation, including in dose-response MR studies and randomized clinical trials, to explore its viability as a preventive agent for COPD. Future research should aim to replicate these findings in diverse populations and to further elucidate the biological mechanisms underlying these differential effects.
Ethical Statement
All data used in this study were obtained from publicly available, de-identified summary-level statistics from previously published genome-wide association studies (GWAS). The original studies that collected the data had already received ethical approval from their respective institutional review boards (IRBs) and obtained informed consent from all participants. Therefore, no additional ethical approval was required for this analysis. This study is in accordance with the ethical principles of the Declaration of Helsinki and qualifies for exemption from ethical review under item 1 of Article 32 of the “Measures for Ethical Review of Life Science and Medical Research Involving Human Subjects” (February 18, 2023, China).
Disclosure
The authors declare no competing interests.
References
1. Jaén-Moreno MJ, Rico-Villademoros F, Ruiz-Rull C, Laguna-Muñoz D, Del Pozo GI, Sarramea F. A systematic review on the association between schizophrenia and bipolar disorder with chronic obstructive pulmonary disease. Copd. 2023;20(1):31–12. doi:10.1080/15412555.2022.2154646
2. Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med. 2001;163(5):1256–1276. doi:10.1164/ajrccm.163.5.2101039
3. Aoshiba K, Tsuji T, Itoh M, Yamaguchi K, Nakamura H. An evolutionary medicine approach to understanding factors that contribute to chronic obstructive pulmonary disease. Resp Int Rev Thoracic Diseases. 2015;89(3):243–252. doi:10.1159/000369861
4. Rodrigues SO, Cunha C, Soares GMV, Silva PL, Silva AR, Gonçalves-de-Albuquerque CF. Mechanisms, pathophysiology and currently proposed treatments of chronic obstructive pulmonary disease. Pharmaceuticals. 2021;14(10):979. doi:10.3390/ph14100979
5. McKeever TM, Lewis SA, Smit HA, Burney P, Britton JR, Cassano PA. The association of Acetaminophen, aspirin, and ibuprofen with respiratory disease and lung function. Am J Respir Crit Care Med. 2005;171(9):966–971. doi:10.1164/rccm.200409-1269OC
6. Lipiec A, Wawrzyniak ZM, Sybilski AJ, et al. The association between paracetamol use and the risk of asthma, rhinitis and eczema in the Polish population. Ann. Agric. Environ. Med. 2018;25(3):428–432. doi:10.26444/aaem/86336
7. Lee RU, Stevenson DD. Aspirin-exacerbated respiratory disease: evaluation and management. Allergy Asthma Immunol Res. 2011;3(1):3–10. doi:10.4168/aair.2011.3.1.3
8. Wang Z, Sun Y. Unraveling the causality between chronic obstructive pulmonary disease and its common comorbidities using bidirectional Mendelian randomization. Eur. J. Med. Res. 2024;29(1):143. doi:10.1186/s40001-024-01686-x
9. Jiang H, Zhang X, Zhang J, Liang J, Wang L. Association between opioid and benzodiazepine use and all-cause mortality in individuals with chronic obstructive pulmonary disease: a prospective cohort study. Int J Chronic Obstr. 2024;19:2181–2192. doi:10.2147/COPD.S467131
10. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Human Molecular Genetics. 2014;23(R1):R89–98. doi:10.1093/hmg/ddu328
11. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–525. doi:10.1093/ije/dyv080
12. de Leeuw C, Savage J, Bucur IG, Heskes T, Posthuma D, de Leeuw C. Understanding the assumptions underlying Mendelian randomization. Eur. J. Hum. Genet. 2022;30(6):653–660. doi:10.1038/s41431-022-01038-5
13. Elsworth B, Lyon M, Alexander T, et al. The MRC IEU OpenGWAS data infrastructure. BioRxiv. 2020:
14. Kurki MI, Karjalainen J, Palta P, et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature. 2023;613(7944):508–518. doi:10.1038/s41586-022-05473-8
15. Liao W, Lin X, Liu K, et al. Association between platelet indices and risk of chronic obstructive pulmonary disease: a bidirectional mendelian randomization study. Int J Chronic Obstr. 2025;20:2967–2977. doi:10.2147/COPD.S531797
16. Wang J, Wang C, Li S, et al. Causal effect between circulating metabolic markers and glioma: a bidirectional, two-sample, Bayesian weighted Mendelian randomization. Discov. Oncol. 2025;16(1):315. doi:10.1007/s12672-025-02050-z
17. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 2016;40(4):304–314. doi:10.1002/gepi.21965
18. Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. 2017;46(6):1985–1998. doi:10.1093/ije/dyx102
19. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nature Genet. 2018;50(5):693–698. doi:10.1038/s41588-018-0099-7
20. Lee S, Lee W. A review of mendelian randomization: assumptions, methods, and application to obesity-related diseases. J Obes Metab Syndr. 2025;34(1):14–26. doi:10.7570/jomes24031
21. Barr RG, Wentowski CC, Curhan GC, et al. Prospective study of Acetaminophen use and newly diagnosed asthma among women. Am J Respir Crit Care Med. 2004;169(7):836–841. doi:10.1164/rccm.200304-596OC
22. Goto T, Faridi MK, Camargo CA, Hasegawa K. The association of aspirin use with severity of acute exacerbation of chronic obstructive pulmonary disease: a retrospective cohort study. NPJ Prim. Care Respir. Med. 2018;28(1):7. doi:10.1038/s41533-018-0074-x
23. Rao P, Knaus EE. Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): cyclooxygenase (COX) inhibition and beyond. J Pharm Pharm Sci. 2008;11(2):81s–110s. doi:10.18433/J3T886
24. Li L, Zhang Y, Liu X, et al. Potential causal association between aspirin use and the reduced risk of hayfever or allergic rhinitis: a Mendelian randomization study. Front Immunol. 2023;14:1232981. doi:10.3389/fimmu.2023.1232981
25. Cai W, Fang Z, Tian Z, Li D, Tang K. Causal relationship between aspirin consumption and heart failure: a Mendelian randomization study. ESC Heart Failure. 2024;11(1):533–540. doi:10.1002/ehf2.14617
26. Mitchell JA, Akarasereenont P, Thiemermann C, Flower RJ, Vane JR. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc Natl Acad Sci USA. 1993;90(24):11693–11697. doi:10.1073/pnas.90.24.11693
27. Friedewald VE, Bennett JS, Christo JP, et al. AJC Editor’s consensus: selective and nonselective nonsteroidal anti-inflammatory drugs and cardiovascular risk. Am J Cardiol. 2010;106(6):873–884. doi:10.1016/j.amjcard.2010.04.006
28. Zurlinden TJ, Reisfeld B. Characterizing the effects of race/ethnicity on acetaminophen pharmacokinetics using physiologically based pharmacokinetic modeling. Eur J Drug Metab Pharmacokinet. 2017;42(1):143–153. doi:10.1007/s13318-016-0329-2
29. Court MH, Zhu Z, Masse G, et al. Race, gender, and genetic polymorphism contribute to variability in acetaminophen pharmacokinetics, metabolism, and protein-adduct concentrations in healthy african-american and european-american volunteers. J Pharmacol Exp Ther. 2017;362(3):431–440. doi:10.1124/jpet.117.242107
30. Yiannakopoulou E. Pharmacogenomics of acetylsalicylic acid and other nonsteroidal anti-inflammatory agents: clinical implications. Eur J Clin Pharmacol. 2013;69(7):1369–1373. doi:10.1007/s00228-013-1477-9
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
NSAID Hypersensitivity in the Pediatric Population: Classification and Diagnostic Strategies
Cavkaytar O, Arga M
Journal of Asthma and Allergy 2022, 15:1383-1399
Published Date: 28 September 2022
A Cross-Sectional Study to Investigate the Prevalence of Self-Medication of Non-Opioid Analgesics Among Medical Students at Qassim University, Saudi Arabia
Elghazaly A, Alsahali S, Farooqui M, Ibrahim N, Alshammari M, Almutairi A, Almutairi M, Almutairi W
Patient Preference and Adherence 2023, 17:1371-1379
Published Date: 7 June 2023
The Causal Relationship Between Gastroesophageal Reflux Disease and Chronic Obstructive Pulmonary Disease: A Bidirectional Two-Sample Mendelian Randomization Study
Liu B, Chen M, You J, Zheng S, Huang M
International Journal of Chronic Obstructive Pulmonary Disease 2024, 19:87-95
Published Date: 10 January 2024
Association of Chronic Obstructive Pulmonary Disease with Risk of Psychiatric Disorders: A Two-Sample Mendelian Randomization Study
Zhang Q, Zhang H, Xu Q
International Journal of Chronic Obstructive Pulmonary Disease 2024, 19:343-351
Published Date: 1 February 2024
Differential Causal Effects of Common Analgesics on Breast Cancer Risk and Survival: Evidence from Mendelian Randomization
Peng Z, Liu Z, Wang G
International Journal of Women's Health 2025, 17:5287-5301
Published Date: 9 December 2025