Back to Journals » Drug Design, Development and Therapy » Volume 12
Genetically engineered drug rhCNB induces apoptosis and cell cycle arrest in both gastric cancer cells and hepatoma cells
Authors Guo Y, Huang Y, Tian S, Xie X, Xing G, Fu J
Received 19 April 2018
Accepted for publication 21 May 2018
Published 20 August 2018 Volume 2018:12 Pages 2567—2575
DOI https://doi.org/10.2147/DDDT.S171675
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Yanzi Guo,1,* Yonghao Huang,2,* Shuhong Tian,1 Xueli Xie,1 Guilan Xing,1 Jian Fu1
1Key Laboratory of Preclinical Pharmacology and Toxicology of Hainan Province, Hainan Medical College, Haikou 571199, China; 2Key Laboratory of Tropical Diseases and Translational Medicine of the Ministry of Education & Hainan Provincial Key Laboratory of Tropical Medicine, Hainan Medical College, Haikou 571199, China
*These authors contributed equally to this work
Objectives: Calcineurin B (CNB) is a regulatory subunit of calcineurin, and it has antitumor activity. In this study, we aimed to investigate the effect of recombinant human calcineurin B (rhCNB) on the proliferation of gastric cancer cells and hepatoma cells both in vitro and in vivo.
Materials and methods: Cell viability and cell proliferation were detected by MTT and BrdU assay. Flow cytometry, Western blot and immunohistochemistry were performed to determine rhCNB-induced apoptosis and cell cycle arrest. The antitumor activities of rhCNB were observed in mice tumor models.
Results: We demonstrated that rhCNB inhibits the proliferation of gastric cancer cells and hepatoma cells both in vitro and in vivo. We showed that the inhibition of cell proliferation by rhCNB is associated with apoptosis and cell cycle arrest in both tumor cell lines. Furthermore, we indicated that rhCNB promotes p53 protein expression, a potent proapoptotic factor. Meanwhile, we also exhibited that rhCNB decreases the expression of both cyclin B1 and CDK1 proteins, two proteins associated with G2/M arrest.
Conclusion: Together, these findings suggest that rhCNB markedly inhibits tumor growth and provides guidance for its drug development.
Keywords: rhCNB, tumor cells, apoptosis, cell cycle arrest, p53
Introduction
Calcineurin (CN), the only known serine/threonine phosphatase regulated by Ca2+/CaM, is made up of two subunits: a catalytic subunit calcineurin A and a regulatory subunit calcineurin B (CNB).1,2 CN plays an important role in activating immune cells primarily due to its dephosphorylating activity, and this activity is inhibited by the immunosuppressive drugs cyclosporin A (CsA) and FK506.3,4 CNB is a regulatory subunit of CN, and its basic role is to regulate the phosphatase activity of CN. However, CNB also has other functions, especially anticancer activity. CNB is involved in apoptosis and the proteasome pathway via its interactions with heat shock protein 60, procaspase-3, proteasome subunit alpha type-7 and other proteins.5–7 Recent studies have indicated that recombinant human calcineurin B (rhCNB) has an anticancer effect in a variety of tumor models. It inhibited the proliferation of human hepatoma HepG2 cells through inducing apoptosis.8 It reduced the growth rate of S180 solid tumors and prolonged the life of the mice by stimulation of immunocytes.9 It had been shown to confer greater protection against tumor-forming E.G7-OVA cells by activation of dendritic cells.10 These results indicate that rhCNB holds promise as an antitumor drug and its antitumor mechanisms are mainly involved in inducing apoptosis and activating immune cells.
Apoptosis, also known as programmed cell death, has been shown to be an attractive strategy for cancer treatment.11,12 There are two main pathways of apoptosis: extrinsic death receptor pathway and intrinsic mitochondrial pathway. The extrinsic death receptor pathway is stimulated by binding of specific ligands to death receptors on the cell membrane, and this triggers a sequential activation of caspase-8 and -3, eventually leading to apoptosis.13,14 The intrinsic apoptotic pathway can be triggered by various intracellular stimuli, such as radiation, viral infections, growth factor starvation and oxidative stress. These insults stimulate mitochondria outer membrane permeabilization, resulting in the leakage of cytochrome C (Cyt-C) from the mitochondria and the formation of the apoptosome complex, which consists of Apaf-1, caspase-9 and Cyt-C. Activation of caspase-9 by apoptosome subsequently cleaves and activates executioner caspases, eventually leading to apoptosis.12,15 Most of other caspases play key roles in apoptosis; caspase-8, -9 and -10 are thought to be the initiator caspases, whereas caspase-3, -6 and -7 serve as effector caspases.16,17 Caspase-3 is the main downstream effector caspase that cleaves the majority of the cellular substrates in apoptotic cells, and it is activated by caspase-8 or -9 in extrinsic or intrinsic pathway.
Dysregulation of cell cycle control and abnormal cellular proliferation is a fundamental aspect of cancer.18,19 Therefore, inducing cell cycle arrest is anticipated to provide an effective approach to cancer therapies for clinical use.20,21 The cell cycle is regulated at different stages by various cyclin–cyclin-dependent kinase (CDK) complexes. CDK1 is the CDK responsible for the entry and exit from G2/M and mitosis. It forms a complex with cyclin B1 or cyclin A.19 Downregulation of both cyclin B1 and CDK1 is often correlative with the G2/M phase of cell cycle arrest.22,23
In this study, we demonstrated that rhCNB inhibits the proliferation of gastric cancer cells and hepatoma cells via inducing apoptosis and cell cycle arrest both in vitro and in vivo, providing the groundwork for the potential application of rhCNB in antitumor therapy.
Materials and methods
Cell culture, agents, and antibodies
Human gastric cancer cell lines (SGC-7901 and MGC-803) and hepatoma cell lines (Bel-7402 and HepG2) were purchased from Sinochrome/Cytogenetics (Shanghai, China). All cell lines were cultured in Roswell Park Memorial Institute-1640 supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA), 100 U/mL penicillin (Sigma-Aldrich Co., St Louis, MO, USA), and 100 μg/mL streptomycin (Sigma-Aldrich Co.) at 37°C in an atmosphere containing 5% CO2. To stably express Gluc and the cyan fluorescent protein (CFP), cells were transduced with the CSCW–Gluc–IRES–CFP lentivirus vector. rhCNB (enzyme unit < 0.25 U) was provided by Haikou Qili Pharmaceutical Co., Ltd (Hainan, China). The Annexin V-fluorescein isothiocyanate (FITC)/PI Apoptosis Detection Kit was purchased from KeyGen Biotech (Jiangsu, China). The Cell Cycle Analysis Kit was purchased from Beyotime (Shanghai, China). The following antibodies were used in this study: caspase-3, p53, cyclin B1 and CDK1 (purchased from Abcam, Cambridge, UK).
Cell viability assay
The cell viabilities were determined by MTT assay. Cells were cultured in 96-well plates and exposed to the tested compounds for 24 h. In all, 10 μL of MTT was added to each well. After 4 h incubation, the medium was removed and 200 μL of dimethylsulfoxide was added to dissolve the crystal formazan dye. The absorbance was measured at 570 nm on an enzyme-linked immunosorbent assay reader.
BrdU assay
BrdU signaling was assayed using a BrdU Cell Proliferation Assay Kit (ab126556; Abcam). Briefly, cells were cultured in 96-well plates and rhCNB treatment for 24 h. Subsequently, 10 μM BrdU was added to each well, and samples were incubated for 12 h at 37°C. BrdU signaling was determined by measuring the absorbance at 450 nm.
Flow cytometry
Cells were cultured and exposed to the tested compounds for 24 h. For apoptosis analysis, cells were harvested and washed once with PBS and then resuspended and incubated in propidium iodide (PI)/Annexin V solution (KeyGen Biotech). At least 10,000 live cells were analyzed on a flow cytometer (BD Accuri™ C6; BD Biosciences, San Jose, CA, USA). For cell cycle analysis, cells were harvested and fixed in 75% cold ethanol at −20°C overnight. Cells were then resuspended and incubated in PI solution. DNA content was determined using a flow cytometer.
Western blot
Cells were harvested and lysed with radioimmunoprecipitation assay (RIPA) buffer. Protein concentrations were quantified by a bicinchoninic acid protein assay kit (23227; Thermo Fisher Scientific). Proteins were size fractionated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to (Polyvinylidene Fluoride) membranes. After blocking, the membranes were incubated with primary antibodies at 4°C overnight and then incubated with secondary antibodies at room temperature for 2 h. Target proteins were examined using enhanced chemiluminescence reagents (WBKLS0100; Merck Millipore, Billerica, MA, USA).
Immunohistochemistry (IHC)
Immunohistochemical analysis was performed as described previously.24,25 Briefly, sections were deparaffinized and rehydrated by incubation with xylene and decreasing concentrations of ethanol. Antigen retrieval was performed by pretreatment of the slides in citrate–EDTA antigen retrieval solution. The slides were immersed into 3% H2O2/methanol for 10 min at room temperature to quench endogenous peroxidase. After rinsing in 0.05% phosphate buffered solution/Tween-20 (PBST) twice, the primary antibody was added and incubated at 4°C overnight. The slides were then washed in PBST and incubated with horseradish peroxidase-conjugated secondary antibody at 37°C for 30 min. After washing, chromogen DAB 3,3’-diaminobenzidine/substrate reagent was added onto the slides and the slides were incubated for a further 10 min. Finally, the slides were dehydrated and mounted. Images were captured using a DM2500 fluorescence microscope (Leica Microsystems, Wetzlar, Germany).
Animal models
All animal experiments complied with the National Institutes of Health (USA)’s Guide for the Care and Use of Laboratory Animals (NIH Publication No 8023, revised 1978), and all studies were approved by the Institutional Animal Care and Treatment Committee of Hainan Medical College. MGC-803–Gluc–CFP or Bel-7402–Gluc–CFP cells (2×106 cells/mouse) were implanted subcutaneously in the flanks of nude mice (BALB/c, 6–8 weeks of age, infertile, 18–20 g of weight). When the tumor volumes reached 100 mm3, mice were divided into two groups and treated with rhCNB (20 mg/kg) and solvent control, respectively. The tumor volumes were monitored every 3 days using a handheld imaging device (TM900; Peira, Flanders, Belgium). The side of tumor was recorded by bioluminescence imaging before or after 15 days treatment (Xenogen IVIS Kinetic; Caliper Life Sciences, Hopkinton, MA, USA). Animals were sacrificed after 3 weeks. Tumor tissues were isolated and frozen in liquid nitrogen or fixed in 10% formalin immediately.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 6.0 (GraphPad Software, Inc., La Jolla, CA, USA). Comparisons between the two groups were performed by the Student’s t-test. Statistical significance was defined as *P<0.05, **P<0.01 and ***P<0.001.
Results
rhCNB inhibits the growth of tumor cells both in vitro and in vivo
To examine the effect of rhCNB in both gastric cancer cells, human gastric cancer cell lines SGC-7901 and MGC-803 were treated with various concentrations of rhCNB for 24 h, followed by MTT assay. As shown in Figure 1A, rhCNB markedly suppressed gastric cancer cell proliferation in a dose-dependent manner in both cell lines. Consistently, BrdU assays showed that there were a significantly lower percentage of BrdU-positive cells in rhCNB-treated cells in a dose-dependent manner (Figure 1B). In addition, the significant decreased cell proliferation by rhCNB was obtained by microscope, as indicated in Figure 1C. Collectively, these results demonstrate that rhCNB inhibits the proliferation of gastric cancer cells in vitro. To evaluate the effect of rhCNB on gastric cancer growth in vivo, BALB/c nude mice were inoculated with MGC-803 cells and treated with rhCNB or vehicle. As shown in Figure 2A, rhCNB treatment significantly decreased rate of tumor growth compared to that in the control group. Consistently, tumor weight was reduced in rhCNB-treated mice compared with those treated with control treatment (Figure 2B). In addition, the size of rhCNB-treated tumors was much smaller than that of the control group (Figure 2C). Furthermore, bioluminescence imaging showed that rhCNB treatment significantly inhibited tumor growth (Figure 2G). Taken together, these data suggest that rhCNB inhibits the growth of gastric cancer both in vitro and in vivo.
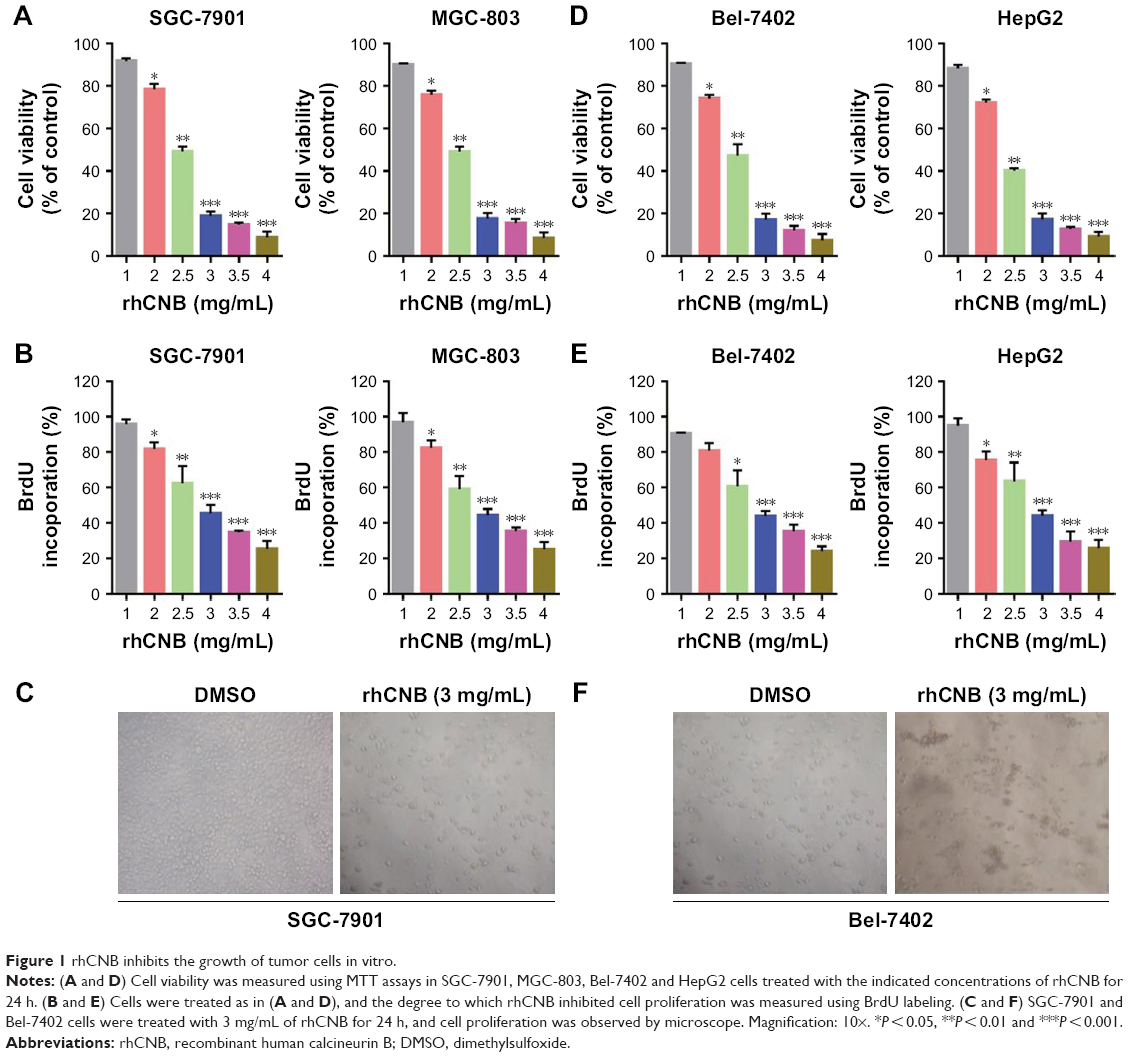 | Figure 1 rhCNB inhibits the growth of tumor cells in vitro. |
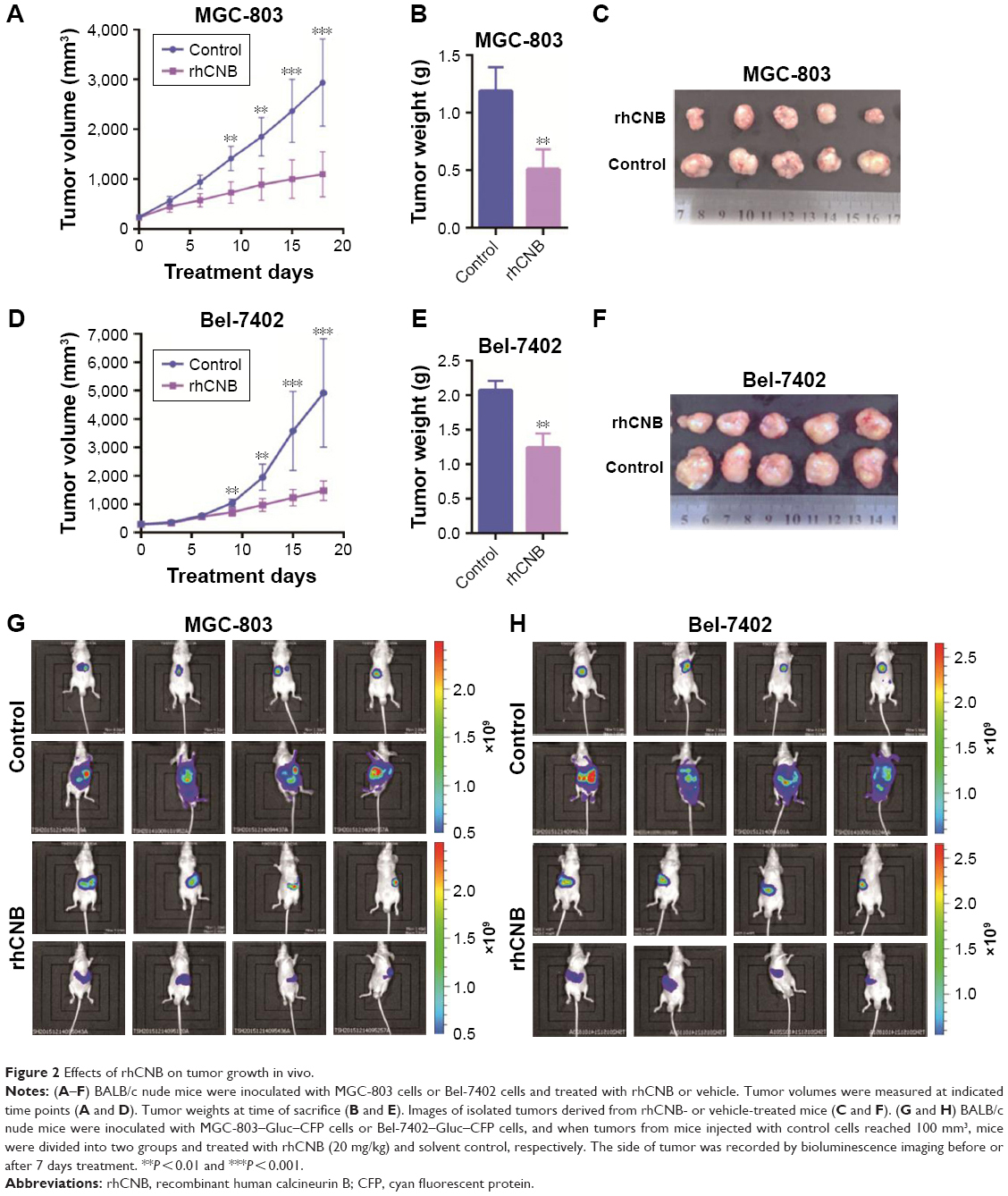 | Figure 2 Effects of rhCNB on tumor growth in vivo. |
We next tested whether rhCNB inhibits the growth of tumor both in vitro and in vivo in another cell model. To this end, we subjected human hepatoma cell lines Bel-7402 and HepG2 to different concentrations of rhCNB treatment for 24 h. As shown in Figure 1D–F, rhCNB markedly inhibits the proliferation of hepatoma cells in vitro. Bel-7402 cells were also implanted subcutaneously in BALB/c nude mice and treated with rhCNB or vehicle. As shown in Figure 2D–F, rhCNB significantly inhibits the growth of hepatoma in vivo. This result was further supported by in vivo bioluminescence imaging assay (Figure 2H). Taken together, consistent with the findings in gastric cancer, rhCNB inhibits the growth of hepatoma both in vitro and in vivo.
rhCNB induces apoptosis in tumor cells
To evaluate whether inhibition of cell proliferation by rhCNB in gastric cancer cells was associated with apoptosis, MGC-803 cells were analyzed by flow cytometry following Annexin V-FITC and PI staining. As shown in Figure 3A, rhCNB treatment obviously increased the percentage of apoptotic cells in MGC-803 cells in a dose-dependent manner. In addition, activation of caspase-3, one of key effector molecules of apoptosis, was detected. As shown in Figure 3C, the level of cleaved caspase-3 was markedly accumulated in MGC-803 cells upon rhCNB treatment. Collectively, these data demonstrated that rhCNB induces apoptosis in gastric cancer cells. It is well-accepted that p53 protein is a critical tumor suppressor and can mediate apoptosis in cancer cells.26–28 Therefore, we next addressed whether p53 is involved in rhCNB-induced apoptosis in gastric cancer cells. First, we determined the expression of p53 in MGC-803 cells upon rhCNB treatment by Western blot. As shown in Figure 3C, rhCNB treatment promoted the expression of p53 in MGC-803 cells. Moreover, IHC assay showed that p53 expression is elevated in MGC-803 cells (Figure 3B). Taken together, these data suggest that p53 may be involved in rhCNB-induced apoptosis in gastric cancer cells.
 | Figure 3 rhCNB induces apoptosis in tumor cells. |
We next tested whether rhCNB also induces apoptosis in hepatoma cells. To this end, Bel-7402 cells were used. As shown in Figure 3A and D, rhCNB induces apoptosis in hepatoma cells. Furthermore, consistent with the findings in gastric cancer cells, rhCNB promoted p53 expression in hepatoma cells by Western blot and IHC assays (Figure 3B and D), speculating rhCNB-mediated apoptosis in p53-dependent manner in hepatoma cells.
rhCNB induces cell cycle arrest in tumor cells
To further investigate the mechanism underlying rhCNB-induced tumor cell proliferation inhibition, we analyzed the effect of rhCNB on cell cycle distribution using flow cytometry. As shown in Figure 4A, rhCNB treatment for 24 h resulted in G2/M arrest in MGC-803 cells, as evidenced by an accumulation of cells in the G2/M phase and a loss of cells in the G0/G1 phase. In addition, both cyclin B1 and CDK1 proteins, two proteins associated with G2/M arrest, were detected. As shown in Figure 5A, rhCNB treatment decreased both cyclin B1 and CDK1 protein expression levels. Furthermore, IHC assay showed that both cyclin B1 and CDK1 expression levels are decreased in MGC-803 cells (Figure 5B). Taken together, these data demonstrated that rhCNB induces G2/M arrest in gastric cancer cells.
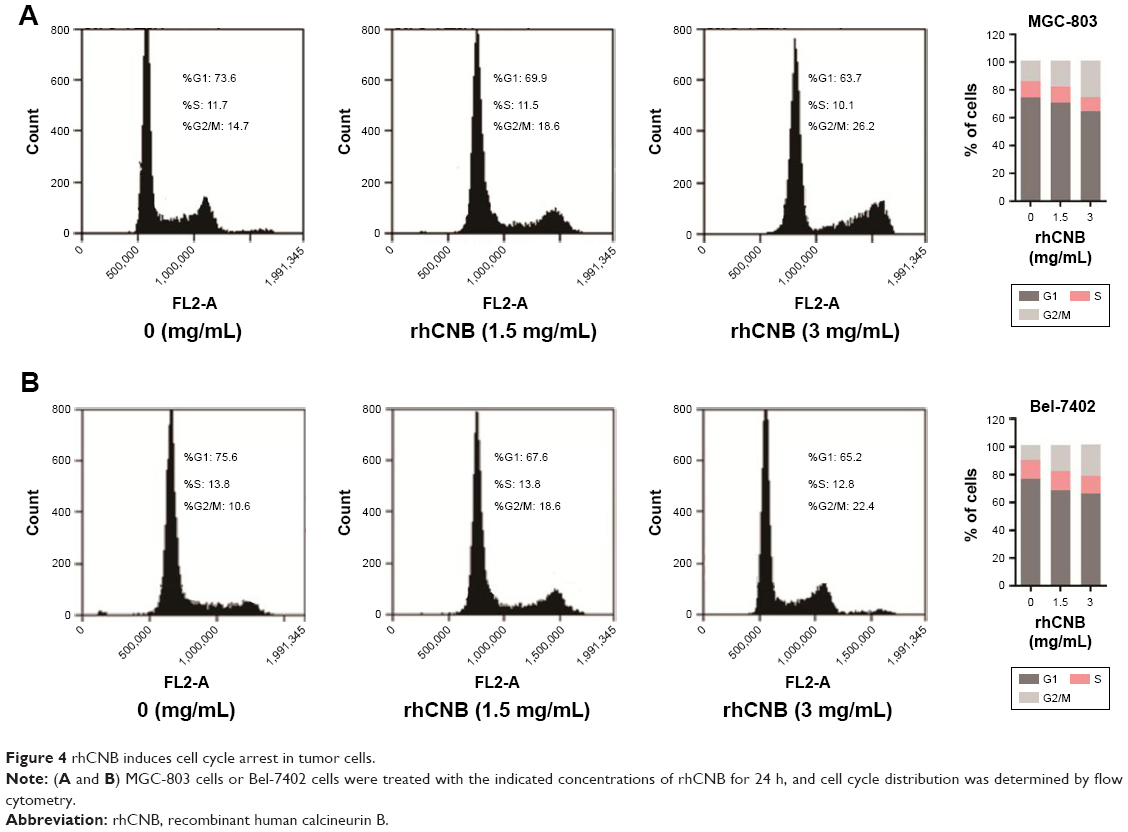 | Figure 4 rhCNB induces cell cycle arrest in tumor cells. |
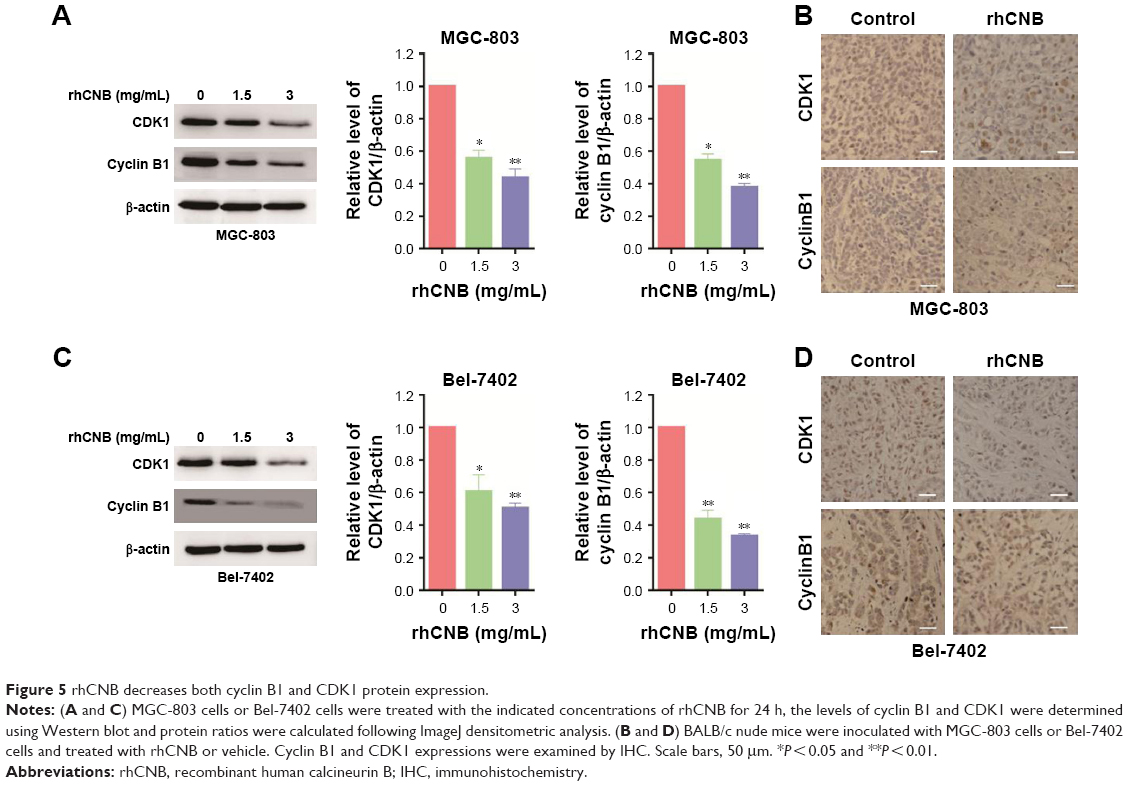 | Figure 5 rhCNB decreases both cyclin B1 and CDK1 protein expression. |
We next examined whether rhCNB also induces cell cycle arrest in hepatoma cells. For this purpose, Bel-7402 cells were used and analyzed by flow cytometry. As shown in Figure 4B, rhCNB induces G2/M arrest in hepatoma cells. Furthermore, consistent with the findings in gastric cancer cells, rhCNB decreased both cyclin B1 and CDK1 expression in hepatoma cells by Western blot and IHC assays (Figure 5C and D). Taken together, these data demonstrated that rhCNB induces G2/M arrest in hepatoma cells.
Discussion
CNB is the regulatory subunit of CN, and it has been shown to have potential as an anticancer agent.2,10 rhCNB also has been shown to have an anticancer effect in a variety of tumor models, which is identical to native CNB.8,9 Many recent studies have indicated that rhCNB inhibits tumor cell proliferation mainly via inducing apoptosis and activating immune cells. Consistently, in the present study, we showed that rhCNB induces apoptosis in both gastric cancer cells and hepatoma cells. In addition to apoptosis, we demonstrated for the first time that rhCNB induces G2/M arrest of cell cycle in both tumor cells.
It is demonstrated that rhCNB has antitumor effect in a variety of tumor models, such as human hepatoma HepG2 cells, S180 cells and E.G7-OVA cells.8–10 In this study, we showed that rhCNB inhibits the proliferation of not only hepatoma cells but also gastric cancer cells. rhCNB induces apoptosis and cell cycle arrest in both gastric cancer cells and hepatoma cells. Moreover, we also proved that rhCNB has antitumor effect in A549 cells, HT-29 cells and Hela cells (data not shown). Therefore, although our studies were focused on gastric cancer cells and hepatoma cells, it is likely that rhCNB induces apoptosis and cell cycle arrest in other tumor types.
It is well accepted that p53 protein is a critical tumor suppressor, and early studies demonstrated that p53 induces apoptosis programs, cell cycle arrest and senescence through its transcription function to regulate a series of target genes that are involved in induction of apoptosis or senescence and inhibition of the cell cycle.26–28 In this study, we showed that rhCNB upregulates p53 expression in both gastric cancer cells and hepatoma cells, suggesting that p53 may be involved in inducing apoptosis or cell cycle arrest in both tumor cells, but this result needs to be further verified.
Conclusion
We demonstrated that rhCNB inhibits the proliferation of gastric cancer cells and hepatoma cells both in vitro and in vivo. We further showed that the inhibition of cell proliferation by rhCNB is associated with apoptosis and cell cycle arrest in both tumor cell lines. Our findings provided new insight on the antitumor mechanism of rhCNB and guidance for its drug development.
Acknowledgment
This work was funded by the National Natural Science Foundation of China (81160408).
Disclosure
The authors report no conflicts of interest in this work.
References
Hemenway CS, Heitman J. Calcineurin. Structure, function, and inhibition. Cell Biochem Biophys. 1999;30(1):115–151. | ||
Musson RE, Smit NP. Regulatory mechanisms of calcineurin phosphatase activity. Curr Med Chem. 2011;18(2):301–315. | ||
Clipstone NA, Crabtree GR. Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature. 1992;357(6380):695–697. | ||
Yu DY, Luo J, Bu F, Zhang W, Wei Q. Effects of cyclosporin A, FK506 and rapamycin on calcineurin phosphatase activity in mouse brain. IUBMB Life. 2006;58(7):429–433. | ||
Li W, Handschumacher RE. Identification of two calcineurin B-binding proteins: tubulin and heat shock protein 60. Biochim Biophys Acta. 2002;1599(1–2):72–81. | ||
Saeki M, Irie Y, Ni L, et al. Calcineurin potentiates the activation of procaspase-3 by accelerating its proteolytic maturation. J Biol Chem. 2007;282(16):11786–11794. | ||
Li N, Zhang Z, Zhang W, Wei Q. Calcineurin B subunit interacts with proteasome subunit alpha type 7 and represses hypoxia-inducible factor-1alpha activity via the proteasome pathway. Biochem Biophys Res Commun. 2011;405(3):468–472. | ||
Yang Y, Yang H, Yang J, Li L, Xiang B, Wei Q. The genetically engineered drug rhCNB induces apoptosis via a mitochondrial route in tumor cells. Oncotarget. 2017;8(39):65876–65888. | ||
Syn N, Wang L, Sethi G, Thiery JP, Goh BC. Exosome-mediated metastasis: from epithelial-mesenchymal transition to escape from immunosurveillance. Trends Pharmacol Sci. 2016;37(7):606–617. | ||
Li J, Guo J, Su Z, Hu M, Liu W, Wei Q. Calcineurin subunit B activates dendritic cells and acts as a cancer vaccine adjuvant. Int Immunol. 2011;23(5):327–334. | ||
Fischer U, Schulze-Osthoff K. Apoptosis-based therapies and drug targets. Cell Death Differ. 2005;12(suppl 1):942–961. | ||
Zhang YS, Shen Q, Li J. Traditional Chinese medicine targeting apoptotic mechanisms for esophageal cancer therapy. Acta Pharmacol Sin. 2016;37(3):295–302. | ||
Wallach D, Kang TB, Kovalenko A. The extrinsic cell death pathway and the elan mortel. Cell Death Differ. 2008;15(10):1533–1541. | ||
Long JS, Ryan KM. New frontiers in promoting tumour cell death: targeting apoptosis, necroptosis and autophagy. Oncogene. 2012;31(49):5045–5060. | ||
Vaux DL. Apoptogenic factors released from mitochondria. Biochim Biophys Acta. 2011;1813(4):546–550. | ||
Kumar S. Caspase function in programmed cell death. Cell Death Differ. 2007;14(1):32–43. | ||
Adrain C, Creagh EM, Cullen SP, Martin SJ. Caspase-dependent inactivation of proteasome function during programmed cell death in Drosophila and man. J Biol Chem. 2004;279(35):36923–36930. | ||
Steinman RA. Cell cycle regulators and hematopoiesis. Oncogene. 2002;21(21):3403–3413. | ||
Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9(3):153–166. | ||
Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov. 2016;6(4):353–367. | ||
Rader J, Russell MR, Hart LS, et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin Cancer Res. 2013;19(22):6173–6182. | ||
Garcia-Morales P, Carrasco-Garcia E, Ruiz-Rico P, et al. Inhibition of Hsp90 function by ansamycins causes downregulation of cdc2 and cdc25c and G(2)/M arrest in glioblastoma cell lines. Oncogene. 2007;26(51):7185–7193. | ||
Garner AP, Weston CR, Todd DE, Balmanno K, Cook SJ. Delta MEKK3:ER* activation induces a p38 alpha/beta 2-dependent cell cycle arrest at the G2 checkpoint. Oncogene. 2002;21(53):8089–8104. | ||
Liu R, Li J, Xie K, et al. FGFR4 promotes stroma-induced epithelial-to-mesenchymal transition in colorectal cancer. Cancer Res. 2013;73(19):5926–5935. | ||
Noman MZ, Buart S, Romero P, et al. Hypoxia-inducible miR-210 regulates the susceptibility of tumor cells to lysis by cytotoxic T cells. Cancer Res. 2012;72(18):4629–4641. | ||
Crighton D, Ryan KM. Splicing DNA-damage responses to tumour cell death. Biochim Biophys Acta. 2004;1705(1):3–15. | ||
Kaiser AM, Attardi LD. Deconstructing networks of p53-mediated tumor suppression in vivo. Cell Death Differ. 2018;25(1):93–103. | ||
Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018;25(1):104–113. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.