Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 16
Genetic Susceptibility to Chronic Kidney Disease: Links, Risks and Management
Authors Sawaf H, Gudura TT, Dorobisz S, Sandy D, Wang X, Bobart SA ![]()
Received 5 October 2022
Accepted for publication 24 December 2022
Published 5 January 2023 Volume 2023:16 Pages 1—15
DOI https://doi.org/10.2147/IJNRD.S363041
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pravin Singhal
Hanny Sawaf,1 Tariku T Gudura,1 Sylvester Dorobisz,1 Dianne Sandy,2 Xiangling Wang,1 Shane A Bobart2
1Department of Kidney Medicine, Cleveland Clinic, Cleveland, OH, USA; 2Department of Kidney Medicine, Cleveland Clinic Florida, Weston, FL, USA
Correspondence: Shane A Bobart, Department of Kidney Medicine, 2950 Cleveland Clinic Blvd, Weston, FL, 33331, USA, Email [email protected]
Abstract: Chronic kidney disease (CKD) is associated with significant morbidity and mortality worldwide. In recent years, our understanding of genetic causes of CKD has expanded significantly with several renal conditions having been identified. This review discusses the current landscape of genetic kidney disease and their potential treatment options. This review will focus on cystic kidney disease, glomerular disease with genetic associations, congenital anomalies of kidneys and urinary tract (CAKUT), autosomal dominant-tubulointerstitial kidney disease (ADTKD), inherited nephrolithiasis and nephrocalcinosis.
Keywords: genetics, chronic kidney disease, end stage kidney disease, cystic kidney disease
Introduction
The most common causes of CKD include hypertension, diabetes mellitus, glomerular disease as well as polycystic kidney disease (PKD), which is a genetic disorder. Other forms of genetic kidney disease include those with glomerular patterns of injury such as genetic nephrotic syndrome, focal segmental glomerulosclerosis, APOL1-related kidney disease and atypical hemolytic uremic syndrome. Some systemic disorders have glomerular patterns of injury such as Alport syndrome and Fabry disease. There are also congenital anomalies of kidneys and urinary tract (CAKUT), as well as tubulointerstitial patterns of kidney disease such as autosomal dominant-tubulointerstitial kidney disease (ADTKD). Finally, channelopathies may cause nephrolithiasis in the setting of primary hyperoxaluria, and Bartter and Gitelman syndromes can result in nephrocalcinosis that leads to progressive kidney disease.
As the ability to test for genetic disease has evolved from Sanger sequencing to next-generation sequencing, gene panel analysis is now widely used to test for specific genes implicated in the development in genetic kidney disease, resulting in more targeted and cost-effective testing. As a result, it is important for clinicians to understand the presentation, risks and management of these genetic kidney diseases to provide comprehensive care to those with genetic kidney disease.1
Cystic Kidney Disease
Autosomal Dominant Polycystic Kidney Disease
Autosomal dominant polycystic kidney disease (ADPKD) is the most common hereditary cystic kidney disease and accounts for 5–10% of end stage kidney disease (ESKD). Most cases are caused by mutations of the PKD 1 gene on chromosome 16 and the PKD2 gene on chromosome 4, which encode the polycystin 1 and 2 proteins, respectively. The PKD 1 mutation accounts for approximately 85% of cases and is associated with more severe disease with onset of renal failure in the 6th decade, compared to those with the PKD 2 mutation who commonly manifest after the 7th decade of life. Patients are often asymptomatic with normal renal function in young adulthood due to compensatory hyperfiltration of the preserved nephrons. However, as the progressive destruction of nephrons occurs, accompanied by severe distortion of the renal architecture, symptoms begin to manifest in the 4th decade of life. Early diagnosis may sometimes be achieved when there is known family history of the disease. Hypertension, polyuria, hematuria, nephrolithiasis, and complications related to cyst burden such as abdominal pain, recurrent infections, and progressive renal failure necessitating kidney replacement therapy (KRT) are common presentations for patients with ADPKD.2,3
Autosomal Recessive Polycystic Kidney Disease
Autosomal recessive polycystic kidney disease (ARPKD) is a rare fibrocystic reno-hepatic genetic disorder mainly affecting infants and children. It occurs due to mutation of PKHD1 gene that encodes the fibrocystin protein, leading to formation of liver and renal cysts primarily affecting the collecting duct, unlike ADPKD that affects any segment of nephron. Clinical manifestations depend on the degree of loss of function of fibrocystin with complete loss causing severe disease. ARPKD may present with oligohydramnios in-utero, congenital hepatomegaly causing pulmonary hypoplasia during infancy and bilateral renal cysts with early onset hypertension during early childhood. About half of affected children progress to ESKD in early childhood.4,5
Diagnosis and Treatment of ADPKD and ARPKD
Kidney ultrasound, computed tomography (CT) and magnetic resonance imaging (MRI) are common modalities used to assess the number and size of cysts for diagnosis, and for measuring the total kidney volume (TKV). The height adjusted TKV is used in the Mayo Clinic ADPKD classification to identify patients at risk of progression who may benefit from treatment. Genetic testing is recommended for those patients with atypical presentations, or those with unknown family history presenting with clinical features suggestive of inherited cystic disease.6,7 Intracranial cerebral aneurysms are common in ADPKD and it is controversial whether specific patients should be screened such as those with a family history of sudden death, and those with a personal or family history of aneurysm. However, given the high risk of rupture, some studies advocate both from a clinical and cost-effectiveness standpoint for universal screening in ADPKD.8
The goals of treatment for patients with ADPKD and ARPKD are similar. Blood pressure control with angiotensin converting enzyme inhibition (ACEi) and angiotensin receptor blockers (ARB), low sodium intake and increased fluid intake with a goal of achieving low urine osmolality are associated with slower rate of decline of renal function. Tolvaptan, which is an oral V2 receptor antagonist, may be considered in those at high risk of disease progression and is approved by the FDA for the treatment of ADPKD only. Tolvaptan has been demonstrated to slow the rate of decline of glomerular filtration rate (GFR), improve blood pressure control and minimize pain from large kidney cysts. Tolvaptan use requires frequent monitoring of patients’ laboratory parameters, especially liver function tests as there is a risk of liver toxicity with its use. Those patients who progress to ESKD require kidney replacement therapy (KRT) with dialysis or kidney transplantation. The overall outcome post replacement is comparable with non ADPKD patients.9,10
Nephronophthisis
Nephronophthisis (NPHP) is a common autosomal recessive cystic kidney disorder leading to ESKD during the first three decades of life. Unlike polycystic kidney diseases, patients with NPHP tend to have normal sized or shrunken kidneys due to predominant tubulointerstitial fibrosis and the fact that cyst concentration is only around the corticomedullary junction. Since the first identification of the NPHP gene as a possible cause for NPHP in 1997 by Hildebrandt et al, more than 20 genetic mutations causing defects in primary cilia and centrosomes have been identified. About 25% of NPHP is due to homozygous deletion of the NPHP1 gene. Extra-renal involvement causing skeletal defects, visual impairment, liver fibrosis and cardiac septal defects are not uncommon.11 Affected infants develop early onset polyuria, severe hypertension and renal failure, unlike juveniles and adolescents who will be normotensive despite progressive renal failure, due to volume depletion. Unlike other cystic kidney disease, ultrasound may not reveal renal cysts but may show hyperechogenic kidneys prenatally. Confirmatory genetic testing may reveal associated mutations in more than 70% of cases. Kidney biopsy is considered for those patients with inconclusive genetic testing. Chronic tubulointerstitial changes and basement membrane thickening are common findings on biopsy. There is no specific therapy for affected patients except supportive care such as control of hypertension and adequate fluid intake.12,13 KRT with dialysis or transplant is the main option for patients progressing to ESKD.
Genetic Diseases/Mutations with Glomerular Manifestations
Nephrotic Syndrome
Hereditary nephrotic syndrome can occur in both the pediatric and adult population. Primary or autoimmune-mediated forms of nephrotic syndrome are often steroid responsive, however the minority that are steroid resistant nephrotic syndrome (SRNS), may have a genetic etiology.14 The majority of hereditary nephrotic syndrome is due to monogenic genetic mutations affecting podocytes and can present as either isolated glomerular involvement or as part of a syndrome with extra-renal manifestations.15–18
Clinically, it is important to recognize when genetic testing for nephrotic syndrome should be offered as doing so can assist with prognostication, assess the potential for immunosuppression success, and risk of recurrence after kidney transplant. Commonly accepted indications to test include: congenital or onset of SRNS when <1 year old, strong family history, onset in an adult <25 years old, absence of response to immunosuppression, presence of extra-renal features consistent with a syndromic etiology, and in certain ethnic groups (eg, to assess for APOL1 nephropathy in the African American population).15–18 To determine when to test patients with FSGS for a genetic etiology, the suggested approach includes assessing clinical and renal pathology findings on electron microscopy – specifically looking for segmental foot process effacement with the absence of response to immunosuppression or with the absence of nephrotic syndrome.19 Recessive mutations will be harder to detect clinically as both parents need to be carriers, while dominant mutations often present with family history, and screening is important for transplantation donor risk assessment.
There are several genetic mutations associated with the development of nephrotic syndrome and these can be differentiated based on the location of the podocyte protein affected such as nuclear, lysosomal, mitochondrial, cytoskeletal, intracellular, slit diaphragm, glomerular basement membrane or endothelial cell gene mutations (see Figure 1 for location, name and mode of inheritance). Clinically, the majority of early onset nephrotic syndrome is due to NPHS1 (nephrin) mutations resulting in Finnish-type nephropathy, and less commonly by NPHS2 (podocin), WT1, PLCE1, LAMB2 or LMX1B mutations.15,20,21 Mutations in CD2AP, MYO1E, TRPC6 can also cause nephrotic syndrome in childhood.
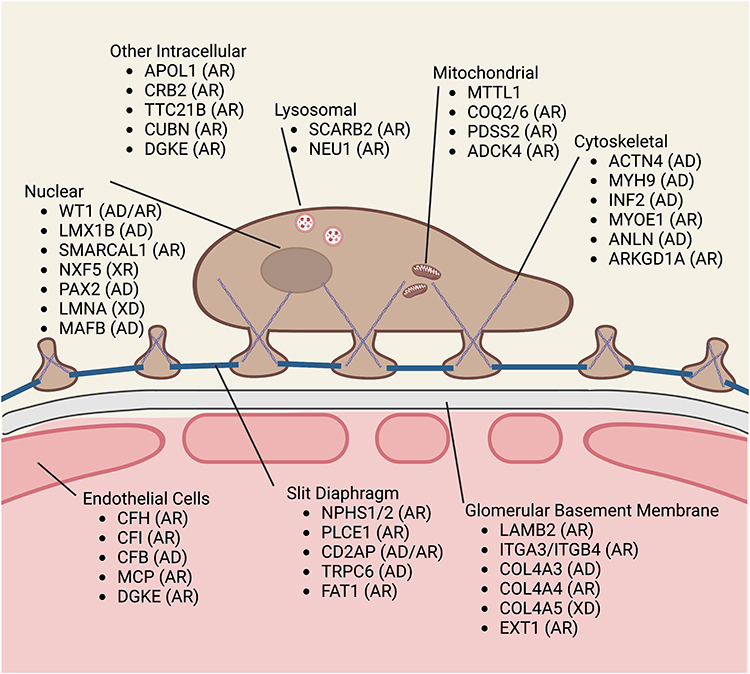 |
Figure 1 Podocyte/Glomerular protein genetic mutations and mode of inheritance. Abbreviations: AD, autosomal dominant; AR, autosomal recessive; WT1, Wilms’ tumor protein; LMX1B, LIM homeobox transcription factor 1 – ß; SMARCAL1, SWI/SNF-related matrix-associated actin-dependent regulator of chromatin sub family A-like protein 1; NXF5, nuclear RNA export factor 5; PAX2, paired box gene 2; LMNA, lamins A and C; MAFB, a transcription factor; APOL1, apolipoprotein L1; CRB2, crumbs cell polarity complex component 2; TTC21B, IFT139 (a component of intraflagellar transport-A); CUBN, cubilin; DGKE, diacylglycerol kinase – ε; SCARB2, scavenger receptor class B member 2; NEU1, sialidase 1 N-Acetyl- α-Neuraminidase; MTTL1, mitochondrially encoded tRNA leucine 1; COQ2, 4-hydorxybenzoate polyprenyltransferase; COQ6, ubiquinone biosynthesis monooxygenase COQ6; PDSS2, decaprenyl-diphosphate synthase subunit 2; ADCK4, aarF domain containing kinase 4; ACTN4, α-actinin-4; MYH9, myosin heavy chain 9; INF2, inverted formin-2; MYOE1, myosin IE; ANLN, anillin; ARKGD1A, rho GDP-dissociation inhibitor (GID) a1; CHF, complement factor H; CFI, complement factor I; CFB, complement factor B; MCP, monocyte chemotactic protein; DGKE, diacylglycerol kinase ε; NPHS1, nephrin; NPHS2, podocin; PLCE1, phospholipase C ε1; CD2AP, CD2-associated protein; TRPC6, canonical transient receptor potential 6; FAT1, FAT atypical cadherin 1; LAMB2, laminin subunit ß2; ITGA3, Integrin – α3; ITGB4, integrin ß4; COLA3,4,5, collagen (IV) α3,α4,α5; EXT1, exostosin glycosyltransferase 1. |
From a histological perspective, patients can present with several patterns of injury such as minimal change disease, focal segmental glomerulosclerosis (FSGS), collapsing glomerulopathy or diffuse mesangial sclerosis.22,23 In the adult population, the majority of cases of FSGS are inherited in autosomal dominant fashion with the most common genes implicated being INF2, TRPC6, ACTN4, SCARB2 and PAX218 and may not always present with nephrotic range proteinuria. FSGS due to COL4A3/4/5 mutations associated with Alport syndrome will be discussed later. Diffuse mesangial sclerosis is more commonly seen in the congenital/infantile setting and usually with mutations in LAMB2, PLCE1, NPHS1/2, WT1.17,21,23,24 Histologically, a clue to differentiate NPHS1 from NPHS2 mutations is the presence of proximal tubule microcytic dilation in those with NPHS1; however, this is not a universal phenomenon.21 Finally, some genes may have several glomerular disease presentations such as DGKE and INF2 mutations which can be associated with membrano-proliferative glomerulonephritis, FSGS and thrombotic microangiopathy (TMA).25
It must be noted that these genetic mutations do not result in purely renal manifestations, but rather can have syndromic presentations. A few key genes for the practicing nephrologist to be aware of include WT1 (Wilm’s Tumor 1 protein) – which can lead to Frasier or Denys Drash syndrome, LAMB2 (laminin-B2) mutations which can cause Pierson syndrome, INF2 mutations which can lead to Charcot-Marie-Tooth disease, LMX1B mutations which can lead to nail-patella syndrome.15,17,18 This highlights the importance of involving genetic counsellors and geneticists in the care of genetic kidney disease so that extra-renal manifestations are either screened for, identified and managed based on the mutation identified.
There are no randomized clinical trials or proven treatments for genetic forms of nephrotic syndrome. Currently, the mainstay of treatment remains RAAS inhibition. In severe forms, in an effort to forgo nephrectomy, the addition of indomethacin may be necessary.26 The majority of cases are resistant to immunosuppression; however, due to its podocyte stabilizing effect, cyclosporine may be considered in select cases. In the setting of COQ2 or COQ6 mutation, supplementation with co-enzyme Q may be beneficial and for ARHGDIA mutations, eplerenone may be considered.19 Future treatments hinge on the development of gene therapy.26
Apolipoprotein L1 (APOL1) Nephropathy
The APOL1 gene is located on chromosome 2227 and originates from sub-Saharan Africa, as these variants provided protection against the trypanosomal transmitted African sleeping sickness.28 APOL1 risk alleles are inherited in a recessive manner as one needs to inherit one of either the G1 or G2 gain of function variants from both parents. High risk allele combinations of G1/G1, G1/G2 or G2/G2 convey increased risk of kidney disease. The likelihood of kidney disease when inheriting only one risk allele is minimal.25
There is a spectrum of kidney disease associated with APOL1, including focal segmental glomerulosclerosis, hypertension-associated end-stage kidney disease (H-ESKD), and collapsing nephropathy in the setting of HIV infection or interferon use. Within this spectrum of kidney disease, it is estimated that two risk alleles convey a 7 to 10-fold increased risk of H-ESKD, 17-fold for FSGS, and 29 to 89-fold for HIV/collapsing nephropathy.29–31 However, it is important to note that not everyone with two risk alleles develop CKD, but rather, require a “second hit” or environmental factor to trigger disease in genetically susceptible individuals.32 In addition to these patterns of injury associated with APOL1 inheritance, lupus nephritis, a disease that disproportionately affects African Americans, can also have an increased risk of collapsing glomerulopathy and poor prognosis in the presence of APOL1 risk alleles.33 The same has also been described in PLA2R associated membranous nephropathy.34
Despite the absence of a clear mechanism of disease, there is ongoing progress towards the development of treatments of APOL1 associated kidney disease. While RAAS inhibition and various immunosuppressive regimens are sub-optimal/ineffective in primary APOL1 associated disease, targeted therapies hold promise. In secondary disease, such as those in the setting of HIV/interferon use, there is often improvement once the underlying trigger is treated. For primary APOL1 disease, APOL1 antisense oligonucleotides, APOL1 small molecule inhibitors and inhibitors of inflammatory pathways implicated in APOL1 disease may be potential available treatment options in the future.35
Atypical Hemolytic Uremic Syndrome
Hemolytic uremic syndrome (HUS) is a life-threatening disorder due to uncontrolled complement pathway activation causing hemolytic anemia, thrombocytopenia, acute kidney failure with thrombotic microangiopathic injury to several organs. The majority of cases occur in a setting of Shiga-toxigenic Escherichia coli (STEC) infection. Atypical HUS (aHUS) which occurs in the absence of an identified infection accounts for about 10% of cases. Inherited or acquired defects in the alternative complement pathway activation and regulation are the main causes for aHUS.36 More than half of patients with aHUS have underlying mutations of complement genes causing loss of function of regulatory proteins such as complement factor (CF) H, CFI, membrane cofactor protein (CD46), complement factor 3 (C3) as well gain of function mutation of effector proteins such as complement factor B37 (Figures 1 and 2). If untreated, about half of patients progress to ESKD and approximately 25% die from the disease. Eculizumab, an anti – C5 monoclonal antibody, is currently used for treatment of aHUS.38 The risk of recurrence after kidney transplant is reported to be very high.39 However, the use of eculizumab during the peri-transplant period may be helpful.
 |
Figure 2 The alternative complement pathway with location of several sites of genetic defects (asterisk) and site of action of Eculizumab. Abbreviations: aHUS, atypical hemolytic uremic syndrome; C3aR, C3a receptor; C5aR, C5a receptor; MAC, membrane attack complex. |
Alport Syndrome
Alport syndrome is a disease of type IV collagen40 that has been described dating back to the 1800s.41 Although the true prevalence of Alport syndrome has been disputed, it is generally estimated to be between 1 in 5000 and 1 in 53,000 individuals42 and accounts for about 1–2% of Europeans with ESKD.43 Type IV collagen is made of 6 genetically distinct alpha (⍺) chains arranged into three triple helical protomers (⍺112, ⍺345 and ⍺556).40 Mutations resulting in Alport syndrome occur in the COL4A3, COL4A4 and COL4A5 genes which code for the ⍺3, ⍺4 and ⍺5 chains, respectively.44 This interferes with the assembly of the ⍺3,4,5 cross-linked network that would normally form in the basement membrane.45
Although originally thought of as an X-linked disease, it is now understood that Alport syndrome has multiple types of inheritance depending on the mutation being passed down. The COL4A3 and COL4A4 genes are coded on chromosome 2q35-37 while COL4A5 is coded on chromosome X.44 X-linked Alport syndrome makes up 80% of cases and results from mutations in COL4A5. Mutations in COL4A3 and COL4A4 give rise to Alport syndrome with autosomal recessive or autosomal dominant inheritance which make up about 15% and 5% of these cases, respectively.46 Autosomal dominant Alport syndrome is increasingly recognized. As expected, these pathologies of type IV collagen result in a diseased renal basement membrane giving Alport syndrome its pathological and phenotypical characteristics. On electron microscopy, thinning of the glomerular basement membrane (GBM) has been well described in patients with Alport syndrome.47 Splitting or lamellation of the GBM can also be found later in the disease’s course48,49 (Figure 3). Findings on light microscopy range from minimal glomerular abnormalities to mesangial proliferation to focal segmental glomerulosclerosis, with prominent interstitial foam cells due to severe proteinuria. On immunofluorescence, findings range from negative staining to non-specific staining of IgG, IgA, IgM, C3 or C1q.50
 |
Figure 3 Electron microscopy highlighting the architectural abnormalities of the glomerular basement membranes in an Alport patient. The GBM ranges from segmentally thinned, to thickened and lamellated (x6000 magnification). |
The characteristic findings of Alport syndrome are those of the kidney, eyes and ear. The renal findings of a patient with Alport syndrome can vary based on the mutation and inheritance but are generally characterized by microscopic hematuria and proteinuria which over time can progress to renal failure.51 Lenticonus results from the conical protrusion of the lens which is found in about 50% of men with X-linked Alport syndrome. Central fleck retinopathy has been described in 60% of men with X-linked Alport syndrome.52 Finally, sensorineural hearing loss can affect as many as 90% of males with X-linked Alport syndrome.53
Diagnosing Alport syndrome has seen some evolution through the years and genetic testing is often offered to clarify the diagnosis. Despite recent advancements in testing and a broader understanding in this disease as a whole, treatment of Alport syndrome remains limited. ACEi and ARB therapy remain the first-line treatment as they have demonstrated the ability to slow the rate of disease progression. Either ACEi or ARB should be started at the time of diagnosis and titrated up to the maximal tolerated dose.54 Recently, a case series has shown promise for the use of SGLT2 inhibitors, but large-scale clinical trials are needed.55 Alport syndrome may progress to kidney failure necessitating kidney replacement therapy or kidney transplantation.56 Future treatment options for Alport syndrome include paricalcitol, epidermal growth factor receptor inhibition, chaperone therapy and stem-cell-based therapies.57
Fabry Disease
Fabry disease is a rare X-linked lysosomal storage disease caused by α-galactosidase A (α-Gal A) deficiency. α-Gal A is a lysosomal hydrolase crucial for glycosphingolipid metabolism. Glycosphingolipids are normal constituents of the cell membrane as well as the membranes of intracellular organelles and in Fabry disease, α-Gal A deficiency leads to a tissue accumulation of trihexosylceramide causing the diseases’ manifestations.58 The incidence of Fabry disease in males is estimated to be between 1 in 50,000 and 1 in 117,000.59 There have been over 1000 different mutations identified of the GLA gene that lead to a deficiency in α-Gal.60–62 This deficiency results in a systemic disorder that can be diagnosed at almost any age ranging from newborns to adults in their 50s.59
The renal defects seen in Fabry disease often present as just mild to moderate proteinuria.63 Nephrotic syndrome and microscopic hematuria can occur but they are not as common.64 Glomerular filtration rate (GFR) tends to be preserved at the time of presentation.65 Urinary lipiduria, when examined under the microscope with polarized light, can reveal oval fat bodies with a Maltese cross configuration due to the glycosphingolipid in the urine.66,67 Patients generally experience a gradual deterioration of their renal function with hypertension and ESKD often developing when the patient is in their 40s-50s.63 From a renal pathology perspective on light microscopy, glomerular epithelial cells are enlarged and packed with small, foamy appearing clear vacuoles that represent glycosphingolipid material68 (Figure 4). Vacuoles can also be seen in endothelial cells and smooth muscle cells of arterioles and arteries.63 This causes a progressive segmental and global glomerulosclerosis.68 Immunofluorescence is generally negative. On electron microscopy, podocytes are filled with osmiophilic, granular-to-lamellated membrane structures often termed “zebra bodies”69,70 (Figure 5).
 |
Figure 4 Renal biopsy from a patient with Fabry disease, confirmed after renal biopsy with genetic testing. The biopsy showed glomeruli with “foamy” podocytes due to the accumulation of lipid material within the cytoplasm. Jones methenamine silver x600 magnification. |
 |
Figure 5 Electron microscopy shows lipid inclusions within podocyte cell bodies with the characteristic appearance of “zebra bodies” and “myelin figures” (x9300 magnification). |
The other systemic manifestations seen in Fabry disease include neurological symptoms such as autonomic dysfunction, acroparesthesia, hearing loss, seizures, transient ischemic attacks, and cerebral hemorrhage. Angiokeratomas distributed in the bathing area, and whorl like corneal opacities are characteristic findings of Fabry disease. Cardiac findings include left ventricular hypertrophy, coronary disease, arrhythmias and congestive heart disease.58 Diagnosing Fabry disease can be challenging due to the non-specific symptoms on presentation, and genetic testing is often needed to confirm the diagnosis particularly in female patients, because the enzyme level is less reliable in females. Enzyme replacement therapy (ERT) has been available for treatment of Fabry disease in Europe since 2001 and USA since 2003 and this treatment option has already demonstrated a beneficial effect on the complications and mortality of Fabry disease.71 The introduction of pharmacologic chaperone therapy with migalastat provides an alternative to ERT for certain individuals with amenable mutations.
CAKUT - Congenital Anomalies of the Kidney and Urinary Tract
Congenital anomalies of the kidney and urinary tract is the most common cause of kidney failure in children. It occurs in more than 40% of children and adolescents starting dialysis, unlike adults where its prevalence was reported to be below 5%.72,73 It occurs due to the aberrant development of the kidney and out flow tract during embryogenesis. It may present as an isolated disorder or as a part of a syndrome affecting multiple organs.74 It accounts for about 20–30% of all congenital malformations among live births.73 The underlying cause is not completely understood due to genetic and phenotypic heterogeneity but it is presumed to be multifactorial involving both genetic mutations and environmental factors. More than 40 monogenic genes are identified as causes for CAKUT if mutated. Mutations of HNF1B and PAX2 account for about 5–15% of patients affected with CAKUT.73,75,76
Clinical presentation depends on the severity of anomaly, ranging from life incompatible bilateral renal agenesis or hypoplasia causing intrauterine fetal death to structural and functional anomalies such as renal dysplasia, horseshoe kidney, pelvic kidney, congenital hydronephrosis, ureteropelvic junction (UPJ) obstruction, vesicoureteral reflux (VUR), bladder exstrophy and asymptomatic posterior urethral valves (PUV) leading to severe hypertension, proteinuria, recurrent infection and progressive kidney failure. Most of the affected patients with unilateral defects will remain asymptomatic. The risk of ESKD is highest among affected live births in the first few months of life and later during adolescence.72,74,77,78
Antenatal screening with obstetric ultrasound may help in the early diagnosis of CAKUT. For those with structural deformities and signs of renal failure during childhood, renal ultrasound and voiding cystourethrography are tests of choice.75 Genetic work up will help identify the underlying etiology which may help determine whether it is an isolated disorder or part of a syndrome affecting multiple organs.72,73,77 Treatment of patients with CAKUT involves a multidisciplinary approach from routine follow up, reconstructive surgery to kidney replacement therapy with dialysis or kidney transplant. Among those patients undergoing KRT, survival is better compared to adults due to lower cardiovascular mortality.79
ADTKD - Autosomal Dominant Tubulointerstitial Kidney Disease
Autosomal dominant tubulointerstitial kidney disease is an under recognized monogenic kidney disease that affects the renal tubules and interstitium leading to progressive kidney fibrosis.80 In 2015, a KDIGO consensus report reclassified ADTKD based on underlying genetic defects and distanced the nephrology community from previous misnomers.80 The most common mutations occur in the tubular cell genes encoding uromodulin (UMOD),81–83 hepatocyte nuclear factor-1b (HNF1B),84 renin (REN),85 and mucin-1 (MUC1)83 (see Table 1). The corresponding terminologies for gene-specific diseases are ADTKD-UMOD, ADTKD-HNF1B, ADTKD-REN, and ADTKD-MUC1.80 If a genetic mutation is not found or is not tested, the umbrella diagnostic term is ADTKD-NOS (not otherwise specified).80 ADTKD-UMOD is the most frequent subtype followed by ADTKD-MUC1.83 Next-generation sequencing has increased diagnosis and awareness of ADTKD.86 Clinical features of ADTKD include variably progressive kidney disease leading to ESKD between the second and seventh decades.87,88
 |
Table 1 Potential Genes Mutations and Clinical Manifestations That Result in Autosomal Dominant Tubulointerstitial Kidney Disease (ADTKD) |
Approximately 3% of ESKD in the United States of America is thought to be due to UMOD mutations.86 A family history of kidney disease is common at diagnosis with an autosomal dominant inheritance pattern.80 Urinalysis is often bland and a renal ultrasound is consistent with normal to small sized kidneys.89–91 Kidney biopsy histology typically reveals interstitial fibrosis with tubular atrophy and intact glomeruli.92,93 Usual histologic findings include thickening and lamellation of the tubular of tubular basement membranes.80,92,93 When family history is absent, kidney biopsy may be helpful as it can detect uromodulin (UMOD) accumulation to help establish the ADTKD-UMOD diagnosis.94 However, kidney biopsy may not always be necessary for diagnosis with the advancement of genetic testing.94,95 Hyperuricemia and gout are associated with UMOD and REN mutations.83,87
Uromodulin is encoded by UMOD in the loop of Henle epithelial cells and the pathophysiology of ADTKD-UMOD is not fully understood.96 The disease is thought to be due to impaired intracellular transport of uromodulin and resultant endoplasmic reticulum accumulation.96 Hyperuricemia in ADTKD-UMOD is thought to be due to a downregulation of the Na+–K +–2Cl− cotransporter and reactive Na+-urate cotransporter upregulation.96,97 Mucin-1 is encoded by MUC1 and functions as a transmembrane protein in the distal tubule luminal epithelium.98,99 Similarly, a frameshift mutation in MUC1 is thought to precipitate intracellular accumulation of mucin-1 negating its luminal barrier protection role.80,100 HNF1B encodes a transcription factor hepatocyte nuclear factor 1b (HNF1b) which regulates UMOD amongst many other genes.101,102 This explains how isolated kidney disease is rare with HNF1B mutations and extrarenal findings involving the liver and pancreas are common.91,103 Preprorenin is encoded by REN and proteolysis leads to renin production.104 Renin is known to lead to angiotensin activation, but it is also expressed in the renal tubules and mutations affecting REN are thought to cause intracellular accumulation of abnormal renin.104,105 It is worth mentioning that the mechanism of the mutations leading to tubulointerstitial fibrosis is unknown.
Family history is helpful in diagnosing ADTKD-related disease based on the autosomal dominant inheritance pattern of these mutations.80,81,87 De novo mutations that cause ADTKD are known to occur.83,87 There is also variable penetrance of disease which is manifested by a wide variation in age when kidney replacement therapy is needed. Unrecognized chronic kidney disease in family members can make the diagnosis difficult and testing for genetic mutations is critical for establishing a diagnosis. ADTKD-MUC1 is the most diagnostically challenging, with diagnosis only achievable by molecular diagnostics in very few labs.80,83,95
There are no available targeted therapies for ADTKD. Kidney transplantation offers the best treatment for advanced kidney disease related to ADTKD with no evidence of recurrence of disease.80,83,95 Urate lowering therapy can manage hyperuricemia associated with UMOD and MUC1 mutations.95 RAAS blockade and drugs with propensity for volume depletion should be avoided in ADTKD-REN.95,106 Novel targeted therapeutics including small molecule inhibitors are currently under investigation.95
Nephrolithiasis and Nephrocalcinosis
While most renal stones are due to complex polygenic derangements, exacerbated by environmental and anatomical factors, up to 15% of patients may have monogenic disorders.107 The detection of these disorders can affect treatment, prognosis, likelihood of recurrence and risk to family members. Factors that identify possible inherited stone disease include early age at onset, positive family history, or history of consanguinity. The presence of a high stone burden, radiologic evidence of nephrocalcinosis, or impaired renal function are also important clues. In addition, the presence of extrarenal factors such as hearing or vision abnormalities, and neurological disorders is strongly suggestive of monogenic disease.108 This review will examine some of the monogenic disorders that lead to the metabolic derangements that characterize renal stone disease.
Primary Hyperoxaluria
The primary hyperoxalurias (PH) is a group of three autosomal recessive disorders in which enzyme defects produce insoluble calcium oxalate that results in renal stones or nephrocalcinosis. These disorders are due to abnormal glyoxylate metabolism in the hepatocyte. PH1 is due to alanine: glyoxylate aminotransferase (AGT) deficiency in the peroxisomes and PH2 is due to deficiency of glyoxylate/hydroxypyruvate reductase (GRHPR) in the cytosol. These conditions both result in excess glyoxylate which is converted to oxalate by the enzyme lactate dehydrogenase. PH3 is due to a defect in the mitochondrial enzyme hydroxyoxoglutarate aldolase that results in metabolism of hydroxyoxoglutarate to oxalate.109 PH1 is the most common type affecting 70–80% of patients.110 It is also the most severe with half of young patients progressing to ESKD by early adulthood.111
The disease may present in early childhood but is sometimes diagnosed in adults with recurrent calcium oxalate stones and marked renal insufficiency. Timely diagnosis and intervention are crucial to prevent progression to ESKD, or recurrence after renal transplantation. Elevated 24-hour urine oxalate levels are common, and the subtypes may be differentiated by measuring specific urinary metabolites. Genetic testing is confirmatory. General management includes increased fluid intake aiming for at least three liters of urine per day, reduced dietary oxalate and urinary alkalinization with potassium citrate. Pyridoxine supplementation in PH1 can reduce oxalate production by acting as a coenzyme of alanine-glyoxylate aminotransferase (AGT) which is most effective in cases with c.508 G>A allele. Lumasiran, an RNA interference (RNAi) therapeutic agent that targets glycolate oxidase, was approved by the Food and Drug Administration in 2020 for PH1. Liver transplantation is curative.
Cystinuria
Cystinuria is an autosomal recessive disorder due to defects in the cystine/dibasic amino acid transporter. There is decreased proximal tubular reabsorption of cystine which results in high levels of urinary cysteine. This leads to the formation of cystine stones because of the low solubility of cystine at normal urinary pH. Cystinuria is caused by mutations in the SLC3A1 gene or in the SLC7A9 gene. Patients typically present with stone disease in childhood, often with staghorn calculi or recurrent stone disease. The finding of the pathognomonic hexagonal cystine crystals on urine microscopy, or stone analysis demonstrating 100% cystine stones, establishes the diagnosis. Genetic testing can confirm the diagnosis but is not required. All patients should have 24-hour urine quantification of cystine. Most patients excrete more than 400 mg per day of cystine, compared to normal urinary excretion of 30 mg/day. Conservative measures for treatment include high fluid intake, alkalinization of urine and decreased dietary sodium and animal protein intake. Definitive treatment with thiol containing compounds such as tiopronin and D-penicillamine results in the formation of more soluble drug-cysteine disulphides. This step is usually reserved for more severe stone disease due to the high incidence of adverse effects.112
Stone Disease Due to Disorders of Purine Metabolism
Deficiency of the enzymes adenine phosphoribosyl transferase (APRT) and xanthine dehydrogenase (XDH) leads to accumulation of insoluble substances that cause radiolucent stones. 2,8-dihydroxyadenine (DHA) and xanthine stones are formed respectively. These are autosomal recessive diseases that result in ESKD. 2–8% of children with renal stones have hyperuricosuria.113 Monogenic metabolic disorders such as Lesch Nyhan Syndrome (hypoxanthine-guanine phosphoribosyltransferase (HPRT) deficiency) and phosphoribosyl pyrophosphate synthase (PRPPS) hyperactivity can cause severe hyperuricosuria and uric acid nephrolithiasis. These rare recessive disorders can also lead to ESKD.
Dent Disease
Dent disease is an X-linked recessive disorder that causes ESKD in adulthood. It is caused by mutations in the CLCN5 gene that encodes the chloride/hydrogen ion exchanger, CIC-5. The disease causes a Fanconi syndrome as well as hypercalciuria, nephrocalcinosis and nephrolithiasis. Other proximal tubule disorders include Lowe syndrome and mutant variants of the sodium phosphate (NaPi-2) transporter.
Bartter Syndromes
Bartter syndromes are autosomal recessive disorders due to mutations in the thick ascending limb transporter proteins. Patients have hypokalemic metabolic alkalosis, low normal blood pressure, and hypercalciuria that often leads to nephrocalcinosis. There are 5 types of Bartter syndrome described. The most recent discovery, Bartter syndrome type 5, is due to a gain of function mutation of the calcium sensing receptor (CaSR). This leads to inhibition of the renal outer medullary potassium channel (ROMK) and inactivation of the Na-K-2Cl cotransporter (NKCC2) with resultant hypercalciuria and salt wasting. Nephrocalcinosis occurs in all types of Bartter syndrome, but may be more severe in types 1, 2 and 5.114 Nephrolithiasis is related to distal RTA can be autosomal dominant or recessive in inheritance. Mutations in the basolateral anion (Cl−/HCO3−) exchanger AE1 or subunits of the H+ATPase ion pump result in disease phenotypes ranging from severe bilateral stone disease with failure to thrive in childhood, to mild metabolic acidosis and nephrocalcinosis in adults.
Single Nucleotide Polymorphisms and Nephrolithiasis
Single nucleotide polymorphisms (SNP) in certain genes are associated with hypercalcemia and nephrolithiasis. Claudins are transmembrane proteins that regulate paracellular transport in tight junctions. Claudin 14 (CLDN14) regulates calcium reabsorption in the thick ascending limb of the Loop of Henle, and its expression is modulated by the calcium sensing receptor. CLDN14 SNP’s, rs219780 and rs219781 have been shown to be associated with nephrolithiasis.115 The gene that encodes an alkaline phosphatase isoenzyme (ALPL) has also been identified as a candidate gene for nephrolithiasis. ALPL located in the proximal tubule, hydrolyses pyrophosphate (an inhibitor of stone formation), to phosphate. The SNP ALPL rs1256238 is associated with a higher risk of stone disease.115
Molecular genetics has the potential to alter our approach to renal stone disease, as there will undoubtedly be more mutations and variants identified that cause nephrolithiasis and nephrocalcinosis. Phenotypic expression is highly variable and may be due to a complex interplay of environmental and genetic factors. Identification of these genetic variants and understanding of their pathophysiological effects can lead to early diagnosis and treatment. This will alleviate the morbidity associated with stone disease and in some cases, reduce the likelihood of progression to ESKD.
Conclusion
Historically, many genetic causes of kidney disease were poorly understood and often various forms of CKD were thought to be related to other modifiable risk factors. Now with the ongoing understanding of these pathologies, diagnosing inheritable disorders is done more effectively and earlier and can potentially have a positive impact on the care of a large segment of the CKD population. As our knowledge and understanding of these conditions continues to grow, proper and timely diagnosis of these conditions may lead to a more patient centered approach of CKD management as well as future novel therapeutics.
Acknowledgments
The authors would like to thank Dr. Leal Herlitz, Director of Medical Kidney Pathology at Cleveland Clinic Ohio, for graciously providing pathology figures for this manuscript.
Figures 1 and 2 created using biorender.com.
Funding
There is no funding to report.
Disclosure
Dr Shane A Bobart reports honorarium for Lecture from Travere Therapeutics, outside the submitted work. The authors state no conflicts of interest.
References
1. Renkema KY, Stokman MF, Giles RH, Knoers NV. Next-generation sequencing for research and diagnostics in kidney disease. Nat Rev Nephrol. 2014;10:433–444. doi:10.1038/nrneph.2014.95
2. Gordon CE, Miskulin DC, Perrone RD. Assessing risk of progression in ADPKD. Clin J Am Soc Nephrol. 2022;17:134–136. doi:10.2215/CJN.13071021
3. Cornec-Le Gall E, Chebib FT, Madsen CD, et al.;. The value of genetic testing in polycystic kidney diseases illustrated by a family with PKD2 and COL4A1 mutations. Am J Kidney Dis. 2018;72:302–308. doi:10.1053/j.ajkd.2017.11.015
4. Liebau MC. Early clinical management of autosomal recessive polycystic kidney disease. Pediatr Nephrol. 2021;36:3561–3570. doi:10.1007/s00467-021-04970-8
5. Wang S, Luo Y, Wilson PD, Witman GB, Zhou J. The autosomal recessive polycystic kidney disease protein is localized to primary cilia, with concentration in the basal body area. J Am Soc Nephrol. 2004;15:592–602. doi:10.1097/01.asn.0000113793.12558.1d
6. Bhutani H, Smith V, Rahbari-Oskoui F, et al. A comparison of ultrasound and magnetic resonance imaging shows that kidney length predicts chronic kidney disease in autosomal dominant polycystic kidney disease. Kidney Int. 2015;88:146–151. doi:10.1038/ki.2015.71
7. Pei Y, Hwang YH, Conklin J, et al. Imaging-based diagnosis of autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2015;26:746–753. doi:10.1681/ASN.2014030297
8. Flahault A, Trystram D, Nataf F, et al. Screening for intracranial aneurysms in autosomal dominant polycystic kidney disease is cost-effective. Kidney Int. 2018;93:716–726. doi:10.1016/j.kint.2017.08.016
9. Alam A, Perrone RD. Management of ESRD in patients with autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17:164–172. doi:10.1053/j.ackd.2009.12.006
10. Chebib FT, Perrone RD, Chapman AB, et al. A practical guide for treatment of rapidly progressive ADPKD with tolvaptan. J Am Soc Nephrol. 2018;29:2458–2470. doi:10.1681/ASN.2018060590
11. Hildebrandt F, Zhou W. Nephronophthisis-associated ciliopathies. J Am Soc Nephrol. 2007;18:1855–1871. doi:10.1681/ASN.2006121344
12. Simms RJ, Hynes AM, Eley L, Sayer JA. Nephronophthisis: a genetically diverse ciliopathy. Int J Nephrol. 2011;2011:527137. doi:10.4061/2011/527137
13. Tory K, Rousset-Rouviere C, Gubler MC, et al. Mutations of NPHP2 and NPHP3 in infantile nephronophthisis. Kidney Int. 2009;75:839–847. doi:10.1038/ki.2008.662
14. Ha T-S. Genetics of hereditary nephrotic syndrome: a clinical review. Korean J Pediatr. 2017;60:55–63. doi:10.3345/kjp.2017.60.3.55
15. Benoit G, Machuca E, Antignac C. Hereditary nephrotic syndrome: a systematic approach for genetic testing and a review of associated podocyte gene mutations. Pediatr Nephrol. 2010;25:1621–1632. doi:10.1007/s00467-010-1495-0
16. Joshi S, Andersen R, Jespersen B, Rittig S. Genetics of steroid-resistant nephrotic syndrome: a review of mutation spectrum and suggested approach for genetic testing. Acta Paediatr. 2013;102:844–856. doi:10.1111/apa.12317
17. Lovric S, Ashraf S, Tan W, Hildebrandt F. Genetic testing in steroid-resistant nephrotic syndrome: when and how? Nephrol Dial Transplant. 2016;31:1802–1813. doi:10.1093/ndt/gfv355
18. Rood IM, Deegens JK, Wetzels JF. Genetic causes of focal segmental glomerulosclerosis: implications for clinical practice. Nephrol Dial Transplant. 2012;27:882–890. doi:10.1093/ndt/gfr771
19. De Vriese AS, Sethi S, Nath KA, Glassock RJ, Fervenza FC. Differentiating primary, genetic, and secondary FSGS in adults: a clinicopathologic approach. J Am Soc Nephrol. 2018;29:759–774. doi:10.1681/asn.2017090958
20. Hinkes BG, Mucha B, Vlangos CN, et al.; Arbeitsgemeinschaft fur Paediatrische Nephrologie Study G. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics. 2007;119:e907–919. doi:10.1542/peds.2006-2164
21. Machuca E, Benoit G, Nevo F, et al. Genotype-phenotype correlations in non-Finnish congenital nephrotic syndrome. J Am Soc Nephrol. 2010;21:1209–1217. doi:10.1681/ASN.2009121309
22. Barisoni L, Schnaper HW, Kopp JB. A proposed taxonomy for the podocytopathies: a reassessment of the primary nephrotic diseases. Clin J Am Soc Nephrol. 2007;2:529–542. doi:10.2215/CJN.04121206
23. Barisoni L, Schnaper HW, Kopp JB. Advances in the biology and genetics of the podocytopathies: implications for diagnosis and therapy. Arch Pathol Lab Med. 2009;133:201–216. doi:10.1043/1543-2165-133.2.201
24. Kari JA, Montini G, Bockenhauer D, et al. Clinico-pathological correlations of congenital and infantile nephrotic syndrome over twenty years. Pediatr Nephrol. 2014;29:2173–2180. doi:10.1007/s00467-014-2856-x
25. Li AS, Ingham JF, Lennon R. Genetic disorders of the glomerular filtration barrier. J Clin Am Soc Nephrol. 2020;15:1818–1828. doi:10.2215/CJN.11440919
26. Kemper MJ, Lemke A. Treatment of genetic forms of nephrotic syndrome. Front Pediatr. 2018;6:72. doi:10.3389/fped.2018.00072
27. Smith EE, Malik HS. The apolipoprotein L family of programmed cell death and immunity genes rapidly evolved in primates at discrete sites of host-pathogen interactions. Genome Res. 2009;19:850–858. doi:10.1101/gr.085647.108
28. Patrakka J, Tryggvason K. Molecular make-up of the glomerular filtration barrier. Biochem Biophys Res Commun. 2010;396:164–169. doi:10.1016/j.bbrc.2010.04.069
29. Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329:841–845. doi:10.1126/science.1193032
30. Kasembeli AN, Duarte R, Ramsay M, et al. APOL1 risk variants are strongly associated with HIV-associated nephropathy in black South Africans. J Am Soc Nephrol. 2015;26:2882–2890. doi:10.1681/ASN.2014050469
31. Kopp JB, Nelson GW, Sampath K, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. 2011;22:2129–2137. doi:10.1681/ASN.2011040388
32. Beckerman P, Susztak K. APOL1: the balance imposed by infection, selection, and kidney disease. Trends Mol Med. 2018;24:682–695. doi:10.1016/j.molmed.2018.05.008
33. Larsen CP, Beggs ML, Saeed M, Walker PD. Apolipoprotein L1 risk variants associate with systemic lupus erythematosus-associated collapsing glomerulopathy. J Am Soc Nephrol. 2013;24:722–725. doi:10.1681/ASN.2012121180
34. Larsen CP, Beggs ML, Walker PD, Saeed M, Ambruzs JM, Messias NC. Histopathologic effect of APOL1 risk alleles in PLA2R-associated membranous glomerulopathy. Am J Kidney Dis. 2014;64:161–163. doi:10.1053/j.ajkd.2014.02.024
35. Friedman DJ, Ma L, Freedman BI. Treatment potential in APOL1-associated nephropathy. Curr Opin Nephrol Hypertens. 2022;31:442–448. doi:10.1097/mnh.0000000000000816
36. Sridharan M, Go RS, Willrich MAV. Atypical hemolytic uremic syndrome: review of clinical presentation, diagnosis and management. J Immunol Methods. 2018;461:15–22. doi:10.1016/j.jim.2018.07.006
37. Bu F, Maga T, Meyer NC, et al. Comprehensive genetic analysis of complement and coagulation genes in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2014;25:55–64. doi:10.1681/ASN.2013050453
38. Cofiell R, Kukreja A, Bedard K, et al. Eculizumab reduces complement activation, inflammation, endothelial damage, thrombosis, and renal injury markers in aHUS. Blood. 2015;125:3253–3262. doi:10.1182/blood-2014-09-600411
39. Bobart SA, Alexander MP, Bentall A. Recurrent glomerulonephritis in the kidney allograft. Indian J Nephrol. 2020;30:359–369. doi:10.4103/ijn.IJN_193_19
40. Sundaramoorthy M, Meiyappan M, Todd P, Hudson BG. Crystal structure of NC1 domains. Structural basis for type IV collagen assembly in basement membranes. J Biol Chem. 2002;277:31142–31153. doi:10.1074/jbc.M201740200
41. Alport AC. Hereditary familial congenital haemorrhagic nephritis. Br Med J. 1927;1:504–506. doi:10.1136/bmj.1.3454.504
42. Gibson J, Fieldhouse R, Chan MMY, et al. Genomics England research c: prevalence estimates of predicted pathogenic COL4A3-COL4A5 variants in a population sequencing database and their implications for alport syndrome. J Am Soc Nephrol. 2021;32:2273–2290. doi:10.1681/ASN.2020071065
43. Wing AJ, Brunner FP. Twenty-three years of dialysis and transplantation in Europe: experiences of the EDTA Registry. Am J Kidney Dis. 1989;14:341–346. doi:10.1016/s0272-6386(89)80165-9
44. Kashtan CE, Ding J, Garosi G, et al. Alport syndrome: a unified classification of genetic disorders of collagen IV alpha345: a position paper of the Alport Syndrome Classification Working Group. Kidney Int. 2018;93:1045–1051. doi:10.1016/j.kint.2017.12.018
45. Hudson BG. The molecular basis of Goodpasture and Alport syndromes: beacons for the discovery of the collagen IV family. J Am Soc Nephrol. 2004;15:2514–2527. doi:10.1097/01.ASN.0000141462.00630.76
46. Kashtan CE, Segal Y. Genetic disorders of glomerular basement membranes. Nephron Clin Pract. 2011;118:c9–c18. doi:10.1159/000320876
47. Rumpelt HJ. Hereditary nephropathy (Alport syndrome): correlation of clinical data with glomerular basement membrane alterations. Clin Nephrol. 1980;13:203–207.
48. Funk SD, Lin MH, Miner JH. Alport syndrome and Pierson syndrome: diseases of the glomerular basement membrane. Matrix Biol. 2018;71:250–261. doi:10.1016/j.matbio.2018.04.008
49. Gubler M, Levy M, Broyer M, et al. Alport’s syndrome. A report of 58 cases and a review of the literature. Am J Med. 1981;70:493–505. doi:10.1016/0002-9343(81)90571-4
50. Kamiyoshi N, Nozu K, Fu XJ, et al. Genetic, clinical, and pathologic backgrounds of patients with autosomal dominant alport syndrome. Clin J Am Soc Nephrol. 2016;11:1441–1449. doi:10.2215/CJN.01000116
51. Savige J, Gregory M, Gross O, Kashtan C, Ding J, Flinter F. Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J Am Soc Nephrol. 2013;24:364–375. doi:10.1681/ASN.2012020148
52. Savige J, Sheth S, Leys A, Nicholson A, Mack HG, Colville D. Ocular features in Alport syndrome: pathogenesis and clinical significance. Clin J Am Soc Nephrol. 2015;10:703–709. doi:10.2215/CJN.10581014
53. Nozu K, Nakanishi K, Abe Y, et al. A review of clinical characteristics and genetic backgrounds in Alport syndrome. Clin Exp Nephrol. 2019;23:158–168. doi:10.1007/s10157-018-1629-4
54. Kashtan CE, Gross O. Clinical practice recommendations for the diagnosis and management of Alport syndrome in children, adolescents, and young adults-an update for 2020. Pediatr Nephrol. 2021;36:711–719. doi:10.1007/s00467-020-04819-6
55. Boeckhaus J, Gross O. Sodium-glucose cotransporter-2 inhibitors in patients with hereditary podocytopathies, alport syndrome, and FSGS: a case series to better plan a large-scale study. Cells. 2021;10. doi:10.3390/cells10071815
56. Kashtan CE. Alport syndrome: achieving early diagnosis and treatment. Am J Kidney Dis. 2021;77:272–279. doi:10.1053/j.ajkd.2020.03.026
57. Torra R, Furlano M. New therapeutic options for Alport syndrome. Nephrol Dial Transplant. 2019;34:1272–1279. doi:10.1093/ndt/gfz131
58. Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30. doi:10.1186/1750-1172-5-30
59. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254. doi:10.1001/jama.281.3.249
60. Genomes Project C, Abecasis GR, Auton A, et al. An integrated map of genetic variation from 1092 human genomes. Nature. 2012;491:56–65. doi:10.1038/nature11632
61. Koulousios K, Stylianou K, Pateinakis P, et al. Fabry disease due to D313Y and novel GLA mutations. BMJ Open. 2017;7:e017098. doi:10.1136/bmjopen-2017-017098
62. Kurschat CE. Fabry disease-what cardiologists can learn from the nephrologist: a narrative review. Cardiovasc Diagn Ther. 2021;11:672–682. doi:10.21037/cdt-20-981
63. Feehally JFJ, Johnson RJ, Tonelli M. Comprehensive Clinical Nephrology.
64. Blom D, Speijer D, Linthorst GE, Donker-Koopman WG, Strijland A, Aerts JM. Recombinant enzyme therapy for Fabry disease: absence of editing of human alpha-galactosidase A mRNA. Am J Hum Genet. 2003;72:23–31. doi:10.1086/345309
65. Sestito S, Falvo F, Sallemi A, et al. Renal involvement in paediatric Fabry disease. J Biol Regul Homeost Agents. 2019;33:59–63.
66. Becker GJ, Nicholls K. Lipiduria--with special relevance to Fabry disease. Clin Chem Lab Med. 2015;53(Suppl 2):s1465–1470. doi:10.1515/cclm-2015-0499
67. Utsumi K, Mitsuhashi F, Katsura K, Iino Y, Katayama Y. ”Maltese crosses” in fabry disease. J Nippon Med Sch. 2010;77:284. doi:10.1272/jnms.77.284
68. Alroy J, Sabnis S, Kopp JB. Renal pathology in Fabry disease. J Am Soc Nephrol. 2002;13(Suppl 2):S134–138.
69. Najafian B, Fogo AB, Lusco MA, Alpers CE. AJKD atlas of renal pathology: fabry nephropathy. Am J Kidney Dis. 2015;66:e35–36. doi:10.1053/j.ajkd.2015.08.006
70. Torra R. Renal manifestations in Fabry disease and therapeutic options. Kidney Int Suppl. 2008;S29–32. doi:10.1038/ki.2008.522
71. Ortiz A, Kanters S, Hamed A, et al. Agalsidase beta treatment slows estimated glomerular filtration rate loss in classic Fabry disease patients: results from an individual patient data meta-analysis. Clin Kidney J. 2021;14:1136–1146. doi:10.1093/ckj/sfaa065
72. Song R, Yosypiv IV. Genetics of congenital anomalies of the kidney and urinary tract. Pediatr Nephrol. 2011;26:353–364. doi:10.1007/s00467-010-1629-4
73. van der Ven AT, Vivante A, Hildebrandt F. Novel insights into the pathogenesis of monogenic congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol. 2018;29:36–50. doi:10.1681/asn.2017050561
74. Mackie GG, Stephens FD. Duplex kidneys: a correlation of renal dysplasia with position of the ureteral orifice. J Urol. 1975;114:274–280. doi:10.1016/s0022-5347(17)67007-1
75. Murugapoopathy V, Gupta IR. A primer on congenital anomalies of the kidneys and urinary tracts (CAKUT). Clin J Am Soc Nephrol. 2020;15:723. doi:10.2215/CJN.12581019
76. Saisawat P, Tasic V, Vega-Warner V, et al. Identification of two novel CAKUT-causing genes by massively parallel exon resequencing of candidate genes in patients with unilateral renal agenesis. Kidney Int. 2012;81:196–200. doi:10.1038/ki.2011.315
77. Nicolaou N, Pulit SL, Nijman IJ, et al. Prioritization and burden analysis of rare variants in 208 candidate genes suggest they do not play a major role in CAKUT. Kidney Int. 2016;89:476–486. doi:10.1038/ki.2015.319
78. Nicolaou N, Renkema KY, Bongers EM, Giles RH, Knoers NV. Genetic, environmental, and epigenetic factors involved in CAKUT. Nat Rev Nephrol. 2015;11:720–731. doi:10.1038/nrneph.2015.140
79. Wuhl E, van Stralen KJ, Verrina E, et al. Timing and outcome of renal replacement therapy in patients with congenital malformations of the kidney and urinary tract. Clin J Am Soc Nephrol. 2013;8:67–74. doi:10.2215/CJN.03310412
80. Eckardt KU, Alper SL, Antignac C, et al. Kidney Disease: improving Global O: autosomal dominant tubulointerstitial kidney disease: diagnosis, classification, and management--A KDIGO consensus report. Kidney Int. 2015;88:676–683. doi:10.1038/ki.2015.28
81. Dahan K, Devuyst O, Smaers M, et al. A cluster of mutations in the UMOD gene causes familial juvenile hyperuricemic nephropathy with abnormal expression of uromodulin. J Am Soc Nephrol. 2003;14:2883–2893. doi:10.1097/01.asn.0000092147.83480.b5
82. Hart TC, Gorry MC, Hart PS, et al. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J Med Genet. 2002;39:882–892. doi:10.1136/jmg.39.12.882
83. Olinger E, Hofmann P, Kidd K, et al. Clinical and genetic spectra of autosomal dominant tubulointerstitial kidney disease due to mutations in UMOD and MUC1. Kidney Int. 2020;98:717–731. doi:10.1016/j.kint.2020.04.038
84. Lindner TH, Njolstad PR, Horikawa Y, Bostad L, Bell GI, Sovik O. A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1beta. Hum Mol Genet. 1999;8:2001–2008. doi:10.1093/hmg/8.11.2001
85. Zivna M, Kidd K, Zaidan M, et al. An international cohort study of autosomal dominant tubulointerstitial kidney disease due to REN mutations identifies distinct clinical subtypes. Kidney Int. 2020;98:1589–1604. doi:10.1016/j.kint.2020.06.041
86. Groopman EE, Marasa M, Cameron-Christie S, et al. Diagnostic utility of exome sequencing for kidney disease. N Engl J Med. 2019;380:142–151. doi:10.1056/NEJMoa1806891
87. Bollee G, Dahan K, Flamant M, et al. Phenotype and outcome in hereditary tubulointerstitial nephritis secondary to UMOD mutations. Clin J Am Soc Nephrol. 2011;6:2429–2438. doi:10.2215/CJN.01220211
88. Moskowitz JL, Piret SE, Lhotta K, et al. Association between genotype and phenotype in uromodulin-associated kidney disease. Clin J Am Soc Nephrol. 2013;8:1349–1357. doi:10.2215/CJN.11151012
89. Bleyer AJ, Kmoch S, Antignac C, et al. Variable clinical presentation of an MUC1 mutation causing medullary cystic kidney disease type 1. Clin J Am Soc Nephrol. 2014;9:527–535. doi:10.2215/CJN.06380613
90. Faguer S, Decramer S, Chassaing N, et al. Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney Int. 2011;80:768–776. doi:10.1038/ki.2011.225
91. Heidet L, Decramer S, Pawtowski A, et al. Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clin J Am Soc Nephrol. 2010;5:1079–1090. doi:10.2215/CJN.06810909
92. Ayasreh N, Bullich G, Miquel R, et al. Autosomal dominant tubulointerstitial kidney disease: clinical presentation of patients with ADTKD-UMOD and ADTKD-MUC1. Am J Kidney Dis. 2018;72:411–418. doi:10.1053/j.ajkd.2018.03.019
93. Ekici AB, Hackenbeck T, Moriniere V, et al. Renal fibrosis is the common feature of autosomal dominant tubulointerstitial kidney diseases caused by mutations in mucin 1 or uromodulin. Kidney Int. 2014;86:589–599. doi:10.1038/ki.2014.72
94. Onoe T, Hara S, Yamada K, et al. Significance of kidney biopsy in autosomal dominant tubulointerstitial kidney disease-UMOD: is kidney biopsy truly nonspecific? BMC Nephrol. 2021;22:1. doi:10.1186/s12882-020-02169-x
95. Mabillard H, Sayer JA, Olinger E. Clinical and genetic spectra of autosomal dominant tubulointerstitial kidney disease. Nephrol Dial Transplant. 2021. doi:10.1093/ndt/gfab268
96. Rampoldi L, Scolari F, Amoroso A, Ghiggeri G, Devuyst O. The rediscovery of uromodulin (Tamm-Horsfall protein): from tubulointerstitial nephropathy to chronic kidney disease. Kidney Int. 2011;80:338–347. doi:10.1038/ki.2011.134
97. Bernascone I, Janas S, Ikehata M, et al. A transgenic mouse model for uromodulin-associated kidney diseases shows specific tubulo-interstitial damage, urinary concentrating defect and renal failure. Hum Mol Genet. 2010;19:2998–3010. doi:10.1093/hmg/ddq205
98. Kraus S, Abel PD, Nachtmann C, et al. MUC1 mucin and trefoil factor 1 protein expression in renal cell carcinoma: correlation with prognosis. Hum Pathol. 2002;33:60–67. doi:10.1053/hupa.2002.29682
99. Leroy X, Zerimech F, Zini L, et al. MUC1 expression is correlated with nuclear grade and tumor progression in pT1 renal clear cell carcinoma. Am J Clin Pathol. 2002;118:47–51. doi:10.1309/1F99-BPDY-7DHH-9G97
100. Kirby A, Gnirke A, Jaffe DB, et al. Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat Genet. 2013;45:299–303. doi:10.1038/ng.2543
101. Coffinier C, Barra J, Babinet C, Yaniv M. Expression of the vHNF1/HNF1beta homeoprotein gene during mouse organogenesis. Mech Dev. 1999;89:211–213. doi:10.1016/s0925-4773(99)00221-x
102. Kolatsi-Joannou M, Bingham C, Ellard S, et al. Hepatocyte nuclear factor-1beta: a new kindred with renal cysts and diabetes and gene expression in normal human development. J Am Soc Nephrol. 2001;12:2175–2180. doi:10.1681/ASN.V12102175
103. Devuyst O, Olinger E, Weber S, et al. Autosomal dominant tubulointerstitial kidney disease. Nat Rev Dis Primers. 2019;5:60. doi:10.1038/s41572-019-0109-9
104. Prieto MC, Gonzalez AA, Navar LG. Evolving concepts on regulation and function of renin in distal nephron. Pflugers Arch. 2013;465:121–132. doi:10.1007/s00424-012-1151-6
105. Schaeffer C, Izzi C, Vettori A, et al. Autosomal dominant tubulointerstitial kidney disease with adult onset due to a novel renin mutation mapping in the mature protein. Sci Rep. 2019;9:11601. doi:10.1038/s41598-019-48014-6
106. Labriola L, Olinger E, Belge H, Pirson Y, Dahan K, Devuyst O. Paradoxical response to furosemide in uromodulin-associated kidney disease. Nephrol Dial Transplant. 2015;30:330–335. doi:10.1093/ndt/gfu389
107. Howles SA, Thakker RV. Genetics of kidney stone disease. Nat Rev Urol. 2020;17:407–421. doi:10.1038/s41585-020-0332-x
108. Ferraro PM, D’Addessi A, Gambaro G. When to suspect a genetic disorder in a patient with renal stones, and why. Nephrol Dial Transplant. 2013;28:811–820. doi:10.1093/ndt/gfs545
109. Rumsby G. Genetic defects underlying renal stone disease. Int J Surg. 2016;36:590–595. doi:10.1016/j.ijsu.2016.11.015
110. Sas DJ, Enders FT, Mehta RA, et al. Clinical features of genetically confirmed patients with primary hyperoxaluria identified by clinical indication versus familial screening. Kidney Int. 2020;97:786–792. doi:10.1016/j.kint.2019.11.023
111. Hopp K, Cogal AG, Bergstralh EJ, et al.; Rare Kidney Stone C. Phenotype-genotype correlations and estimated carrier frequencies of primary hyperoxaluria. J Am Soc Nephrol. 2015;26:2559–2570. doi:10.1681/ASN.2014070698
112. Servais A, Thomas K, Dello Strologo L, et al. Metabolic nephropathy workgroup of the European reference network for rare kidney D, eUrogen: cystinuria: clinical practice recommendation. Kidney Int. 2021;99:48–58. doi:10.1016/j.kint.2020.06.035
113. Sayer JA. The genetics of nephrolithiasis. Nephron Exp Nephrol. 2008;110:e37–43. doi:10.1159/000151730
114. Oliveira B, Kleta R, Bockenhauer D, Walsh SB. Genetic, pathophysiological, and clinical aspects of nephrocalcinosis. Am J Physiol Renal Physiol. 2016;311:F1243–F1252. doi:10.1152/ajprenal.00211.2016
115. Elshamaa MF, Fadel FI, Kamel S, Farouk H, Alahmady M, Ramadan Y. Genetic polymorphisms in CLDN14 (rs219780) and ALP (rs1256328) genes are associated with risk of nephrolithiasis in Egyptian children. Turk J Urol. 2021;47:73–80. doi:10.5152/tud.2020.20141
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Bardet-Biedl Syndrome: Current Perspectives and Clinical Outlook
Melluso A, Secondulfo F, Capolongo G, Capasso G, Zacchia M
Therapeutics and Clinical Risk Management 2023, 19:115-132
Published Date: 30 January 2023
Level of Depression and Anxiety on Quality of Life Among Patients Undergoing Hemodialysis
Alshelleh S, Alhawari H, Alhouri A, Abu-Hussein B, Oweis A
International Journal of General Medicine 2023, 16:1783-1795
Published Date: 10 May 2023