Back to Journals » The Application of Clinical Genetics » Volume 14
Genetic Testing in CYLD Cutaneous Syndrome: An Update
Authors Nagy N, Dubois A, Szell M, Rajan N ![]()
Received 4 July 2021
Accepted for publication 5 October 2021
Published 29 October 2021 Volume 2021:14 Pages 427—444
DOI https://doi.org/10.2147/TACG.S288274
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Nikoletta Nagy,1,2 Anna Dubois,3 Marta Szell,1,2 Neil Rajan3,4
1Department of Medical Genetics, University of Szeged, Szeged, Hungary; 2Dermatological Research Group of the Eotvos Lorand Research Network, University of Szeged, Szeged, Hungary; 3Department of Dermatology, Royal Victoria Infirmary, Newcastle upon Tyne, NE1 4LP, UK; 4Translational and Clinical Research Institute, Newcastle University, Newcastle upon Tyne, NE1 3BZ, UK
Correspondence: Neil Rajan
Translational and Clinical Research Institute, Newcastle University, Newcastle upon Tyne, NE1 3BZ, UK
Email [email protected]
Abstract: CYLD cutaneous syndrome (CCS) is an inclusive label for the inherited skin adnexal tumour syndromes Brooke–Spiegler Syndrome (BSS-OMIM 605041), familial cylindromatosis (FC – OMIM 132700) and multiple familial trichoepitheliomas (MFT-OMIM 601606). All three syndromes arise due to germline pathogenic variants in CYLD, a tumour suppressor gene (OMIM 605018). CCS is transmitted in an autosomal dominant pattern, and has variable expressivity, both of the three syndromic phenotypes, and of the severity of tumour burden. Age-related penetrance figures are not precisely reported. The first tumours typically appear during puberty and progressively accumulate through adulthood. Penetrance is typically high, with equal numbers of males and females affected. Genetic testing is important for confirmation of the clinical diagnosis, genetic counselling and family planning, including preimplantation diagnosis. Additionally, identified CCS patients may be eligible for future clinical trials of non-surgical pre-emptive interventions that aim to prevent tumour growth. In this update, we review the clinical presentations of germline and mosaic CCS. An overview of the germline pathogenic variant spectrum of patients with CCS reveals more than 100 single nucleotide variants and small insertions and deletions in coding exons, most frequently resulting in predicted truncation. In addition, a minority of patients have large deletions involving the CYLD gene, intronic pathogenic variants that affect splicing, or inversions. We discuss germline and somatic testing approaches. Somatic testing of tumour tissue, relevant in mosaic CCS, can reveal recurrently detected pathogenic variants when two or more tumours are tested. This can influence genetic testing of children, who may inherit this as a germline variant, and inform genetic counselling and prenatal diagnosis. Finally, we discuss testing technologies that are currently used, their benefits and limitations, and future directions for genetic testing in CCS.
Keywords: CYLD gene testing
Introduction
CYLD cutaneous syndrome (CCS) is an inclusive label for the inherited skin adnexal tumour syndromes Brooke–Spiegler Syndrome (BSS-OMIM 605041), familial cylindromatosis (FC – OMIM 132700) and multiple familial trichoepitheliomas (MFT-OMIM 601606). All three syndromes arise due to germline pathogenic variants in CYLD. CCS patients develop multiple benign hair follicle tumours on the head and torso,1 which grow from puberty and accumulate throughout adulthood2 (Figure 1A).3 The most frequent tumours seen in CCS are cylindromas, spiradenomas and trichoepitheliomas. The presentation of multiple cylindromas, and/or the related tumour spiradenoma, is exclusively seen in CCS, as is the presentation of multiple trichoepitheliomas in combination with cylindromas. In severe cases, tumours may cover most of the scalp or face, and sun-protected hair-bearing sites such as pubic skin can be affected. Despite an autosomal dominant pattern of inheritance, female CCS patients may be more severely affected, and whilst this has been reported in several families, the underlying mechanism for this disparity is not fully understood.4
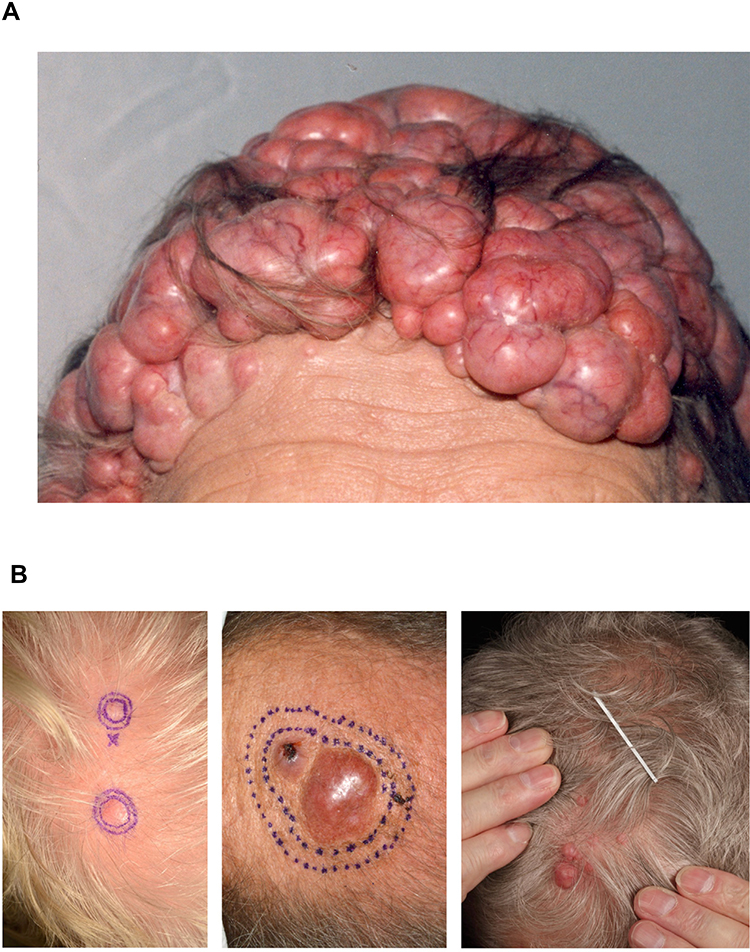 |
Figure 1 Cylindromas: germline and mosaic presentations. (A) Cylindromas and spiradenomas progressively grow and form a confluent mass, as seen in this severely affected patient with CCS. (B) Mosaic presentations of unilateral cylindromas, that may warrant skin biopsy and genetic testing to determine a recurrently detected pathogenic variant across multiple tumours. Image (B) reprinted from J Am Acad Dermatol, 81, Arefi M, Wilson V, Muthiah S et al. Diverse presentations of cutaneous mosaicism occur in CYLD cutaneous syndrome and may result in parent-to-child transmission. 1300–1307, Copyright (2019), with permission from Elsevier.24 |
CYLD is a tumour suppressor gene. Pathogenic variants in CYLD that give rise to CCS result in loss of function of the encoded protein. CCS patients are heterozygous for pathogenic variants in CYLD in all normal cells, ie, one copy of CYLD is mutated, but the remaining normal copy is functional. When the normal copy is mutated, the result is loss of functional CYLD, which can lead to the affected cell becoming neoplastic. It is believed that this “two-hit” mechanism5 occurs in hair follicle stem cells leading to the different skin adnexal tumours seen frequently in CCS. Recent genetic studies have confirmed that in cylindroma cells, both copies of CYLD are inactivated, and biallelic CYLD pathogenic variants alone appear sufficient to drive tumourigenesis.6 Malignant transformation of skin tumours has been described, where additional genetic changes in addition to biallelic CYLD mutations are reported.6
Clinical Phenotype of CCS
Cylindromas are smooth nodular tumours which are typically pink in colour, although can have a translucent appearance. Blood vessels may be seen across the surface of the tumour (Figure 2A). Cylindromas grow progressively over many years, and at presentation skin tumours may be several centimetres in size.7 Spiradenomas (Figure 2B) are also nodular and may have a striking blue/black colour; sometimes this is only noticed intraoperatively. Spiradenomas are usually reported to be painful and can grow quickly compared to cylindromas. Features of both lesions are also seen to occur in a single tumour, in keeping with the histological finding of cylindrospiradenoma discussed below. Trichoepitheliomas (Figure 2C) are skin-coloured, small, papular tumours, usually found on the perinasal, melo-labial and glabellar skin. In patients with European ancestry, they are usually 3–4mm across. In patients with African, Indian, and Chinese ancestry, trichoepitheliomas may be larger and the face may be the predominant tumour site, with few cases of confluent scalp tumours reported in the literature. Milia may also be seen in CCS patients and are sometimes the only indication of carrier status in a pedigree with CCS.8
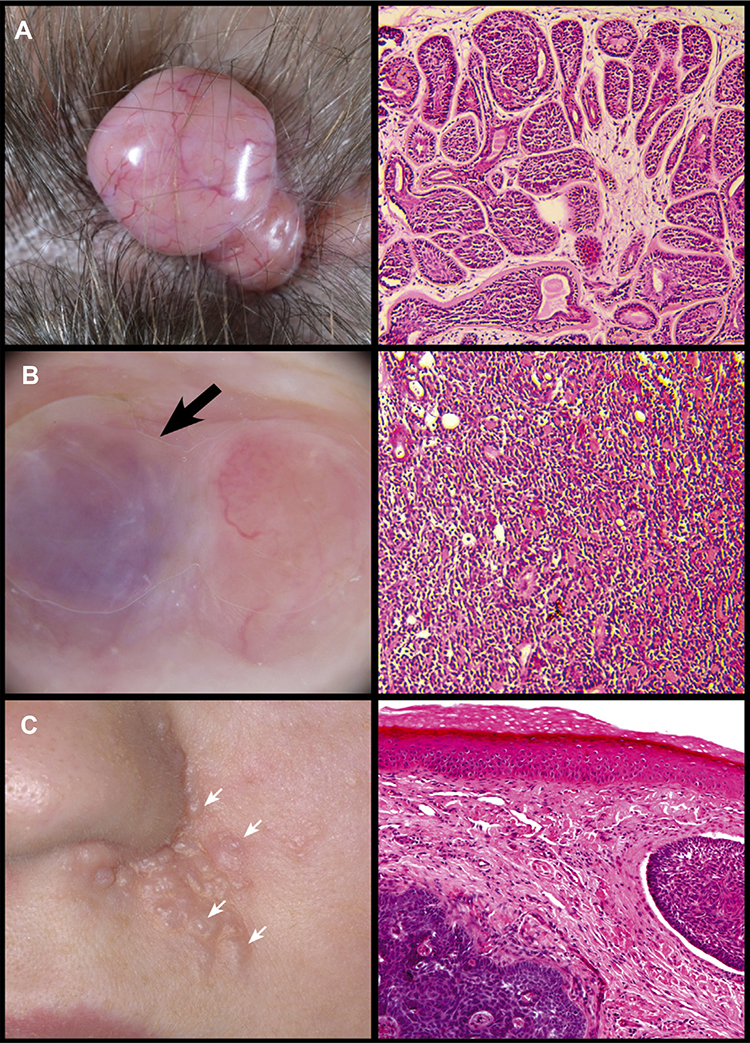 |
Figure 2 Skin tumours frequently seen in CCS. (A) Cylindroma. (B) Spiradenoma indicated by black arrow. (C) Trichoepithelioma indicated by white arrows. Reprinted from Dermatol Clin, 35 (1), Dubois A, Hodgson K, Rajan N. Understanding Inherited Cylindromas: Clinical Implications of Gene Discovery. 61–71, Copyright (2017), with permission from Elsevier.116 |
Histologic and Genetic Features of CCS Skin Tumours
CCS skin tumours are postulated to arise from hair follicle stem cells, due to clinical features such as presentation at hair-bearing sites and expression of histological markers seen in hair follicles (Figure 2A–C).9–11 Cylindromas are non-encapsulated nodular tumours that extend into the dermis. Basaloid tumour cells are usually arranged in a cylindrical pattern, apparent when the tumour is sectioned, which originally inspired the name cylindroma. Each “cylinder” is arranged in a jigsaw pattern, separated by a thickened basement membrane.12 Compared to cylindroma, tumour cells in spiradenomas are disorganised. A dense basophilic cellular proliferation is seen and typically lymphocytes are present. Skin tumours in CCS patients may demonstrate clinical and histological features consistent with both cylindroma and spiradenoma. Spiradenocylindroma is a term that has been coined to capture this frequent histophenotype.13,14 It is now recognised that cylindroma and spiradenoma represent different levels of organisation of the same tumour type. Epigenetic dysregulation of the Wnt signalling pathway has been shown to be associated with the development of spiradenoma.15 Recent studies of CCS tumours using whole-genome sequencing have shown that additional epigenetic modifier genes DNMT3A and BCOR are somatically mutated in these tumours, and may also explain the transition to spiradenoma.6 Notably, sporadic spiradenomas do not usually carry CYLD mutations, but instead have mutations in ALPK1.16 Trichoepitheliomas are comprised of basaloid cells, with palisading evident peripherally. Mesenchymal papillary bodies may also be seen. Again, biallelic CYLD mutations have been described in these tumours recently.6
The Clinical Burden of Disease in CCS
The “benign” histological label applied to CCS frequently fails to capture the clinical burden of disease faced by CYLD pathogenic variant carriers. The progressive growth of these tumours is not widely documented in the existing literature, and small studies have shown that the majority of CCS tumours grow over time, making early surgery a useful intervention. The severity of the phenotype may be assessed by multiple factors including the total number of skin tumours, tumour size, tumour symptoms and the presence of malignant tumours. In some cases, the need for repeated surgery from an early age and complete scalp removal serve as proxy markers of tumour burden. The tumours and the surgical procedures themselves can be disfiguring in patients who are severely affected. Total scalp excision is considered a last resort to address severely affected cases, where tumours form a confluent mass affecting large areas of the scalp.17 It is thought that early pre-emptive surgery may reduce the need for, or delay, total scalp excision. CCS tumours favour the ear canal, and the patency and consequently hearing of CCS patients may be impaired. This is a challenging site for surgical intervention, and tumours are preferably removed before they cause deafness.18 Exceptionally, patients with severe periocular trichoepitheliomas may experience impairment of visual fields and eyelid opening. The pubic and perineal skin is another favoured site of CCS tumour development. Painful spiradenomas at this site may impair sexual function. Patients with CCS have been reported with depression and social withdrawal due to their skin tumour burden.
Mosaic Presentations of CCS
Patients may present with only a cluster of CCS tumours arranged in a linear, often unilateral pattern.19,20 These lines are recognised to correspond to the lines of Blaschko (Figure 1B),21 which are linear bands of skin that are thought to develop from a single epidermal progenitor cell during fetal development.22 Such presentations reflect genetic mosaicism, where some cells carry a different genetic sequence from the rest of the person. There are two genetic scenarios relating to mosaicism to consider in CCS. The first is in an individual whose parent has CCS and has transmitted a heterozygous pathogenic variant in CYLD in the germline of the fetus. A further random somatic mutation in the remaining normal allele of CYLD occurring during early fetal development will result in both copies of the CYLD gene being inactivated in that progenitor cell. Daughter cells from this progenitor cell contributing to the line of Blaschko will be predisposed to develop cutaneous tumours, sometimes from childhood.23 Alternatively, a somatic mutation in CYLD may occur at during early fetal development in an individual whose parent does not have CCS, and in such cases the daughter cells contributing to a line of Blaschko will still have one working copy of the CYLD gene. These patients typically develop tumours in a unilateral cluster later in adult life, when additional second hits affecting the CYLD gene in this band occur. Genetic analysis of two or more such skin tumours should demonstrate a recurrently detected CYLD pathogenic variant. These patients may also demonstrate a low level of the same pathogenic variant in blood leucocyte DNA, demonstrable with next-generation sequencing techniques sensitive to detect low-level mosaicism, if the post-zygotic mutational event occurred in a cell that contributed to both skin and blood lineages.24
Malignancy in CCS
Cutaneous malignancy is well recognised in CCS, although it occurs relatively infrequently.25 Cylindroma and spiradenoma may both undergo malignant transformation, or apparent de novo skin malignancies may arise in CCS patients. Clinical features that should be reported by patients as they are associated with cutaneous malignancy are as follows: tumour ulceration; rapid tumour growth; tumour pain; intermittent bleeding from a tumour, colour change in the surface of the tumour; tethering of the tumour to underlying bone.26 Invasion through the skull plate has been observed,27,28 supporting the use of radiological imaging in selected advanced CCS cases pre-operatively. Malignant CCS tumours may metastasise to other tissues including the liver, lungs and bones.25 In patients with malignant metastatic disease, death from metastatic CCS disease has been reported in patients as young as 42 years of age.29
The histological features seen in malignant CCS tumours have been reported29 to include salivary gland type basal cell adenocarcinoma-like pattern, low-grade (BCAC-LG) and high-grade BCAC. Sarcomatoid (metaplastic) carcinoma and invasive adenocarcinoma are also recognised. Comprehensive genomic analyses, performed in just a small number of cases so far, have provided new insights, and suggest that molecular analysis of these tumours may aid classification.6 A case of poorly differentiated adenocarcinoma from the skin of a CCS patient that underwent whole-exome sequencing demonstrated somatic mutations in TP53 and EP300. In a case of BCAC-LG, a mutation in BCOR in addition to biallelic mutations in CYLD was found. Finally, a case of spiradenocarcinoma was found to have homozygous MBD4 mutations, and this tumour also had a mutation in the gene that encodes the epigenetic modifier KDM6A.
In addition to the adnexal carcinomas above, cutaneous squamous cell carcinoma (cSCC) arising in patients with CCS has been described in isolated reports30,31 as well as follicular cSCC.32 Trichoblastic carcinoma has been reported to arise from preexisting trichoblastoma in CCS,33 which can metastasise. BCC has been reported to arise from trichoepithelioma,34 and has been reported to be associated with mutations in PTCH1 in addition to CYLD. In a recent study of a South American family with CCS, BCC was reported in 25% of affected patients.35
Salivary Gland Tumours
A benign salivary gland tumour termed membranous basal cell adenoma (MBCA)36 is recognised to develop infrequently in CCS, usually after the age of 40. Presentation of MBCA in the parotid gland may be unilateral or bilateral. MBCA can be managed surgically, but recurs in up to 25% of cases.37 MBCA may transform to adenocarcinoma in CCS, but this is rare.38,39
Pulmonary Cylindromas
Lung metastases resulting from cutaneous cylindromas are recognised (Figure 3).40–42 These pulmonary tumours may be single or multiple and have been considered to be “benign” metastases, as there is no history of a primary malignant cutaneous cylindroma in the skin, an absence of lymph node disease, and benign histology in pulmonary cylindromas. Pulmonary tumours may present with breathlessness, and at times warrant surgical interventions such as endoscopic laser ablation or surgery to maintain lung function, as they may be present for several years. The true rate of these tumours in CCS patients is not currently known, as some of these tumours are asymptomatic. Recently, whole-genome sequencing of pulmonary cylindromas demonstrated a pathogenic variant in AKT1 in addition to biallelic mutations in CYLD. Mutational signature and clonality analysis of multiple tumours demonstrated the presence of a UV mutation signature, confirming origin from a cutaneous skin tumour.6
 |
Figure 3 Pulmonary and cutaneous cylindromas visualised radiologically and endoscopically. (A) Spatial location of cutaneous CCS tumours seen on a CT with contrast indicated in green, and pulmonary CCS tumours indicated in yellow. Adapted from (B) Intra bronchial CCS tumour visualised during bronchoscopy. Adapted with permission from Brown SM, Arefi M, Stones R, et al. Inherited pulmonary cylindromas: extending the phenotype of CYLD mutation carriers. Br J Dermatol. 2018;179:662–668. © 2018 The Authors. British Journal of Dermatology published by John Wiley & Sons Ltd on behalf of British Association of Dermatologists.41 |
CYLD as a Causative Gene for CCS
CYLD was discovered using DNA samples from carefully phenotyped pedigrees of families with FC, BSS and MFT. The CYLD locus was mapped to chromosome 1643 using linkage analysis of large CCS families from the North of England, and the CYLD gene was subsequently found to be mutated in patients with BSS, FC and MFT phenotypes.44–46 Linkage studies suggest that CCS is a single locus disease,45 and supports the encompassing term CCS. A minority of cases deemed “mutation-negative” by Sanger sequencing have been subsequently shown to have large rearrangements that disrupt CYLD,47 intronic variants that impact on CYLD splicing48 or large contiguous deletions that include CYLD and adjacent genes.49 Of note, whilst CYLD mutations in CCS cause loss of function, gain of function mutations in CYLD have recently been reported in familial frontotemporal dementia – amyotrophic lateral sclerosis,50 extending the roles of the CYLD gene in human disease.
CYLD spans a 56kb genomic footprint and has 20 exons (Figure 4A).51 It encodes an ubiquitin hydrolase enzyme that is involved in removing ubiquitin molecules that are “tagged” on a range of protein substrates as post-translational modifications, influencing the function, localisation and docking of ubiquitin-tagged proteins. CYLD demonstrates specificity for protein substrates tagged with lysine 63 linked and Met 1 ubiquitin chains.52,53 CYLD function has been extensively reviewed.54–57 Importantly in the context of CCS, CYLD negatively regulates several key cell survival pathways important in hair development, growth and maintenance, including NF-κB,58 Wnt,59 Notch60 and TGF-β,61 which are also important in inflammation and cancer. Deregulation of NF-KB and Wnt signalling pathways following loss of functional CYLD in CCS tumour cells has been demonstrated, and may play a central role in tumorigenesis.6,62
 |
Figure 4 CYLD gene pathogenic variants identified to date. (A) Exonic locations of CYLD pathogenic variants in CCS patients; note a predisposition to the 3’ end of the gene, from which the catalytic domains are encoded. (B) Frameshift and nonsense pathogenic variants resulting in a predicted truncating protein are the most frequent mutation type seen. |
Germline Pathogenic Variants in CCS
The first 21 pathogenic variants in CYLD were identified in 2000.44 Fifteen years later the number had risen to 95,63 increasing further to 107 in the last six years.64–69 Pathogenic variants are named according to Human Genome Variation Society (HGVS) nomenclature guidelines (www.HGVS.org). These are numbered with respect to the CYLD gene reference sequence (ENSG00000083799 corresponding to the CYLD gene and ENST00000311559 corresponding to the CYLD transcript). Here, we review previously published CYLD pathogenic variants (Table 1) and also report two which are novel: a heterozygous four-base deletion (c.1806_1809delCTTA) located in exon 12 detected in a Hungarian patient affected by CCS, and a nonsense pathogenic variant (c.2841C > G) in exon 20 identified in a CCS patient from the UK (Figure 4A). For the two novel reported mutations, written informed consents were obtained from the enrolled patients according to a protocol (Ref ID: BSS-GENET-001, BSS-GENET-002) approved by the Local Ethics Committee and the National Public Health and Medical Officer Service in adherence to the Helsinki guidelines.
 |
 |
 |
Table 1 List of Published Pathogenic Variants in CCS |
Across the 107 published pathogenic variants of the CYLD gene, almost all (99%) identified pathogenic variants are located between exon 9 and 20, most frequently in exon 16, 17 or 20. The encoded CYLD protein has two known functional domains: three cytoskeleton-associated glycine-rich domains (CAP-GLY) connecting CYLD to the microtubules and the ubiquitin-specific protease domain (USP) responsible for the deubiquitinase activity of the protein.70 The USP domain is coded by exons 12–20, the region in which exists the majority (80%) of identified CYLD pathogenic variants in CCS patients (Figure 4).63–69
Regarding mutation types (Figure 4B), the most common are frameshift mutations (39%), which are responsible for approximately half of the reported CYLD disease--causing variants.20,34,44,46–48,64–66,71–87 Nonsense mutations (22%) account for one quarter of the total identified mutations.44,47,68,69,71,73,74,76,83,85,88–94 Missense mutations are represented with a relatively low number, just 10% of the CYLD pathogenic variants,47,48,67,71,74,85,88,95–99 and splice site mutations 14%.44,66,69,82,83,97,100–104 A small proportion of reported pathogenic variants are due to large deletions24,49 and rearrangements.47 It is of interest to note that more than 10 large deletions including CYLD and adjacent genes have been described in children in an online database of developmental disorders, DECIPHER.105 Renal hypoplasia and intellectual disability have been reported in these cases, and only a single case currently reports a skin tumour, but no histological data is available for this case. In other cases with contiguous deletions involving CYLD, external ear abnormalities (pinnae), anal atresia and hypospadias in males has also been reported.24
Eleven percent of the CYLD pathogenic variants are recurrent.44,71–74,89–93,106,107 Haplotype analyses suggest that some of these recurrent pathogenic variants such as c.2806C > T and c.2272C > T, reported in geographically distant patients including Japanese, Chinese, Spanish, Dutch, Austrian, Canadian, Irish, Czech and Hungarian, are located in regions that are likely to be mutational hotspots in CYLD.44,48,73,74,90–93,106
Genotype–phenotype correlation has not been established, and mutations do not appear to predict severity. The phenotypes seen with each mutation type vary even within families, both in terms of phenotypic presentation and tumour number.93,106 Besides several other presumed yet unidentified genetic, environmental or lifestyle causes, the observed high phenotypic diversity in CCS patients may also be explained by the presence of genetic variants located outside the CYLD gene locus that could modify the phenotype.108 Genetic variants identified in STAT3, TRAF3 and in NBR1 may alter the effect of the loss of CYLD function on NF-κB activity in CCS tumour cells.108 The results of recent functional analyses support the role of these genes in the modulation of CYLD function,109 and support larger scale investigations to discover phenotype modifier genes.
Genetic Testing in CCS
Before considering genetic testing for CCS, genetic disorders where multiple facial papules may be a presenting sign should also be considered. The differential diagnosis, in addition to the MFT phenotype of CCS for multiple facial papules, includes the following conditions: Birt–Hogg–Dubé syndrome: presents with multiple facial fibrofolliculomas and trichodiscomas due to germline pathogenic variants in FLCN. Tuberous sclerosis: presents with facial angiofibromas that may mimic trichoepitheliomas, and is due to pathogenic variants in TSC1 and TSC2. Neurofibromatosis, due to pathogenic variants in NF1, has also been recognised as a mimic of CCS.110 Cowden syndrome may present with periauricular trichilemmomas, and is due to pathogenic variants in PTEN. Multiple facial trichoepitheliomas and severe hair loss should raise the possibility of Marie-Unna hypotrichosis, recently linked to a pathogenic variant in an untranslated inhibitory region in the open reading frame of HR.111 Multiple scalp tumours may represent trichilemmal cysts, and these are associated with pathogenic variants in PLCD1. These examples highlight the importance of dermatopathology assessment of these skin tumours. In some healthcare settings, these disorders are tested as a panel of genes, as discussed below.
Genetic testing to establish CYLD pathogenic variant status can be performed in patients satisfying clinical diagnostic criteria for CCS: 1) A patient with two or more cylindromas, spiradenomas or trichoepitheliomas (with at least one tumour being histologically confirmed); 2) A patient with a single (ideally histologically confirmed) cylindroma, spiradenoma or trichoepithelioma who has a family history of confirmed CCS (either based on genetic or histological information).
Germline Testing for CCS
The aim of germline DNA testing is to obtain sequence data covering the CYLD locus, typically of the coding exons where the majority of pathogenic variants lie (Figure 5). Peripheral blood leucocytes are the preferred DNA source but, in some cases, buccal swabs are used. It is helpful to check requirements with the receiving laboratory. Extracted DNA can be subject to a range of different assays, including PCR of the coding exons of CYLD, targeted capture of CYLD followed by next-generation sequencing, either alone, as part of a panel of genes, or as part of a whole-exome capture, with analysis of selected genes (virtual panel). In England, for example, CYLD testing is currently available as part of panel genes used for the clinical presentation of “Multiple benign monogenic skin tumours” (https://panelapp.genomicsengland.co.uk/panels/558/). Such sequence data from coding CYLD exons allow detection of single nucleotide missense, nonsense and splice site variants, as well as small indels that disrupt the reading frame. The study of RNA sequencing data derived from peripheral blood leucocytes can inform the reclassification of novel splice site variants of uncertain significance. The yield of such tests in patients with multiple scalp cylindromas ranges from 85% to 100%.74 In patients with the MFT phenotype alone, the yield may be lower, and in one study was 44%. Overall, the rate from this study of all CCS phenotypes was 72%.74 This aligns with our experience as a UK test centre receiving national referrals for CCS over a 5-year period, and detecting a pathogenic variant in 69% of the 56 pedigrees submitted.112 In mutation-negative cases, additional testing to determine copy number changes and large deletions may be achieved via MLPA or SNP arrays (Figure 5). In some settings, RNA sequencing data of mutation-negative cases can lead to the detection of deep intronic mutations.48 Increasingly, as whole-genome sequencing reduces in cost, this may be a means to obtain comprehensive information on genetic variation involving CYLD, including structural changes such as inversions or gene fusions. Providing CYLD coverage is of sufficient depth, there is the additional potential to detect mosaicism.
 |
Figure 5 A suggested testing strategy for CCS that addresses germline and mosaic presentations. *RNA from blood leucocytes require special collection tubes; **in principle such a patient may still yet harbour a germline pathogenic variant, and it remains the clinician’s decision if she would prefer to pursue GERMLINE testing first. Abbreviations: PV, pathogenic variant; SNV, single nucleotide variant. |
Risk to Immediate Family in a Genotyped Individual with Germline CCS
In most cases of germline CCS, the affected parent is clinically obvious. It can be helpful to determine the parental origin by confirmatory cascade testing where parental DNA is accessible and parents appear to be unaffected. CCS can present with subtle features, however, and some patients can be asymptomatic. If an unaffected parent of a proband is confirmed to have the proband’s pathogenic variant, the proband’s siblings have a 50% risk of also having inherited the familial pathogenic variant. In the scenario where neither parent is affected and does not carry the proband’s pathogenic variant, this may represent a de novo pathogenic mutation. It is important to recognise that the risk to the proband’s siblings in this case is still higher than the background population as either parent may have gonadal mosaicism. Cascade testing in the proband’s siblings can determine their genetic status.
Children of the proband themselves also have a 50% risk of inheriting the familial pathogenic variant. Importantly, mutation analysis cannot currently prognosticate severity. Because intrafamilial clinical variability is observed in CCS, offspring who inherit a CYLD pathogenic variant may be more or less severely affected than the transmitting parent.
Testing of Suspected Mosaic CCS Cases
Unilateral clustered CCS skin tumours should lead the physician to consider a diagnosis of mosaic CCS. Suspected mosaic CCS patients can have two different genetic mechanisms for the presentation of unilateral CCS tumours as discussed above. Unilateral tumours and a family history of CCS should lead to germline testing of blood leucocyte DNA.113 In the absence of a family history, and a negative blood DNA result, CYLD sequencing of DNA from two CCS tumours from such a cluster should reveal a recurring pathogenic CYLD variant. This should ideally be done on fresh biopsies, as paraffin-embedded samples can be challenging to sequence due to DNA fragmentation. This approach may also benefit some cases with evidence of bilateral or multiple clusters where blood DNA testing is negative (Figure 6). Gonadal mosaicism in such cases is possible as discussed below.24
 |
Figure 6 An overview of risk of transmission of germline and mosaic variants in CCS. Notes: Reprinted from J Am Acad Dermatol, 81, Arefi M, Wilson V, Muthiah S et al. Diverse presentations of cutaneous mosaicism occur in CYLD cutaneous syndrome and may result in parent-to-child transmission. 1300–1307, Copyright (2019), with permission from Elsevier.24 |
Genetic Counselling Issues for Genotyped CCS Patients
Family Planning
Genetic counselling can be informed by knowledge of genetic status, particularly in individuals that have not developed a clinical phenotype. Genetic testing is often discussed when a patient with CCS is considering family planning. A confirmed absence of the familial pathogenic variant in an at risk unaffected patient can assure the prevention of transmission of CCS to the next generation. The lack of ability to currently prognosticate severity in CCS based on genetic information makes a positive result less useful. In the context of prenatal testing, when a CYLD pathogenic variant is established in a family, prenatal testing and preimplantation genetic diagnosis (PGD) for pregnancies at increased risk are possible. A limitation of the ability to currently prognosticate severity and the variability of severity between generations should be discussed with the family when exploring such options.
Sporadic mosaic CCS cases may also have gonadal mosaicism, and there is a risk of parent-to-child transmission that is lower than in germline CCS.24 Knowledge of the mosaic pathogenic variant from skin tumour genetic assessment may allow techniques such DNA analysis of sperm in males to determine the level of gonadal mosaicism. Otherwise, PGD or other strategies may be considered in mosaic individuals who wish to use them when family planning.
Surveillance of CCS Patients
An annual full skin examination by a dermatologist is recommended in individuals with a clinical and/or genetic diagnosis of CCS. The frequency of follow-up can be tailored to the individual patient, as some patients require repeated surgery every three to four months if tumours continue to appear and grow. In patients with stable disease, or those without skin tumours, it may be reasonable to offer ad hoc follow-up. All CCS patients should be asked to report change in existing tumours such as rapid growth, tumours that appear different to existing lesions, or bleeding or ulcerated tumours. Dermatological assessment should be rapidly available for these patients, to determine if urgent excision is warranted. Clinical salivary gland examination may also be performed on an annual basis. Patients over 40 years of age reporting new-onset breathlessness should have pulmonary radiological imaging due to the potential risk of pulmonary cylindromas.
Clinical Trials for Genotyped CCS Patients
Treatment for CCS is predominantly surgical and is reviewed elsewhere.114 A placebo-controlled, phase 1b/2a clinical trial of a topical targeted kinase inhibitor (tropomyosin receptor kinase-Trk) has been reported and showed short-term safety.115 Additional research is needed to determine the utility of targeting Trk in CCS. There are isolated single-case reports of topical, intralesional or systemic interventions in CCS, but the generalisability of these are limited as they are not placebo controlled, they lack objective measures used to assess improvement and so far, only short-term follow-up is reported. Resources where new clinical trials are registered, and are regularly updated include www.clinicaltrials.gov and www.clinicaltrialsregister.eu.
Concluding Statements
Genetic testing in CCS has been advanced by next-generation sequencing technologies, in particular for mosaic presentations. Determining the pathogenic variant in CYLD in an affected patient with CCS influences genetic counselling and family planning decisions. Challenges ahead include the comprehensive delineation of disease modifying genes, such that clinicians are better placed to prognosticate regarding severity of future disease. In addition, the progressive molecular dissection of CCS skin tumours may yield targetable pathways that are amenable to pre-emptive therapeutic interventions in future clinical trials. Continued research in partnership with patients with CCS is essential to address these challenges.
Acknowledgments
NR’s research is also supported by the Newcastle NIHR Biomedical Research Centre (BRC). The project received funding from the Hungarian Research Development and Innovation Office grants GINOP-2.3.2-15-2016-00039 and EFOP-3.6.1-16-2016-00008. We thank the patients for granting permission to publish this information. We are grateful to the patients for allowing the use of their clinical images.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Rajan N, Langtry JA, Ashworth A, et al. Tumor mapping in 2 large multigenerational families with CYLD mutations: implications for disease management and tumor induction. Arch Dermatol. 2009;145:1277–1284. doi:10.1001/archdermatol.2009.262
2. Blake PW, Toro JR. Update of cylindromatosis gene (CYLD) mutations in Brooke-Spiegler syndrome: novel insights into the role of deubiquitination in cell signaling. Hum Mutat. 2009;30:1025–1036. doi:10.1002/humu.21024
3. Brown S, Worthy SA, Langtry JAA, et al. Tracking tumor kinetics in patients with germline CYLD mutations. J Am Acad Dermatol. 2018;79:949–951. doi:10.1016/j.jaad.2018.04.014
4. van Balkom ID, Hennekam RC. Dermal eccrine cylindromatosis. J Med Genet. 1994;31:321–324. doi:10.1136/jmg.31.4.321
5. Knudson AG. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–823. doi:10.1073/pnas.68.4.820
6. Davies HR, Hodgson K, Schwalbe E, et al. Epigenetic modifiers DNMT3A and BCOR are recurrently mutated in CYLD cutaneous syndrome. Nat Commun. 2019;10:4717. doi:10.1038/s41467-019-12746-w
7. Evans CD. Turban tumour. Br J Dermatol. 1954;66:434–443. doi:10.1111/j.1365-2133.1954.tb12575.x
8. Bajwa DS, Nasr B, Carmichael AJ, et al. Milia: a useful clinical marker of CYLD mutation carrier status. Clin Exp Dermatol. 2018;43:193–195. doi:10.1111/ced.13296
9. Massoumi R, Podda M, Fassler R, et al. Cylindroma as tumor of hair follicle origin. J Invest Dermatol. 2006;126:1182–1184. doi:10.1038/sj.jid.5700218
10. Sellheyer K. Spiradenoma and cylindroma originate from the hair follicle bulge and not from the eccrine sweat gland: an immunohistochemical study with CD200 and other stem cell markers. J Cutan Pathol. 2015;42:90–101. doi:10.1111/cup.12406
11. Cotton DW, Braye SG. Dermal cylindromas originate from the eccrine sweat gland. Br J Dermatol. 1984;111:53–61. doi:10.1111/j.1365-2133.1984.tb04016.x
12. Bilroth T. Die cylindergeschwulst (cylindroma) in Untersuchungenuber die Entwicklung der Blutgefasse, nebstBeobachtungenaus der koniglichenchirugischenUniverstats-Klink zu Berlin. Berlin: Riemer Germany; 1856:1855–1869.
13. Michal M, Lamovec J, Mukensnabl P, et al. Spiradenocylindromas of the skin: tumors with morphological features of spiradenoma and cylindroma in the same lesion: report of 12 cases. Pathol Int. 1999;49:419–425. doi:10.1046/j.1440-1827.1999.00890.x
14. Kazakov DV, Magro G, Kutzner H, et al. Spiradenoma and spiradenocylindroma with an adenomatous or atypical adenomatous component: a clinicopathological study of 6 cases. Am J Dermatopathol. 2008;30:436–441. doi:10.1097/DAD.0b013e3181812729
15. Rajan N, Burn J, Langtry J, et al. Transition from cylindroma to spiradenoma in CYLD-defective tumours is associated with reduced DKK2 expression. J Pathol. 2011;224:309–321. doi:10.1002/path.2896
16. Rashid M, van der Horst M, Mentzel T, et al. ALPK1 hotspot mutation as a driver of human spiradenoma and spiradenocarcinoma. Nat Commun. 2019;10:2213. doi:10.1038/s41467-019-09979-0
17. Freedman AM, Woods JE. Total scalp excision and auricular resurfacing for dermal cylindroma (turban tumor). Ann Plast Surg. 1989;22:50–57. doi:10.1097/00000637-198901000-00010
18. Langtry J, Rajan N. Pre-emptive intra-auricular electrosurgical resurfacing of cylindromas to prevent conductive deafness in CYLD cutaneous syndrome. Br J Dermatol. 2020;183:e188. doi:10.1111/bjd.19331
19. Chang YC, Colome-Grimmer M, Kelly E. Multiple trichoepitheliomas in the lines of Blaschko. Pediatr Dermatol. 2006;23:149–151. doi:10.1111/j.1525-1470.2006.00202.x
20. Furuichi M, Makino T, Yamakoshi T, et al. Blaschkoid distribution of cylindromas in a germline CYLD mutation carrier. Br J Dermatol. 2012;166:1376–1378. doi:10.1111/j.1365-2133.2012.10869.x
21. Blaschko A. Die Nevenverteilung in derhant in ihrer Beziehung zuden Ekrankungender Haut. In: Verhand Deust Dermatol Gessel. Breslan: VH Congress; 1901.
22. Moss C, Larkins S, Stacey M, et al. Epidermal mosaicism and Blaschko’s lines. J Med Genet. 1993;30:752–755. doi:10.1136/jmg.30.9.752
23. Happle R. Segmental forms of autosomal dominant skin disorders: different types of severity reflect different states of zygosity. Am J Med Genet. 1996;66:241–242. doi:10.1002/(SICI)1096-8628(19961211)66:2<241::AID-AJMG24>3.0.CO;2-S
24. Arefi M, Wilson V, Muthiah S, et al. Diverse presentations of cutaneous mosaicism occur in CYLD cutaneous syndrome and may result in parent-to-child transmission. J Am Acad Dermatol. 2019;81:1300–1307. doi:10.1016/j.jaad.2019.05.021
25. Gerretsen AL, van der Putte SC, Deenstra W, et al. Cutaneous cylindroma with malignant transformation. Cancer. 1993;72:1618–1623. doi:10.1002/1097-0142(19930901)72:5<1618::AID-CNCR2820720521>3.0.CO;2-5
26. Durani BK, Kurzen H, Jaeckel A, et al. Malignant transformation of multiple dermal cylindromas. Br J Dermatol. 2001;145:653–656. doi:10.1046/j.1365-2133.2001.04460.x
27. Serracino HS, Kleinschmidt-Demasters BK. Skull invaders: when surgical pathology and neuropathology worlds collide. J Neuropathol Exp Neurol. 2013;72:600–613. doi:10.1097/NEN.0b013e318299c40f
28. Gildea JH, Lillehei KO, Golitz LE, et al. Benign cylindroma causing transcalvarial invasion in a patient with familial cylindromatosis. Clin Neuropathol. 2007;26:125–130. doi:10.5414/NPP26125
29. Kazakov DV, Zelger B, Rutten A, et al. Morphologic diversity of malignant neoplasms arising in preexisting spiradenoma, cylindroma, and spiradenocylindroma based on the study of 24 cases, sporadic or occurring in the setting of Brooke-Spiegler syndrome. Am J Surg Pathol. 2009;33:705–719. doi:10.1097/PAS.0b013e3181966762
30. Ma H, Feng S, Pei W, et al. Twelve years’ observation of multiple familial trichoepithelioma with squamous carcinoma. Indian J Dermatol. 2016;61:348. doi:10.4103/0019-5154.182464
31. Ganguly S, Jaykar KC, Banerjee PK, et al. Multiple familial trichoepitheliomas in association with squamous cell carcinoma. Indian Dermatol Online J. 2012;3:151–153. doi:10.4103/2229-5178.96726
32. Dubois A, Mestre T, Oliphant T, et al. Squamous cell carcinoma and multiple familial trichoepitheliomas: a recurrent association. Acta Derm Venereol. 2018;98:910–911. doi:10.2340/00015555-2988
33. Schulz T, Proske S, Hartschuh W, et al. High-grade trichoblastic carcinoma arising in trichoblastoma: a rare adnexal neoplasm often showing metastatic spread. Am J Dermatopathol. 2005;27:9–16. doi:10.1097/01.dad.0000142240.93956.cb
34. Melly L, Lawton G, Rajan N. Basal cell carcinoma arising in association with trichoepithelioma in a case of Brooke-Spiegler syndrome with a novel genetic mutation in CYLD. J Cutan Pathol. 2012;39:977–978. doi:10.1111/j.1600-0560.2012.01952.x
35. Arruda AP, Cardoso-Dos-Santos AC, Mariath LM, et al. A large family with CYLD cutaneous syndrome: medical genetics at the community level. J Community Genet. 2020;11:279–284. doi:10.1007/s12687-019-00447-2
36. Jungehulsing M, Wagner M, Damm M. Turban tumour with involvement of the parotid gland. J Laryngol Otol. 1999;113:779–783. doi:10.1017/S0022215100145190
37. Zarbo RJ. Salivary gland neoplasia: a review for the practicing pathologist. Mod Pathol. 2002;15:298–323. doi:10.1038/modpathol.3880525
38. Hyman BA, Scheithauer BW, Weiland LH, et al. Membranous basal cell adenoma of the parotid gland. Malignant transformation in a patient with multiple dermal cylindromas. Arch Pathol Lab Med. 1988;112:209–211.
39. Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update. Head Neck Pathol. 2016;10:125–130. doi:10.1007/s12105-016-0705-x
40. Vernon HJ, Olsen EA, Vollmer RT. Autosomal dominant multiple cylindromas associated with solitary lung cylindroma. J Am Acad Dermatol. 1988;19:397–400. doi:10.1016/S0190-9622(88)70186-3
41. Brown SM, Arefi M, Stones R, et al. Inherited pulmonary cylindromas: extending the phenotype of CYLD mutation carriers. Br J Dermatol. 2018;179:662–668. doi:10.1111/bjd.16573
42. Bulau JE, Kreipe HH, Jessen E, et al. Pulmonary cylindromas in CYLD cutaneous syndrome: a rare differential diagnosis of pulmonary adenoid cystic carcinoma. Clin Lung Cancer. 2021. doi:10.1016/j.cllc.2021.03.016
43. Biggs PJ, Wooster R, Ford D, et al. Familial cylindromatosis (turban tumour syndrome) gene localised to chromosome 16q12-q13: evidence for its role as a tumour suppressor gene. Nat Genet. 1995;11:441–443. doi:10.1038/ng1295-441
44. Bignell GR, Warren W, Seal S, et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet. 2000;25:160–165. doi:10.1038/76006
45. Takahashi M, Rapley E, Biggs PJ, et al. Linkage and LOH studies in 19 cylindromatosis families show no evidence of genetic heterogeneity and refine the CYLD locus on chromosome 16q12-q13. Hum Genet. 2000;106:58–65. doi:10.1007/s004399900227
46. Zhang XJ, Liang YH, He PP, et al. Identification of the cylindromatosis tumor-suppressor gene responsible for multiple familial trichoepithelioma. J Invest Dermatol. 2004;122:658–664. doi:10.1111/j.0022-202X.2004.22321.x
47. van den Ouweland AM, Elfferich P, Lamping R, et al. Identification of a large rearrangement in CYLD as a cause of familial cylindromatosis. Fam Cancer. 2011;10:127–132. doi:10.1007/s10689-010-9393-y
48. Kazakov DV, Thoma-Uszynski S, Vanecek T, et al. A case of Brooke-Spiegler syndrome with a novel germline deep intronic mutation in the CYLD gene leading to intronic exonization, diverse somatic mutations, and unusual histology. Am J Dermatopathol. 2009;31:664–673. doi:10.1097/DAD.0b013e3181a05dad
49. Vanecek T, Halbhuber Z, Kacerovska D, et al. Large germline deletions of the CYLD gene in patients with Brooke-Spiegler syndrome and multiple familial trichoepithelioma. Am J Dermatopathol. 2014;36:868–874. doi:10.1097/DAD.0000000000000068
50. Dobson-Stone C, Hallupp M, Shahheydari H, et al. CYLD is a causative gene for frontotemporal dementia - amyotrophic lateral sclerosis. Brain. 2020;143:783–799. doi:10.1093/brain/awaa039
51. Cunningham F, Amode MR, Barrell D, et al. Ensembl 2015. Nucleic Acids Res. 2015;43:D662–9. doi:10.1093/nar/gku1010
52. Kupka S, De Miguel D, Draber P, et al. SPATA2-mediated binding of CYLD to HOIP enables CYLD recruitment to signaling complexes. Cell Rep. 2016;16:2271–2280. doi:10.1016/j.celrep.2016.07.086
53. Elliott PR, Leske D, Hrdinka M, et al. SPATA2 links CYLD to LUBAC, activates CYLD, and controls LUBAC signaling. Mol Cell. 2016;63:990–1005. doi:10.1016/j.molcel.2016.08.001
54. Lork M, Verhelst K, Beyaert R. CYLD, A20 and OTULIN deubiquitinases in NF-kappaB signaling and cell death: so similar, yet so different. Cell Death Differ. 2017;24:1172–1183. doi:10.1038/cdd.2017.46
55. Hellerbrand C, Massoumi R. Cylindromatosis–a protective molecule against liver diseases. Med Res Rev. 2016;36:342–359. doi:10.1002/med.21381
56. Masoumi KC, Shaw-Hallgren G, Massoumi R. Tumor suppressor function of CYLD in nonmelanoma skin cancer. J Skin Cancer. 2011;2011:614097. doi:10.1155/2011/614097
57. Massoumi R. Ubiquitin chain cleavage: CYLD at work. Trends Biochem Sci. 2010;35:392–399. doi:10.1016/j.tibs.2010.02.007
58. Kloepper JE, Ernst N, Krieger K, et al. NF-κB activity is required for anagen maintenance in human hair follicles in vitro. J Invest Dermatol. 2014;134:2036–2038. doi:10.1038/jid.2014.82
59. Tauriello DV, Haegebarth A, Kuper I, et al. Loss of the tumor suppressor CYLD enhances Wnt/β-catenin signaling through K63-linked ubiquitination of Dvl. Mol Cell. 2010;37:607–619. doi:10.1016/j.molcel.2010.01.035
60. Rajan N, Elliott RJ, Smith A, et al. The cylindromatosis gene product, CYLD, interacts with MIB2 to regulate notch signalling. Oncotarget. 2014;5:12126–12140. doi:10.18632/oncotarget.2573
61. Lim JH, Jono H, Komatsu K, et al. CYLD negatively regulates transforming growth factor-β-signalling via deubiquitinating Akt. Nat Commun. 2012;3:771. doi:10.1038/ncomms1776
62. Rajan N, Elliott R, Clewes O, et al. Dysregulated TRK signalling is a therapeutic target in CYLD defective tumours. Oncogene. 2011;30:4243–4260. doi:10.1038/onc.2011.133
63. Nagy N, Farkas K, Kemeny L, et al. Phenotype-genotype correlations for clinical variants caused by CYLD mutations. Eur J Med Genet. 2015;58:271–278. doi:10.1016/j.ejmg.2015.02.010
64. Dubois A, Wilson V, Bourn D, et al. CYLD GeneticTesting for Brooke-Spiegler syndrome, familial cylindromatosis and multiple familial trichoepitheliomas. PLoS Curr. 2015;7. doi:10.1371/currents.eogt.45c4e63dd43d62e12228cc5264d6a0db
65. Hunstig F, Schulz S, Nieten I, et al. A case of Brooke-Spiegler syndrome with a novel mutation in the CYLD gene in a patient with aggressive non-Hodgkin’s lymphoma. J Cancer Res Clin Oncol. 2016;142:845–848. doi:10.1007/s00432-015-2079-y
66. Tantcheva-Poor I, Vanecek T, Lurati MC, et al. Report of three novel germline CYLD mutations in unrelated patients with Brooke-Spiegler syndrome, including classic phenotype, multiple familial trichoepitheliomas and malignant transformation. Dermatology. 2016;232:30–37. doi:10.1159/000437303
67. Shiver M, Hughes M, Naylor M, et al. A novel CYLD gene mutation and multiple basal cell carcinomas in a patient with Brooke-Spiegler syndrome. Clin Exp Dermatol. 2016;41:98–100. doi:10.1111/ced.12669
68. Pinho AC, Gouveia MJ, Gameiro AR, et al. Brooke-Spiegler Syndrome - an underrecognized cause of multiple familial scalp tumors: report of a new germline mutation. J Dermatol Case Rep. 2015;9:67–70. doi:10.3315/jdcr.2015.1208
69. Malzone MG, Campanile AC, Losito NS, et al. Brooke-Spiegler syndrome presenting multiple concurrent cutaneous and parotid gland neoplasms: cytologic findings on fine-needle sample and description of a novel mutation of the CYLD gene. Diagn Cytopathol. 2015;43:654–658. doi:10.1002/dc.23275
70. Gao J, Huo L, Sun X, et al. The tumor suppressor CYLD regulates microtubule dynamics and plays a role in cell migration. J Biol Chem. 2008;283:8802–8809. doi:10.1074/jbc.M708470200
71. Sima R, Vanecek T, Kacerovska D, et al. Brooke-Spiegler syndrome: report of 10 patients from 8 families with novel germline mutations: evidence of diverse somatic mutations in the same patient regardless of tumor type. Diagn Mol Pathol. 2010;19:83–91. doi:10.1097/PDM.0b013e3181ba2d96
72. Cakmak Genc G, Dursun A, Nagy N, et al. Brooke-Spiegler syndrome: two patients from a Turkish family with multiple familial trichoepithelioma. Am J Dermatopathol. 2019;41:778–780. doi:10.1097/DAD.0000000000001265
73. Bowen S, Gill M, Lee DA, et al. Mutations in the CYLD gene in Brooke-Spiegler syndrome, familial cylindromatosis, and multiple familial trichoepithelioma: lack of genotype-phenotype correlation. J Invest Dermatol. 2005;124:919–920. doi:10.1111/j.0022-202X.2005.23688.x
74. Saggar S, Chernoff KA, Lodha S, et al. CYLD mutations in familial skin appendage tumours. J Med Genet. 2008;45:298–302. doi:10.1136/jmg.2007.056127
75. Wu JW, Xiao SX, Huo J, et al. A novel frameshift mutation in the cylindromatosis (CYLD) gene in a Chinese family with multiple familial trichoepithelioma. Arch Dermatol Res. 2014;306:857–860. doi:10.1007/s00403-014-1499-x
76. Grossmann P, Vanecek T, Steiner P, et al. Novel and recurrent germline and somatic mutations in a cohort of 67 patients from 48 families with Brooke-Spiegler syndrome including the phenotypic variant of multiple familial trichoepitheliomas and correlation with the histopathologic findings in 379 biopsy specimens. Am J Dermatopathol. 2013;35:34–44.
77. Hongli J, Lifeng Z, Zhenzhen F, et al. Detection of grass carp reovirus (GCRV) with monoclonal antibodies. Arch Virol. 2014;159:649–655. doi:10.1007/s00705-013-1864-7
78. Reuven B, Margarita I, Dov H, et al. Multiple trichoepitheliomas associated with a novel heterozygous mutation in the CYLD gene as an adjunct to the histopathological diagnosis. Am J Dermatopathol. 2013;35:445–447. doi:10.1097/DAD.0b013e31827132af
79. Hester CC, Moscato EE, Kazakov DV, et al. A new Cylindromatosis (CYLD) gene mutation in a case of Brooke-Spiegler syndrome masquerading as basal cell carcinoma of the eyelids. Ophthalmic Plast Reconstr Surg. 2013;29:e10–1. doi:10.1097/IOP.0b013e3182565c41
80. Ying ZX, Ma HQ, Liu Y, et al. A novel mutation of CYLD in a Chinese family with multiple familial trichoepithelioma. J Eur Acad Dermatol Venereol. 2012;26:1420–1423. doi:10.1111/j.1468-3083.2011.04309.x
81. Chen M, Liu H, Fu X, et al. Mutation analysis of the CYLD gene in two Chinese families with multiple familial Trichoepithelioma. Australas J Dermatol. 2011;52:146–147. doi:10.1111/j.1440-0960.2011.00763.x
82. Kazakov DV, Vanecek T, Zelger B, et al. Multiple (familial) trichoepitheliomas: a clinicopathological and molecular biological study, including CYLD and PTCH gene analysis, of a series of 16 patients. Am J Dermatopathol. 2011;33:251–265. doi:10.1097/DAD.0b013e3181f7d373
83. Liang YH, Sun CS, Ye XY, et al. Novel substitution and frameshift mutations of CYLD in two Chinese families with multiple familial trichoepithelioma. Br J Dermatol. 2008;158:1156–1158. doi:10.1111/j.1365-2133.2008.08491.x
84. Heinritz W, Grunewald S, Strenge S, et al. A case of Brooke-Spiegler syndrome with a new mutation in the CYLD gene. Br J Dermatol. 2006;154:992–994. doi:10.1111/j.1365-2133.2006.07142.x
85. Zheng G, Hu L, Huang W, et al. CYLD mutation causes multiple familial trichoepithelioma in three Chinese families. Hum Mutat. 2004;23:400. doi:10.1002/humu.9231
86. Salhi A, Bornholdt D, Oeffner F, et al. Multiple familial trichoepithelioma caused by mutations in the cylindromatosis tumor suppressor gene. Cancer Res. 2004;64:5113–5117. doi:10.1158/0008-5472.CAN-04-0307
87. Poblete Gutierrez P, Eggermann T, Holler D, et al. Phenotype diversity in familial cylindromatosis: a frameshift mutation in the tumor suppressor gene CYLD underlies different tumors of skin appendages. J Invest Dermatol. 2002;119:527–531. doi:10.1046/j.1523-1747.2002.01839.x
88. Almeida S, Maillard C, Itin P, et al. Five new CYLD mutations in skin appendage tumors and evidence that aspartic acid 681 in CYLD is essential for deubiquitinase activity. J Invest Dermatol. 2008;128:587–593. doi:10.1038/sj.jid.5701045
89. Oranje AP, Halley D, den Hollander JC, et al. Multiple familial trichoepithelioma and familial cylindroma: one cause! J Eur Acad Dermatol Venereol. 2008;22:1395–1396. doi:10.1111/j.1468-3083.2008.02648.x
90. Young AL, Kellermayer R, Szigeti R, et al. CYLD mutations underlie Brooke-Spiegler, familial cylindromatosis, and multiple familial trichoepithelioma syndromes. Clin Genet. 2006;70:246–249. doi:10.1111/j.1399-0004.2006.00667.x
91. Oiso N, Mizuno N, Fukai K, et al. Mild phenotype of familial cylindromatosis associated with an R758X nonsense mutation in the CYLD tumour suppressor gene. Br J Dermatol. 2004;151:1084–1086. doi:10.1111/j.1365-2133.2004.06231.x
92. Zhang G, Huang Y, Yan K, et al. Diverse phenotype of Brooke-Spiegler syndrome associated with a nonsense mutation in the CYLD tumor suppressor gene. Exp Dermatol. 2006;15:966–970. doi:10.1111/j.1600-0625.2006.00501.x
93. Farkas K, Deak BK, Sanchez LC, et al. The CYLD p.R758X worldwide recurrent nonsense mutation detected in patients with multiple familial trichoepithelioma type 1, Brooke-Spiegler syndrome and familial cylindromatosis represents a mutational hotspot in the gene. BMC Genet. 2016;17:36. doi:10.1186/s12863-016-0346-9
94. Amaro C, Freitas I, Lamarao P, et al. Multiple trichoepitheliomas–a novel mutation in the CYLD gene. J Eur Acad Dermatol Venereol. 2010;24:844–846. doi:10.1111/j.1468-3083.2009.03497.x
95. Espana A, Garcia-Amigot F, Aguado L, et al. A novel missense mutation in the CYLD gene in a Spanish family with multiple familial trichoepithelioma. Arch Dermatol. 2007;143:1209–1210. doi:10.1001/archderm.143.9.1209
96. Zuo YG, Xu Y, Wang B, et al. A novel mutation of CYLD in a Chinese family with multiple familial trichoepithelioma and no CYLD protein expression in the tumour tissue. Br J Dermatol. 2007;157:818–821. doi:10.1111/j.1365-2133.2007.08081.x
97. Ly H, Black MM, Robson A. Case of the Brooke-Spiegler syndrome. Australas J Dermatol. 2004;45:220–222. doi:10.1111/j.1440-0960.2004.00101.x
98. Wang FX, Yang LJ, Li M, et al. A novel missense mutation of CYLD gene in a Chinese family with multiple familial trichoepithelioma. Arch Dermatol Res. 2010;302:67–70. doi:10.1007/s00403-009-1003-1
99. Nagy N, Farkas K, Kinyo A, et al. A novel missense mutation of the CYLD gene identified in a Hungarian family with Brooke-Spiegler syndrome. Exp Dermatol. 2012;21:967–969. doi:10.1111/exd.12040
100. Huang TM, Chao SC, Lee JY. A novel splicing mutation of the CYLD gene in a Taiwanese family with multiple familial trichoepithelioma. Clin Exp Dermatol. 2009;34:77–80. doi:10.1111/j.1365-2230.2008.02870.x
101. Nasti S, Pastorino L, Bruno W, et al. Five novel germline function-impairing mutations of CYLD in Italian patients with multiple cylindromas. Clin Genet. 2009;76:481–485. doi:10.1111/j.1399-0004.2009.01259.x
102. Kazakov DV, Schaller J, Vanecek T, et al. Brooke-Spiegler syndrome: report of a case with a novel mutation in the CYLD gene and different types of somatic mutations in benign and malignant tumors. J Cutan Pathol. 2010;37:886–890. doi:10.1111/j.1600-0560.2010.01511.x
103. Schmidt A, Schmitz R, Giefing M, et al. Rare occurrence of biallelic CYLD gene mutations in classical Hodgkin lymphoma. Genes Chromosomes Cancer. 2010;49:803–809.
104. Kacerovska D, Szep Z, Kollarikova L, et al. A novel germline mutation in the CYLD gene in a Slovak patient with Brooke-Spiegler syndrome. Cesk Patol. 2013;49:89–92.
105. Firth HV, Richards SM, Bevan AP, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Hum Genet. 2009;84:524–533. doi:10.1016/j.ajhg.2009.03.010
106. Nagy N, Rajan N, Farkas K, et al. A mutational hotspot in CYLD causing cylindromas: a comparison of phenotypes arising in different genetic backgrounds. Acta Derm Venereol. 2013;93:743–745. doi:10.2340/00015555-1590
107. Hu G, Onder M, Gill M, et al. A novel missense mutation in CYLD in a family with Brooke-Spiegler syndrome. J Invest Dermatol. 2003;121:732–734. doi:10.1046/j.1523-1747.2003.12514.x
108. Pap EM, Farkas K, Szell M, et al. Identification of putative phenotype-modifying genetic factors associated with phenotypic diversity in Brooke-Spiegler syndrome. Exp Dermatol. 2020;29:1017–1020. doi:10.1111/exd.14161
109. Danis J, Kelemen E, Rajan N, et al. TRAF3 and NBR1 both influence the effect of the disease-causing CYLD(Arg936X) mutation on NF-κB activity. Exp Dermatol. 2021. doi:10.1111/exd.14365
110. Baden HP. Cylindromatosis simulating neurofibromatosis. N Engl J Med. 1962;267:296–297. doi:10.1056/NEJM196208092670608
111. Huang Y, Cai C, Ren L, et al. Marie Unna hereditary hypotrichosis accompanied by multiple familial trichoepithelioma in a Chinese family. J Dermatol. 2019;46:413–417. doi:10.1111/1346-8138.14811
112. Dubois A, Rajan N. CYLD cutaneous syndrome. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews((R)). Seattle: University of Washington, Seattle Copyright 1993–2020; 1993.
113. Saunders H, Tucker P, Saurine T, et al. Pedigree of multiple benign adnexal tumours of Brooke-Spiegler type. Australas J Dermatol. 2003;44:144–148. doi:10.1046/j.1440-0960.2003.00663.x
114. Rajan N, Trainer AH, Burn J, et al. Familial cylindromatosis and Brooke-Spiegler syndrome: a review of current therapeutic approaches and the surgical challenges posed by two affected families. Dermatol Surg. 2009;35:845–852. doi:10.1111/j.1524-4725.2009.01142.x
115. Danilenko M, Stamp E, Stocken DD, et al. Targeting tropomyosin receptor kinase in cutaneous CYLD defective tumors with pegcantratinib: the TRAC randomized clinical trial. JAMA Dermatol. 2018;154:913–921. doi:10.1001/jamadermatol.2018.1610
116. Dubois A, Hodgson K, Rajan N. Understanding inherited cylindromas: clinical implications of gene discovery. Dermatol Clin. 2017;35(1):61–71. doi:10.1016/j.det.2016.08.002
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.