Back to Journals » The Application of Clinical Genetics » Volume 14
Genetic Factors Underlying Sudden Infant Death Syndrome
Authors Keywan C, Poduri AH, Goldstein RD, Holm IA
Received 2 November 2020
Accepted for publication 24 January 2021
Published 15 February 2021 Volume 2021:14 Pages 61—76
DOI https://doi.org/10.2147/TACG.S239478
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Christine Keywan,1 Annapurna H Poduri,1– 5 Richard D Goldstein,1,6,7 Ingrid A Holm1,6,8
1Robert’s Program for Sudden Unexpected Death in Pediatrics, Boston Children’s Hospital, Boston, MA, USA; 2F.M. Kirby Neurobiology Center, Boston Children’s Hospital, Boston, MA, USA; 3Epilepsy Genetics Program, Department of Neurology, Boston Children’s Hospital and Harvard Medical School, Boston, MA, USA; 4Broad Institute of MIT and Harvard, Cambridge, MA, USA; 5Department of Neurology, Harvard Medical School, Boston, MA, USA; 6Department of Pediatrics, Harvard Medical School, Boston, MA, USA; 7Division of General Pediatrics, Department of Pediatrics, Boston Children’s Hospital, Boston, MA, USA; 8Division of Genetics and Genomics, Department of Pediatrics, and Manton Center for Orphan Diseases Research, Boston Children’s Hospital, Boston, MA, USA
Correspondence: Ingrid A Holm
Boston Children’s Hospital, Division of Genetics and Genomics, 3 Blackfan Circle, Mailstop BCH3150, Boston, MA 02115, USA
Email [email protected]
Abstract: Sudden Infant Death syndrome (SIDS) is a diagnosis of exclusion. Decades of research have made steady gains in understanding plausible mechanisms of terminal events. Current evidence suggests SIDS includes heterogeneous biological conditions, such as metabolic, cardiac, neurologic, respiratory, and infectious conditions. Here we review genetic studies that address each of these areas in SIDS cases and cohorts, providing a broad view of the genetic underpinnings of this devastating phenomenon. The current literature has established a role for monogenic genetic causes of SIDS mortality in a subset of cases. To expand upon our current knowledge of disease-causing genetic variants in SIDS cohorts and their mechanisms, future genetic studies may employ functional assessments of implicated variants, broader genetic tests, and the inclusion of parental genetic data and family history information.
Keywords: SIDS, SUID, sudden infant death, gene, genetic, review
Background
Sudden Infant Death syndrome (SIDS) is defined as
the sudden unexpected death of an apparently healthy infant under one year of age that remains unexplained after a thorough case investigation, including performance of a complete autopsy with ancillary testing, examination of the death scene, and review of the clinical history
according to the consensus definition from the 3rd International Congress on Sudden Infant and Child Death.1 SIDS occurs in approximately 1/1000 live-born infants2 and is the leading cause of post-neonatal mortality in developed countries. There are several well-described extrinsic and intrinsic risk factors that raise the risk of SIDS, such as male sex,3 prematurity,4 maternal alcohol or tobacco exposure,5,6 prone sleep position,7 and sleeping on soft bedding or on a shared sleep surface.8 Rates of SIDS have significantly declined in recent decades, a change widely attributed to the “Back to Sleep” campaign initiated in 1994 which promotes safe infant sleep practices. Yet, importantly, SIDS rates declined alongside other causes of neonatal and infant mortality, suggesting the decline may have also been influenced by broad improvements in medical care for pregnant women and infants.9,10 Despite the considerable attention to infant sleep position and environment, mortality rates have remained essentially unchanged since 1996, further suggesting that unidentified and unaddressed factors beyond their reach may contribute to SIDS.
The leading etiological model of SIDS is the “triple risk” model which postulates that SIDS occurs in a biologically vulnerable infant during a critical developmental period, when triggered by a stressor.11 Intrinsic factors leading to biological vulnerabilities, including genetic factors, could lead an infant to be susceptible to certain conditions that would otherwise not be lethal, such as illness, fever, or environmental factors such as sleep position or ambient temperature. Indeed, several lines of evidence suggest that SIDS has genetic underpinnings, including a 4- to 5-fold relative risk of SIDS in subsequent siblings12 and an increased SIDS risk in monozygotic twins compared to dizygotic.13,14
Understanding the genetic factors predisposing some infants to SIDS is a substantially difficult task. In most disciplines of medicine, a biological condition is first discovered and thoroughly described prior to a genetic cause being successfully identified. However, in SIDS genetic research this has occurred in reverse: genetic testing and research has helped elucidate plausible mechanisms of death in SIDS. SIDS is understood as a heterogeneous condition, and we only have a crude understanding of what conditions contribute to the mechanisms responsible for its fatal outcome. We will review various lines of evidence in this rapidly evolving field related to confirmed and suspected biological mechanisms implicated in SIDS, and their underlying genetic basis (Table 1). While there are other review articles that review the genetic factors in SIDS, we are not aware of any as comprehensive, detailed, or current. The majority of reviews focus on individual areas of research, such as cardiogenetics in SIDS and immunogenetics in SIDS, reflective of how the considerations in individual areas of SIDS research tend to be isolated from one another. Additionally, this review includes a discussion of specific limitations and challenges that exist in SIDS research while also providing suggestions for future research directions.
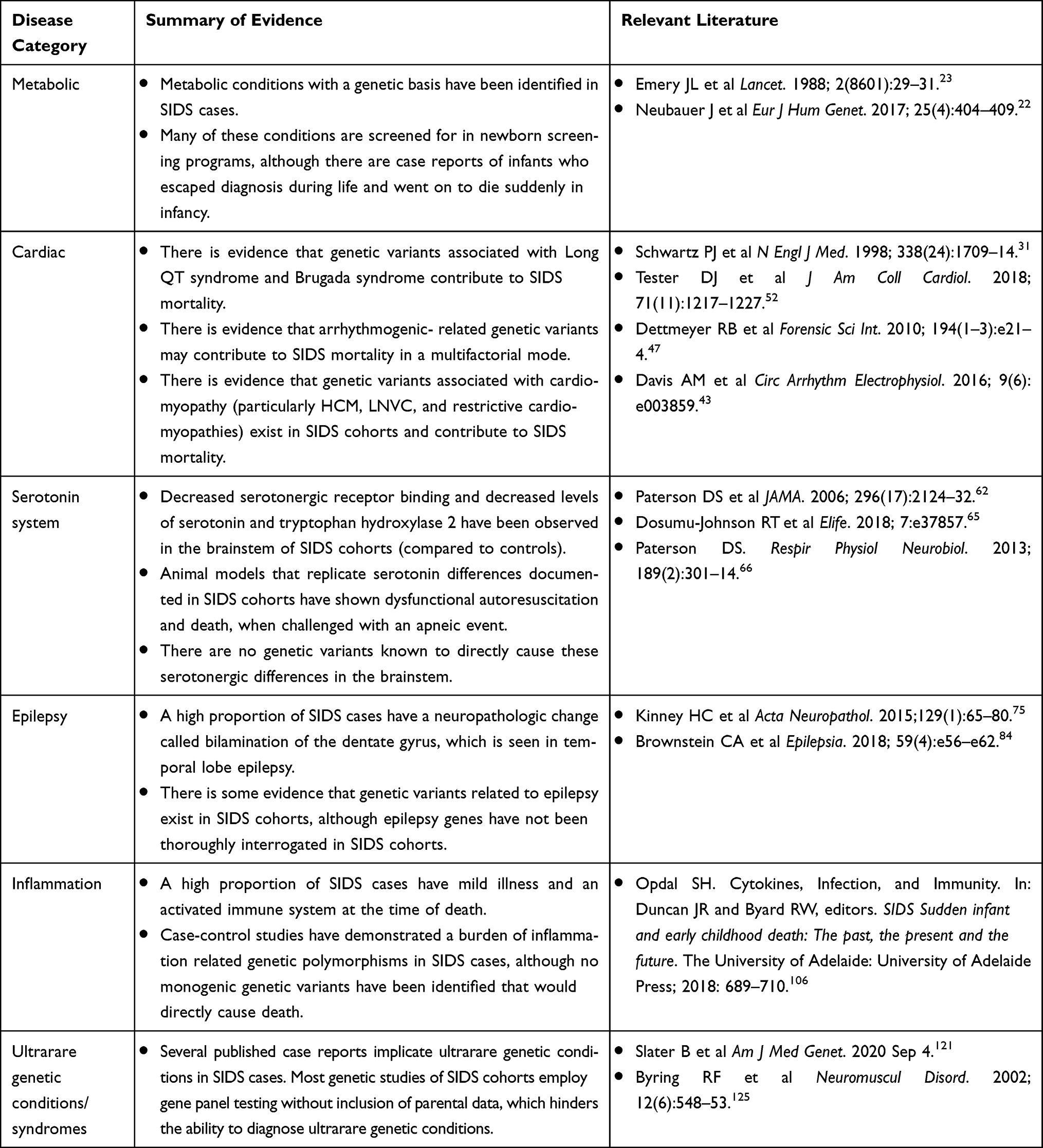 |
Table 1 Summary of Genetic Evidence by Disease Category |
Inborn Errors of Metabolism
Metabolic conditions were the first genetic conditions to be reported in SIDS cases, and the ability to diagnose and treat them reduced the number of infants dying of what had previously been considered SIDS. During the 1970s and ’80s several case reports were published describing infants who had died suddenly from various metabolic conditions, such as glutaric aciduria type II,15 mitochondrial phosphoenolpyruvate carboxykinase deficiency,16 maple syrup urine disease,17 and 21-hydroxylase deficiency.18 One cohort study of SIDS in 1985 identified abnormalities in glycogenosis, the urea cycle, and carnitine palmityl transferase in 5/28 SIDS cases.19 In 1985, a retrospective pathology review of 200 SIDS cases was conducted and reported that 7% had severe diffuse panlobular fatty change of the liver, consistent with disorders of fatty acid oxidation.20 Several SIDS cases had fatty changes in the liver and were found to have defects in medium-chain acyl-coenzyme A dehydrogenase on biochemical assays, indicative of medium-chain acyl-coA deficiency (MCAD). MCAD is a fatty acid oxidation disorder that causes life-threatening episodes of hypoglycemia during periods of fasting, which can be prevented by frequent feedings.21 MCAD is caused by autosomal recessive variants in the ACADM gene, and this singular condition accounted for 1% of the total cohort studied by Howat and colleagues in 1985. One genetic study of 161 SIDS cases published in 2017 reported two infants who carried variants in genes that cause congenital disorder of glycosylation type IM and systemic primary carnitine deficiency, accounting for 1% of their total cohort.22
Given the mounting evidence of metabolic conditions contributing to a substantial proportion of SIDS cases, recommendations were created to aid in more accurate post-mortem diagnoses during the late 1980s.23 These conditions are now largely diagnosed and treated as part of routine newborn screening in many countries, although not all countries have widespread newborn screening programs. Even so, there are reported cases of metabolic conditions that evaded diagnosis during life yet led to sudden unexpected deaths even when newborn screening had been performed.24–26
Cardiac Arrhythmia and Cardiomyopathy
Cardiac disorders have been extensively investigated as potential causes of SIDS, yet there is still much that is unknown in this area. In 1976, it was first formally postulated that cardiac arrhythmias, in particular Long QT syndrome (LQTS), was a plausible mechanism contributing to SIDS deaths.27 LQTS is characterized by prolonged QTc intervals on electrocardiograms (ECG)s when secondary causes have been excluded. Patients with LQTS are at increased risk of syncope and sudden cardiac death (SCD).28 LQTS is inherited in an autosomal dominant fashion. Loss-of-function variants in KCNQ1 and KCNH2 cause LQTS types 1 and 2, respectively. Gain of function variants in SCN5A causes LQTS type 3. Together, these three genes account for 70% of LQTS, while 5% are accounted for by more rare genetic alterations in other genes, and 25% do not have an identifiable genetic cause.29 Shortly after LQTS was postulated to cause SIDS, the hypothesis was supported by a study in which ECG data were collected on parents who had previously lost a child to SIDS, in which 26% of the parent dyads had either a mother or father with a prolonged QT interval.30 Two decades later, a prospective study that collected ECG data on newborn infants over a 19-year period was published.31 Twenty four of 34,442 infants later died of SIDS, and 12 of these 24 had prolonged QTc intervals (at or above the 97.5th percentile of the group as a whole) during the first week of life. In the years following, two case reports of a SIDS case and a “near miss” SIDS case32,33 harboring de novo SCN5A and KCNQ1 variants spurred many candidate gene studies of SIDS cases with a focus on genes causing LQTS and other arrhythmia syndromes.
Brugada syndrome is characterized by ventricular arrhythmias and risk of SCD. Patients are diagnosed when they have “type 1” ECG patterns, ie ≥2 mm coved ST-segment elevation in one or more right precordial lead, either spontaneously, or provoked by a sodium channel blocker along with clinical histories consistent with Brugada syndrome.28 Brugada syndrome is primarily associated with autosomal dominant loss of function variants in SCN5A, although causative variants exist in other genes.29 Arrhythmias in Brugada syndrome typically occur during sleep or rest and in children, they can be provoked by fever.34 Similarly, SIDS occurs overwhelmingly during sleep, and frequently in infants with concurrent fevers. One interesting case report from 2008 describes male monozygotic twins who simultaneously died of SIDS and were found to carry a likely pathogenic nonsense SCN5A variant, W822X; the identical timing of death in these cases defies obvious explanation.35 There are several other case reports describing SIDS cases with family histories of Brugada syndrome, genetic variants associated with Brugada syndrome, and supportive functional evidence, including patch-clamp-derived electrophysiological evidence supporting pathogenicity of some of those variants.36–38
Catecholaminergic Polymorphic Ventricular Tachycardiac (CPVT) is characterized by bidirectional and polymorphic ventricular tachycardia typically prompted by physical activity or emotional stress. CPVT1 is caused by autosomal dominant variants in RYR2, and CPVT2, which is less common, is caused by autosomal recessive variants in CASQ2.28 One study of 134 SIDS cases in 2007 reported 2 variants in RYR2, which displayed gain of function effects consistent with variants observed in CPVT.39 RYR2 variants causing CPVT are largely de novo,40 and a broad lack of parental DNA in SIDS cohorts limits our understanding of the potential contribution of this condition to SIDS. One RYR2 variant previously implicated in SIDS with supportive functional studies was more recently reported in a family of five carriers who have had negative exercise stress tests, echocardiograms and Holter monitors.41 This example demonstrates that in vitro studies do not always directly translate to human disease and emphasizes the importance of segregation data when implicating genetic variants as causative of SIDS.
It is clear that a very small number of “SIDS” deaths can be attributed to de novo variants in arrhythmia genes that cause severe and early onset arrhythmic events or can be attributed to inherited variants in families with family histories consistent with hereditary cardiac arrhythmia syndromes. Controversy remains when inherited variants in arrhythmia genes are implicated as contributory to a SIDS death in cases without a family history consistent with a hereditary arrhythmia syndrome or in which lack of parental genetic data limits our ability to determine if the variant is de novo or inherited. Some of these variants are being over-interpreted, but it is also possible that these variants are contributory and follow multifactorial or polygenic inheritance patterns, particularly when functional studies show a disrupted protein product. There is evidence that the penetrance and expressivity of some arrhythmia syndromes are influenced by other genetic modifiers and may follow oligogenic or polygenic inheritance patterns.42 There is also the possibility that these variants are influenced by environmental or other biological factors and thus more closely follow a multifactorial inheritance pattern, which would fit the triple-risk model of SIDS.43
The hypothesis that hereditary arrhythmia syndromes may play a multifactorial role in SIDS deaths is supported by several lines of evidence. In vitro studies of SCN5A variants identified in SIDS cases have demonstrated normal sodium channel function under resting conditions with abnormal sodium leak under acidosis, suggesting a latent dysfunctional phenotype.44,45 Further, neonatal mouse models have shown that hypoxic environments result in lower expression levels of potassium and sodium channels resulting in an increased risk of sudden cardiac death.46 The authors of the Neary et al study suggest that hypoxia may exacerbate existing genetic predispositions to arrhythmias in humans and cause a sudden death. Both of these studies support the possibility that an arrhythmia phenotype may be provoked under hypoxic circumstances.
Cardiomyopathy may also contribute to the pathogenesis of SIDS. Several infants whose deaths had been classified as SIDS were subsequently diagnosed with cardiomyopathy after a more detailed investigation followed a complete autopsy.47 Dettmeyer et al reported a SIDS case found to have left ventricular noncompaction (LVNC) and another case with hypertrophic cardiomyopathy (HCM), both discovered on detailed histological examinations that are not routinely performed during autopsy. Approximately one third of pediatric LVNC cases have familial disease, and an additional 9% have syndromic or metabolic conditions,48 and several autosomal dominant (MYH7, MYBPC3, ACTC1, TNNI3, MIB1, and others) and X-linked (TAZ) genes have been associated with LVNC.49 Similarly, up to 80% of pediatric cases of hypertrophic cardiomyopathy have an identifiable genetic cause, most commonly variants in sarcomere proteins MYH7 and MYBPC3.50 These findings have led investigators to focus on genes associated with cardiomyopathy; in one study of 286 SIDS cases, seven variants of interest were found in autosomal dominant cardiomyopathy genes (MYBPC3 and TNNI3). Some of the deceased infants had a family history of cardiomyopathy, and most of the variants had been reported in other cases of cardiomyopathy.51
The largest genetic study to date focused on cardiac causes of SIDS included 419 SIDS cases and studied 90 cardiac arrhythmia and cardiomyopathy genes.52 This study used appropriately stringent criteria when assigning pathogenicity to genomic variants thus avoiding over-interpreting genetic variants, which has been an issue in previous studies.53 Tester et al reported 12.6% of their SIDS cases having a “potentially informative” genetic variant in an arrhythmia or cardiomyopathy gene and 4.3% carried variants classified as likely pathogenic or pathogenic based on American College of Genetics and Genomics (ACMG) criteria.54 More routine in vitro studies of genetic variants implicated in SIDS cases, which have been taken on by some centers, will be a key factor in understanding the true impact of variants identified.55,56
Although SIDS has historically been studied as an exclusive diagnosis, cardiogenetic research suggests SIDS exists as part of a spectrum of sudden death between fetal life and adulthood. The hereditary arrhythmia and cardiomyopathy syndromes are generally known to affect older children and adults, but recently, some of these cardiac-related genes have been implicated in cases of stillbirth.57
In summary, current data related to SIDS and cardiac genetics suggest that: 1) monogenic hereditary cardiovascular conditions represent an important but small portion of SIDS cases; 2) cardiovascular genetics may contribute to polygenic and multifactorial causes of SIDS; 3) SIDS may be understood within a continuum between fetal and adult sudden death; and 4) caution should be exercised when implicating genetic variants in cardiac conditions, ideally taking clinical history, family history, segregation testing correlated with cardiac evaluation of family members, and functional data into consideration.
The Brainstem and the Serotonin System
The brainstem originally became a focus in SIDS research because abnormal respiratory patterning,58 ineffective auto-resuscitation (gasping),59 episodic apnea during sleep,60 and arousal deficits61 have been observed in SIDS cases, all involving systems controlled by the brainstem. Supporting the importance of brainstem control of respiration and its potential disruption in at least some cases of SIDS are several lines of evidence from neuropathological case–control studies of SIDS. Studies have reported in SIDS the following abnormalities: decreased serotonergic receptor binding within the medulla oblongata,62 decreased levels of serotonin and its key biosynthetic enzyme, tryptophan hydroxylase 2, in the raphé obscurus,63 and increased levels of serotonin in peripheral blood serum.64 The connection between abnormal serotonin systems and SIDS has been demonstrated in a recent mouse model study.65 Pet1, a transcription factor critical to the function of the serotonin system, was acutely disrupted in transgenic mice pups to test the effects of a dysfunctional serotonergic system during apneic events. When challenged by apnea, the pups’ heart rate and respiration “uncoupled”, ie did not follow the expected linear relationship, and ultimately caused dysfunctional autoresuscitation and death, suggesting a role for serotonin-related brainstem dysfunction in SIDS deaths.
This body of evidence has led researchers to interrogate genes related to the serotonin system in SIDS cases, including FEV (the human homolog to Pet1), TPH2 (tryptophan hydroxylase, the rate-limiting enzyme for serotonin), and HTR1A and HTR2A (serotonin receptors). To date, no genetic variants have been reported in these genes that are rare or damaging enough to cause a sudden death in a case, nor have polymorphisms (variants that are relatively common in the general population but may pose multifactorial risks for biological conditions) in these genes demonstrated robust statistical significance in SIDS populations versus control cohorts.66
Three specific polymorphisms in two genes within the serotonin pathway have received much attention in SIDS research. In SLC6A4, a gene encoding a transporter protein that controls synaptic levels of serotonin,67 there are two polymorphisms that have been studied, one in the promoter region and the other in the second intron. The promoter region polymorphism is a 22–23 base pair insertion/deletion, referred to as the “S”, short, allele. The alternate allele is the “L”, long, allele. The S allele causes reduced transcription of the protein product compared to the L allele, and the S allele asserts dominance over the L allele.68 The other SLC6A4 polymorphism is a tandem repeat within intron 2, consisting of either 9, 10, or 12 sets of 17 bp segment repeats,69 with the 12-repeat allele conferring increased transcription compared to other alleles. The third polymorphism studied in SIDS is in the MAOA gene, which encodes a mitochondrial enzyme that degrades intracellular serotonin.70 The polymorphism consists of either 3, 3.5, 4, or 5 repeats of a 30 base pair sequence within the promoter region of MAOA, with the 3.5 or 4 repeats associated with 2–10 times increased protein expression rates than alternative alleles.71
A number of case–control studies have been conducted to determine the association of these specific polymorphisms with SIDS. The genotypes most commonly associated with SIDS in the literature are the “L/L” SLC6A4 genotype, the “12/12” SLC6A4 genotype, and the “4/4” MAOA genotype. However, over a dozen studies that have been published to determine if there is an association have been conflicting; most studies that have claimed significance are using cohort sizes that are underpowered, and larger cohorts have not found significance. Only one study has moved away from a case–control statistical model towards a genotype-phenotype model to study these polymorphisms in individual cases to understand their relevance.66 This study hypothesized that the L/L promoter polymorphism of SLC6A4, the 12/12intronic polymorphism of SLC6A4, and the 4/4 promoter polymorphism of MAOA would be associated with higher serotonin binding density, lower serotonin receptor binding density, and lower TPH2 and serotonin tissue levels in the medulla (respectively) compared to SIDS cases with other genotypes. Their findings were uniformly contradictory to what was expected and demonstrated that these polymorphisms do not confer the predicted effects on the serotonin system. Based on this evidence, these polymorphisms do not incur a substantial SIDS risk, and testing for these genotypes in patients would not provide meaningful SIDS risk information at this time. However, it is possible other serotonin-related polymorphisms impact SIDS risk, but larger cohorts and less targeted genetic testing will be needed to truly attempt to address this possibility.
Epilepsy-Related Hypotheses
A growing body of literature implicates epilepsy-related mechanisms contributing to SIDS mortality. A connection between febrile seizures and SIDS was first published in 1981, postulating that febrile convulsions and some inexplicable sudden infant deaths may be equivalent responses to a febrile stimulus, but with a different outcome because of the difference in age of occurrence.72
The authors arrived at this hypothesis because several infants who had died of SIDS were admitted to the hospital in cardiac arrest with elevated rectal temperatures. The precise role of febrile seizures and the risk of sudden death in infants and young children are not clear, and large cohorts of children with simple febrile seizures have not been shown to have an increased risk of sudden death, suggesting that a history of simple febrile seizures in isolation may not be sufficient to lead to sudden death.73 In 2016, it was reported that 62% of SIDS cases had a family history of febrile seizures,74 and 41% of SIDS cases have neuropathologic changes in the hippocampus, specifically bilamination of the dentate gyrus,75 otherwise seen in temporal lobe epilepsy.76 This neuropathologic change is much less commonly observed in controls (children who died of known trauma, for example) and is plausibly related to the morphological changes observed in some children after prolonged febrile seizures.77
While evidence regarding the relationship between SIDS and epilepsy is still accruing, there is a clear relationship between sudden death and epilepsy in patients who are known to have a diagnosis of epilepsy. Individuals with epilepsy have a 23-fold increased risk of sudden death compared to the risk of sudden death in age-matched members of the general population, which translates to a ~1/1000 chance of death per year for an adult with epilepsy.78 Patients with Sudden Unexpected Death in Epilepsy (SUDEP) die suddenly, typically discovered following a sleep period in the prone position, without an identifiable cause of death, similar to SIDS.79 SUDEP also applies to individuals with known epilepsy who died following a seizure. In one case report from 2013, a previously healthy 8-month old infant had his first seizure, which was lethal and witnessed by his parents and emergency personnel.80 The suggestion has arisen in the field-based on cases like this and based on the shared neuropathological features among many cases of SIDS and epilepsy – that a tendency to epilepsy may manifest with the very first seizure resulting in death, such that the individual would not have previously been diagnosed with epilepsy and the cause of death thus not considered to be SUDEP. Sudden Death in Childhood (SUDC), the death of a child over the age of 1 year that is unexpected and without a known cause of death after complete autopsy and scene investigation, has also an association with a personal and/or family history of seizures (48.8% of SUDC cases),81 and bilamination of the dentate gyrus (48.1% of SUDC cases).82 Some research groups have adopted the approach of studying these types of deaths as a continuum, with SIDS and SUDC representing a common phenomenon presenting across the age spectrum and SUDEP occurring in patients at any age with known epilepsy, which may yield new insights.74,83,85
In addition to evidence for epileptic-related neuropathological findings present in a substantial subset of SIDS and SUDC cases, epilepsy-related genes have been recently identified in SIDS cohorts. SCN1A encodes a voltage-gated sodium channel, Nav1.1, and pathogenic variants are associated with Dravet syndrome and Genetic Epilepsy with Febrile Seizures Plus (GEFS+), with a wide phenotypic spectrum from mildly affected with febrile seizures to early infantile encephalopathy (OMIM 182389). Two SIDS cases without a clinical history of epilepsy have been reported with variants in the SCN1A gene shown to be damaging via patch-clamp studies of variants in heterologous cell culture.84 SCN1A is one of the genes most commonly implicated in SUDEP.85 A disease-causing variant in SCN1A has also been recently implicated in an SUDC case, with a sibling with the same variant affected by Dravet syndrome.86 SCN1A and other epilepsy-associated genes should be more deeply investigated in future genetic studies on SIDS populations.
Both SCN5A and SCN1A are voltage-gated sodium channels that have been implicated in sudden death and SIDS. The voltage-gated sodium channels generate and propagate action potentials responsible for nerve and muscle excitation. They are a highly conserved family of proteins expressed in excitable tissue in the heart, nervous system, and muscle. Nine sodium channel genes in humans have been identified, referred to as Nav1.1–1.9,87 and the gene family as a whole has been implicated in SIDS and sudden death research. One study considered genes across the entire sodium channel family in a cohort of 73 cases of sudden death in pediatrics, and SIDS cases published in the literature.38 The study identified damaging variants in SCN1A, SCN3A, SCN10A, SCN1B, and SCN4A, in five SIDS patients, none of whom had a personal history of epilepsy or cardiac arrhythmia. This study demonstrates that variants in sodium channel genes, as an entire class, may contribute to the pathogenesis of SIDS.
One recent study focused on five SCN10A variants identified in six cases of a sudden unexpected death, three of whom were infants.88 SCN10A has been associated with cardiac conduction abnormalities, Brugada syndrome, and sudden death.89–91 All three variants identified in the SIDS cases displayed evidence of pathogenicity via patch-clamp studies. The finding of four functionally disruptive SCN10A variants in a cohort of 330 individuals with sudden death suggests SCN10A may be involved in some cases of sudden death, and that SCN10A is deserving of further study in SIDS cohorts.
Another report details four SIDS cases with variants shown to be functionally disruptive in the SCN4A gene via patch-clamp heterologous expression studies.92 SCN4A encodes a sodium channel and pathogenic variants are associated with a range of neuromuscular phenotypes (OMIM 603967). Infants with SCN4A‐related myotonia can present acutely with recurrent episodes of generalized stiffening, apnea, and laryngospasm which may be accompanied by bradycardia and loss of consciousness.93 One infant with an SCN4A variant has been reported to have epilepsy with concurrent apneas, onset at 35 days old.94 The authors propose that damaging SCN4A variants may cause upper airway obstruction via laryngeal and respiratory muscle myotonia in infants when having a seizure. Importantly, this has also been a proposed mechanism of death in SUDEP, and there are rat models of SUDEP which support a role of laryngospasm and upper airway obstruction in the mechanism of death.95
There are important connections to draw between epilepsy and other disease categories previously implicated in SIDS. Some of the cardiac channelopathy genes are expressed in the brain, specifically in the hippocampus,96,97 and many patients with hereditary cardiac arrhythmia syndromes have personal histories of seizures, particularly LQTS type 2.98 One study of 68 SUDEP cases identified variants in KCNH2 and SCN5A that had been previously identified in patients with LQTS and were absent in controls.99 Knock-in mouse models with known pathogenic variants in KCNQ1 (causative of LQTS1) have been shown to develop epilepsy along with cardiac arrhythmias.97
Similarly, the serotonin system has been implicated in SIDS (as described above) and epilepsy. In humans with epilepsy, there is a decrease in serotonin receptor binding,100–102 and animal models have shown that a decrease in extracellular serotonin increases susceptibility to many types of seizures in mice with a genetic susceptibility to epilepsy.103 It has been hypothesized that post-ictal depression of breathing and arousal in some epilepsy patients is caused by dysfunction of the serotonin system, and this mechanism can lead to SUDEP.104 Mouse models specifically addressing the role of genes such as SCN1A in epilepsy and sudden death have demonstrated that seizures lead to respiratory compromise, resulting secondarily in asystole, which has also been observed in a patient.105
In conclusion, current evidence suggests that patients with epilepsy syndromes, or with a propensity to them, exist within SIDS cohorts and that epilepsy-related genes should be more thoroughly investigated with specific attention to sodium channel genes and genes previously implicated in SUDEP. Further studies focused on the inter-relatedness of epilepsy, cardiac arrhythmias, and the serotonin system may yield new insights into mechanisms and their genetic drivers.
Inflammation
The concept of an infectious mechanism contributing to SIDS deaths first emerged in the late 1800s, when a report demonstrated inflammatory changes in the bronchioles of SIDS cases.106 Much more recently, an epidemiology study in the United Kingdom (UK) reported that 26% of all SIDS cases had been ill during the week prior to their deaths.107 Opdal recently wrote, There is convincing evidence that slight infection and an activated immune system are involved in SIDS. One common thread may be the dysregulation of inflammatory responses to apparently mild infections, leading to a cytokine storm that contributes to death.
This review emphasized that inflammatory dysregulation most likely contributes to a SIDS death in conjunction with other risks, such as other biological or environmental predispositions and risks.106
Cytokines are proteins involved in cell signaling and play a critical role in a coordinated immune response. Cytokines include Interferons (INF), interleukins (IL), chemokines, lymphokines, and tumor necrosis factors (TNF).106 Several polymorphisms in interleukins IL-6, IL-10, and IL-1B have been studied in many SIDS cohorts, but none have demonstrated consistent statistical significance in SIDS cases.108–111 Two promoter polymorphisms of a tumor necrosis factor, TNF, have been investigated in SIDS cohorts with conflicting results and functional characterizations of the polymorphisms have not been supportive of a role in SIDS.108,112,113
Two polymorphisms in the IL1A gene, one tandem repeat variant in intron 6, “A1”, and the +4845G/T (rs17561) variant have been shown to be over-represented in SIDS cases compared to controls, when they exist together,114 and has shown functional significance in immune response.115 Another variable repeat variant in intron 2 of a different interleukin gene, IL1RA, “A2” has been studied in SIDS populations, and the A2/A2 haplotype has shown to be statistically significantly increased in SIDS cases compared to controls, in several cohorts116 and functional significance.117 Four promoter polymorphisms in TNF have been investigated in a Norwegian cohort, −1031C/T (rs1799964), −857C/T (rs1799724), −308A/G, and −238A/G, and the combination of −1031CT/-238GG/-857CC/-308GG and −1031TT/-238GG/-857CC/-308AA were also over-represented in SIDS cases, although these polymorphisms have not been studied functionally.118 One recent study showed an association of three polymorphisms in interferon genes, INFG (rs2069705 and rs2069727) and INFA8 (rs1330321), with SIDS compared to controls, although these polymorphisms have also not been functionally studied.119
In summary, there is emerging evidence that some polymorphisms in cytokine genes may play a role in SIDS pathogenesis, while others have not been definitively shown to be over-represented in SIDS vs controls. The polymorphisms currently implicated exist in healthy control populations with frequencies inconsistent with monogenic disease, but current evidence suggests that these polymorphisms may contribute to multifactorial SIDS risk, meaning their presence alone is not sufficient to be a cause of death but their presence may compound other intrinsic or extrinsic risk factors. Larger cohorts and functional assessments will be required to definitively associate any inflammatory polymorphisms with SIDS risk.
Ultra-Rare Genetic Conditions and Syndromes
Several ultra-rare genetic conditions have been identified in SIDS cases and implicated in their deaths. For example, a 2004 case series details an Amish family with 21 related infants, from 9 sibships, affected by a disorder characterized by dysgenesis of the testis, neurologic dysfunction, and sudden death prior to 12 months of age caused by biallelic truncating variants in the TSPYL1 gene; an unrelated non-Amish infant with the same condition was reported in 2020.120,121 This represents an ultrarare recessive genetic condition that can cause sudden deaths in an apparently healthy infant.
Congenital myasthenic syndrome was first proposed as a potential cause of SIDS in 1975, supported by a report in 1980 suggesting infantile myasthenia was the most lethal type of the myasthenic syndromes.122,123 Congenital myasthenic syndromes are a group of disorders caused by impaired neuromuscular transmission and characterized by abnormal fatiguability, which can be transient or permanent. Features include weakness of the facial, extraocular, truncal, respiratory, or limb muscles, and onset can be infantile, childhood, or adolescence. Acute deteriorations can occur triggered by infections, fever, or emotional stress. Pharmaceuticals exist to manage symptoms, although there is no cure.124 There are 31 genes that cause myasthenic syndromes and the majority are autosomal recessive. There is one published case report of the congenital myasthenic syndrome in two siblings who died of SIDS and carried biallelic CHAT variants.125 One sibling had a history of mild ptosis, the other was asymptomatic and both died during febrile episodes, one at age 2 months and the other at age 11 months. The biallelic CHAT variants causative of the myasthenic syndrome were elucidated in this family because they were identified in a living affected sibling. Although this is the only published case report of congenital myasthenic syndrome causing SIDS, it is plausible that biallelic variants in CHAT or other genes causing myasthenic syndromes could contribute to the pathogenesis of SIDS. Due to the autosomal recessive nature of this condition, and the lack of available parental data in the vast majority of SIDS cohorts studied, this condition has not been routinely investigated.
Congenital Central Hypoventilation syndrome (CCHS) is characterized by low ventilation during sleep. Some patients have isolated respiratory dysfunction while others have syndromic CCHS, which most commonly includes Hirschsprung’s disease, neuroblastoma, or other symptoms of autonomic dysregulation such as reduced temperature control or differences in pain perception.126 CCHS is most commonly caused by autosomal dominant variants in PHOX2B, and more rarely by variants in RET, EDN3, or ASCL1, and can be a feature of MECP2 duplication syndrome.127–130 CCHS was first proposed as a potential cause of SIDS in a case series published in 1982 by Guilleminault et al.131 The case series documents the clinical course of six infants with CCHS, one of which was originally referred to their center because of a “near miss SIDS event”. The infant was born with hypotonia, but her respiration appeared normal until she suddenly stopped breathing during her sleep at 6 weeks old and became cyanotic. She resumed respiration without intervention, but this event prompted further evaluation and led to the diagnosis of CCHS. The authors point out that if she had not been able to auto resuscitate, the death would have been labeled “SIDS”.
PHOX2B is a gene encoding a transcription factor crucial to the development of the autonomic nervous system (ANS), specifically the part of the ANS which regulates a response to CO2 levels and coordinates autonomic respiration during sleep.132,133 The most common variants which cause CCHS are PolyAlanine Repeat Mutations (PARM) which expand the number of Ala repeats in exon3 of PHOX2B, although other variant types such as missense, nonsense, and frameshift are well documented in CCHS.134 Some single nucleotide variants in PHOX2B have been identified in SIDS cases, but none have enough evidence to implicate them as a cause of CCHS. One particular type of variant has more recently received attention in the SIDS literature: PolyAlanine Repeat contractions, as opposed to the well-understood expansion variants. One 2014 study reported that these PolyAlanine contractions were over-represented in a Dutch SIDS cohort compared to a control population, and one case report from 2018 implicated a PolyAla contraction as a contributing factor in the sudden death of a 28-day-old infant.135,136 The authors of the case report wrote the sudden death of the infant was attributed to the underlying genetic predisposition to congenital central hypoventilation (i.e. low-penetrance PHOX2B gene), manifesting clinically as transient hypoventilation, correlated to prone position during sleep, whose combined effects likely resulted in a critical impairment of autonomic control of respiration at a susceptible stage of development.
This is a stretch based on available evidence. There have not been any reports of individuals living with CCHS who carry this variant type, and polyalanine contractions within this region exist in the general healthy population. These contraction variants have been reported to be statistically more common in patients with Hirschsprung’s disease, and functional studies have shown that contraction variants cause reduced transactivation of the RET promoter, which is consistent with known molecular mechanisms of increased risk of Hirschsprung’s disease.137,138 Further, Hirschsprung’s disease is known to display complex inheritance, whereas CCHS is understood to be mendelian.139 Current evidence suggests these polyalanine contractions may be risk factors for Hirschsprung’s disease, but additional studies are needed to understand whether they can be similarly implicated in CCHS or SIDS.
Mitochondrial conditions have been implicated as a potential contributor to SIDS mortality over the past several decades. Mitochondrial disorders are a heterogenous group of conditions that are typically multisystemic and challenging to diagnose. Mitochondrial DNA (mtDNA) is maternally inherited and codes for genes responsible for oxidative phosphorylation and the electron transport chain. Tissues with high energy expenditure are most commonly impacted by mitochondrial conditions, such as the brain, skeletal muscles, and heart. There are also nuclear genes that are associated with mitochondrial conditions, which are inherited in a Mendelian fashion.140 There have been several case–control studies that have focused on polymorphisms in mtDNA. Some have shown differences in SIDS populations and have suggested a multifactorial role of these polymorphisms.141 Several mtDNA variants have been reported in SIDS cases, although none have been classified as pathogenic.142 There have been case reports of families affected by mitochondrial disorders with family histories of sudden infant death, following a seemingly maternal inheritance pattern.143,144 It is possible that some SIDS cases are caused by a mitochondrial disorder that is exacerbated by other stressors, such as a minor illness, but more research is needed in this area to clarify this possibility. Genetic sequencing for mitochondrial disorders requires both a blood sample and muscle tissue sample, which are difficult for research studies to systematically obtain.
Genetic Conditions Incompatible with Life
It is reasonable to consider the impact of genes without any known association with a human condition in SIDS cases, as damaging variation in such genes may be incompatible with life. There has been one published exome study of 427 SIDS cases that sought a monogenic basis of SIDS.145 This is the only study that has taken an unbiased approach to the exome to seek the significance of any gene, regardless of disease association or function. This study did not yield any statistically significant results. Importantly, this does not suggest a lack of genetic contribution to SIDS, rather, supports an extreme heterogeneity in the genetic causes which cannot be singled out by these methods in a cohort of this size. Extremely heterogenic causes are more readily identified through very stringent individual phenotyping, the inclusion of parental genetic data, or with extremely large data sets. Exome or genome-wide studies will be the key to implicating new genotype-phenotype correlations with SIDS and promote the discovery of unidentified genetic conditions contributing to SIDS pathogenesis.
Copy Number Variation
There is only one published study of copy number variation (CNV) in a SIDS cohort. In this study, three SIDS cases out of 27 carried a large de novo CNV,146 although the clinical significance of the reported CNVs are not clear. Testing for CNVs is routine in other areas of genomic medicine, such as during evaluations for developmental delay, autism, and epilepsy.147–149 One recent study published by a commercial genetic testing lab reported that 4.7% of all pathogenic variants reported in cardiovascular disorders were CNVs, and 7.7% of all pathogenic variants in rare pediatric conditions were CNVs, with the highest rates specifically observed in cases of early infantile epileptic encephalopathy.150 Genetic conditions implicated in SIDS are sometimes caused by CNVs; therefore, more studies should be conducted to understand the burden and clinical significance of CNVs in SIDS cohorts. Importantly, the clinical interpretation of CNVs is even more difficult than the clinical interpretation of sequence variants due to limited population-level data, and the inclusion of parental data is key to the interpretation of CNVs.
Discussion
Emerging evidence for genetic variants in a variety of genes playing a role in SIDS suggests that genetic testing should be considered routinely in SIDS cases. Before this practice becomes widely adopted, additional research will be required to identify specific genetic causes and to demonstrate the utility of genetic information (eg in counseling on recurrence risk, in screening living family members who may be at risk). There are, however, unique barriers to genomic research in SIDS cases. Systematically identifying cases that qualify for SIDS research has been complicated by inconsistencies in the classification of SIDS deaths. There has been a well-documented diagnostic shift away from “SIDS” and increasingly towards both “Undetermined” and “accidental suffocation”. Unfortunately, cases of infant death due to accidental suffocation exist and more systematic death scene investigations have the potential to uncover important factors. However, there are also many cases where sleep environmental risks, such as sleeping in the prone position, are attributed as causes of death without additional evidence of suffocation. Further complicating research recruitment and family engagement is the fact that some members of the medical examiner community reject the term “SIDS” as a diagnosis. There was a recent effort to rectify this by the 3rd International Congress on Sudden Infant and Child Death in 2019, which suggested clear definitions and exclusion criteria to help guide death certifiers.1 If death certifications become more consistent, cases that should be included in SIDS research can be more readily identified and epidemiologic data will be more precise.
Once a SIDS case has been identified, obtaining and storing appropriate samples for DNA extraction does not occur predictably. The National Association of Medical Examiners (NAME) published a position paper in 2013 that includes SIDS in the list of clinical indications for post-mortem genetic testing, and details what biological tissues to store for genetic testing.151
These recommendations have broadly improved storage practices, but there are still limitations that medical examiners deal with during autopsy, as well as capacity issues that impact storage capability. Genomic technology is increasingly allowing for genomic sequencing on new sample types. For example, dried blood spot cards are more easily stored than tissue samples and can now be used by some labs for whole-exome sequencing, which was not an option even just a few years ago.
The type of genetic testing historically chosen in SIDS research has had severe limitations. The vast majority of published studies have only had access to DNA from the proband and have relied on autopsy and investigative reports for all medical information. Lack of parental DNA and sequencing hinders accurate variant interpretation and stunts the discovery of novel genetic causes of SIDS. Given that all of the genetic factors plausibly related to SIDS are not currently known, a large genetic test for research purposes would be ideal to promote discovery. However, large genetic tests, such as whole exome or whole genome sequencing, require parental data for optimal interpretation. Most available genetics studies have done “panel testing” either by only sequencing a specific subset of genes, or by performing whole-exome sequencing but only interpreting and reporting variants in a given subset of genes. This has been done without parental DNA and in the setting of limited clinical and family history, due to the sole reliance on autopsy and investigative reports for this information, further complicating variant interpretation. Both of these issues would be resolved by more direct involvement of parents and families in SIDS research studies. If parents could provide DNA samples along with personal and family medical history information, researchers could more readily employ broader genetic tests and have the ability to interpret genetic variants more accurately. This would open up more potential for discovering new genetic causes of SIDS and may be psychologically beneficial for parents to directly participate in research.
A notable limitation of current genetics research in SIDS is a lack of diversity. Although ethnic differences have been described in SIDS, very few studies include a high proportion of non-European participants which limits our understanding of the genetic burden of other populations. Further, genetic testing is not as accessible on a clinical or research basis in many non-Western countries.
The current literature has established a role for monogenic genetic causes of SIDS in a small but important subset of cases. There is much more work to do to expand upon our current knowledge of disease-causing genetic variants and their mechanisms. Functional studies of variants identified in SIDS cases should be conducted with more regularity to validate or invalidate their effect on the protein product, along with segregation studies and deep phenotyping of affected families. Ideally, parental data should be incorporated, and more broad genetic tests, such as exome or genome, should be more regularly employed in the research setting. As research groups expand their SIDS cohorts or increase collaborations, they may be powered to better address multifactorial or polygenic causes of SIDS.
Disclosure
The authors report no conflicts of interest related to this work.
References
1. Goldstein RD, Blair PS, Sens MA, et al. Inconsistent classification of unexplained sudden deaths in infants and children hinders surveillance, prevention and research: recommendations from the 3rd international congress on sudden infant and child death. Forensic Sci Med Pathol. 2019;15:622–628. doi:10.1007/s12024-019-00156-9
2. Center for Disease Control. Sudden unexpected infant death and sudden infant death syndrome. [updated September 18, 2020]. Available from: https://www.cdc.gov/sids/data.htm#:~:text=In%202018%2C%20the%20SUID%20rate,100%2C000%20live%20births%20in%202018.
3. Lewak N, van den Berg BJ, Beckwith JB. Sudden infant death syndrome risk factors. Clin Pediatr. 1979;18(7):404–411. doi:10.1177/000992287901800704
4. Horne RSC. Effects of prematurity on heart rate control: implications for sudden infant death syndrome. Expert Rev Cardiovascular Ther. 2006;4(3):335–343. doi:10.1586/14779072.4.3.335
5. Strandberg-Larsen K, Gronboek M, Andersen AN, Andersen PK, Olsen J. Alcohol drinking pattern during pregnancy and risk of infant mortality. Epidemiology. 2009;20(6):884–891. doi:10.1097/EDE.0b013e3181bbd46c
6. Schoendorf KC, Kiely JL. Relationship of sudden infant death syndrome to maternal smoking during and after pregnancy. Pediatrics. 1992;90(6):905–908.
7. Willinger M, Hoffman HJ, Hartford RB. Infant sleep position and risk for sudden infant death syndrome: report of meeting held January 13 and 14, 1994, National Institutes of Health, Bethesda, MD. Pediatrics. 1994;93(5):814–819.
8. Hauck FR, Herman SM, Donovan M, et al. Sleep environment and the risk of sudden infant death syndrome in an urban population: the Chicago infant mortality study. Pediatrics. 2003;111(5):1207–1214.
9. Goldstein RD, Trachtenberg FL, Sens MA, Harty BJ, Kinney HC. Overall postneonatal mortality and rates of SIDS. Pediatrics. 2016;137(1):e20152298. doi:10.1542/peds.2015-2298
10. Hauck FR, Tanabe KO. Beyond “Back to Sleep”: ways to further reduce the risk of sudden infant death syndrome. Pediatr Ann. 2017;46(8):284–290. doi:10.3928/19382359-20170721-01
11. Filiano JJ, Kinney HC. A perspective on neuropathologic findings in victims of the sudden infant death syndrome: the triple-risk model. Biol Neonate. 1994;65(3–4):194–197. doi:10.1159/000244052
12. Christensen ED, Berger J, Alashari MM, et al. Sudden infant death “Syndrome”- insights and future directions from a Utah population database analysis. Am J Med Genet A. 2017;173(1):177–182. doi:10.1002/ajmg.a.37994
13. Rhoades ER, Brenneman G, Lyle J, Handler A. Mortality of American Indian and Alaska native infants. Annu Rev Public Health. 1992;13:269–285. doi:10.1146/annurev.pu.13.050192.001413
14. Pharoah PO, Platt MJ. Sudden infant death syndrome in twins and singletons. Twin Res Hum Genet. 2007;10(4):644–648. doi:10.1375/twin.10.4.644
15. Bennet MJ, Curnock DA, Engel PC, et al. Glutaric aciduria type ii: biochemical investigation and treatment of a child diagnosed prenatally. J Inher Metab Dis. 1984;7(2):57–61. doi:10.1007/BF01805802
16. Clayton PT, Hyland K, Brand M, Leonard JV. Mitochondrial phosphoenolpyruvate carboxykinase deficiency. Eur J Pediatr. 1986;145:46–50. doi:10.1007/BF00441851
17. Hallock J, Morrow G, Karp LA, Barness LA. Postmortem diagnosis of metabolic disorders. The finding of maple syrup urine disease in a case of sudden and unexpected death in infancy. Am J Dis Child. 1969;118(4):649–651. doi:10.1001/archpedi.1969.02100040651022
18. Russell MA, Opitz JM, Viseskul C, Gilbert EF, Bargman GJ. Sudden infant death due to congenital adrenal hypoplasia. Arch Pathol Lab Med. 1977;101(4):168–169.
19. Vawter GF, McGraw CA, Hug G, Kozakewich HP, McNaulty J, Mandell F. An hepatic metabolic profile in sudden infant death (SIDS). Forensic Sci Int. 1986;30(2–3):93–98. doi:10.1016/0379-0738(86)90003-4
20. Howat AJ, Bennett MJ, Variend S, Shaw L, Engel PC. Defects of metabolism of fatty acids in the sudden infant death syndrome. Br Med J (Clin Res Ed). 1985;290(6484):1771–1773. doi:10.1136/bmj.290.6484.1771
21. Egidio RJ, Francis GL, Coates PM, Hale DE, Roesel A. Medium-chain acyl-CoA dehydrogenase deficiency. Am Fam Physician. 1989;39(5):221–226.
22. Neubauer J, Lecca MR, Russo G, et al. Post-mortem whole-exome analysis in a large sudden infant death syndrome cohort with a focus on cardiovascular and metabolic genetic diseases. Eur J Hum Genet. 2017;25(4):404–409. doi:10.1038/ejhg.2016.199
23. Emery JL, Howat AJ, Variend S, Vawter GF. Investigation of inborn errors of metabolism in unexpected infant deaths. Lancet. 1988;2(8601):29–31. doi:10.1016/S0140-6736(88)92955-8
24. Jequier Gygax M, Roulet-Perez E, Meagher-Villemure K, et al. Sudden unexpected death in an infant with L-2-hydroxyglutaric aciduria. Eur J Pediatr. 2009;168(8):957–962. doi:10.1007/s00431-008-0869-9
25. Du SH, Zhang F, Yu YG, Chen CX, Wang HJ, Li DR. Sudden infant death from neonate carnitine palmitoyl transferase II deficiency. Forensic Sci Int. 2017;278:e41–e44. doi:10.1016/j.forsciint.2017.06.020
26. Krizova A, Herath JC. Death of a neonate with a negative autopsy and ketoacidosis: a case report of propionic acidemia. Am J Forensic Med Pathol. 2015;36(3):193–195. doi:10.1097/PAF.0000000000000156
27. Schwartz PJ. Cardiac sympathetic innervation and the sudden infant death syndrome. A possible pathogenetic link. Am J Med. 1976;60(2):167–172. doi:10.1016/0002-9343(76)90425-3
28. Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10(12):1932–1963.
29. Gray B, Behr ER. New insights into the genetic basis of inherited arrhythmia syndromes. Circ Cardiovasc Genet. 2016;9(6):569–577. doi:10.1161/CIRCGENETICS.116.001571
30. Maron BJ, Clark CE, Goldstein RE, Epstein SE. Potential role of QT interval prolongation in sudden infant death syndrome. Circulation. 1976;54(3):423–430. doi:10.1161/01.CIR.54.3.423
31. Schwartz PJ, Stramba-Badiale M, Segantini A, et al. Prolongation of the QT interval and the sudden infant death syndrome. N Engl J Med. 1998;338(24):1709–1714. doi:10.1056/NEJM199806113382401
32. Schwartz PJ, Priori SG, Dumaine R, et al. A molecular link between the sudden infant death syndrome and the long-QT syndrome. N Engl J Med. 2000;343(4):262–267. doi:10.1056/NEJM200007273430405
33. Schwartz PJ, Priori SG, Bloise R, et al. Molecular diagnosis in a child with sudden infant death syndrome. Lancet. 2001;358(9290):1342–1343. doi:10.1016/S0140-6736(01)06450-9
34. Michowitz Y, Milman A, Sarquella-Brugada G, et al. Fever-related arrhythmic events in the multicenter survey on arrhythmic events in Brugada syndrome. Heart Rhythm. 2018;15(9):1394–1401. doi:10.1016/j.hrthm.2018.04.007
35. Turillazzi E, La Rocca G, Anzalone R, et al. Heterozygous nonsense SCN5A mutation W822X explains a simultaneous sudden infant death syndrome. Virchows Arch. 2008;453(2):209–216. doi:10.1007/s00428-008-0632-7
36. Priori SG, Napolitano C, Giordano U, Collisani G, Memmi M. Brugada syndrome and sudden cardiac death in children. Lancet. 2000;355(9206):808–809. doi:10.1016/S0140-6736(99)05277-0
37. Gando I, Morganstein J, Jana K, McDonald TV, Tang Y, Coetzee WA. Infant sudden death: mutations responsible for impaired Nav1.5 channel trafficking and function. Pacing Clin Electrophysiol. 2017;40(6):703–712. doi:10.1111/pace.13087
38. Rochtus AM, Goldstein RD, Holm IA, et al. The role of sodium channels in sudden unexpected death in pediatrics. Mol Genet Genomic Med. 2020;8(8):e1309. doi:10.1002/mgg3.1309
39. Tester DJ, Dura M, Carturan E, et al. A mechanism for sudden infant death syndrome (SIDS): stress-induced leak via ryanodine receptors. Heart Rhythm. 2007;4(6):733–739. doi:10.1016/j.hrthm.2007.02.026
40. Ohno S, Hasegawa K, Horie M. Gender differences in the inheritance mode of RYR2 mutations in catecholaminergic polymorphic ventricular tachycardia patients. PLoS One. 2015;10(6):e0131517. doi:10.1371/journal.pone.0131517
41. Kohli U, Nayak HM. SIDS associated RYR2 p.Arg2267His variant may lack pathogenicity. J Electrocardiol. 2020;60:23–26. doi:10.1016/j.jelectrocard.2020.03.007
42. Cerrone M, Remme CA, Tadros R, Bezzina CR, Delmar M. Beyond the one gene-one disease paradigm: complex genetics and pleiotropy in inheritable cardiac disorders. Circulation. 2019;140(7):595–610. doi:10.1161/CIRCULATIONAHA.118.035954
43. Davis AM, Glengarry J, Skinner JR. Sudden infant death: QT or not QT? that is no longer the question. Circ Arrhythm Electrophysiol. 2016;9(6):e003859. doi:10.1161/CIRCEP.115.003859
44. Wang DW, Desai RR, Crotti L, et al. Cardiac sodium channel dysfunction in sudden infant death syndrome. Circulation. 2007;115(3):368–376. doi:10.1161/CIRCULATIONAHA.106.646513
45. Plant LD, Bowers PN, Liu Q, et al. A common cardiac sodium channel variant associated with sudden infant death in African Americans, SCN5A S1103Y. J Clin Invest. 2006;116(2):430–435. doi:10.1172/JCI25618
46. Neary MT, Mohun TJ, Breckenridge RA. A mouse model to study the link between hypoxia, long QT interval and sudden infant death syndrome. Dis Model Mech. 2013;6(2):503–507. doi:10.1242/dmm.010587
47. Dettmeyer RB, Kandolf R. Cardiomyopathies–misdiagnosed as Sudden Infant Death Syndrome (SIDS). Forensic Sci Int. 2010;194(1–3):e21–4. doi:10.1016/j.forsciint.2009.10.010
48. Miller EM, Hinton RB, Czosek R, et al. Genetic testing in pediatric left ventricular noncompaction. Circ Cardiovasc Genet. 2017;10(6):e001735. doi:10.1161/CIRCGENETICS.117.001735
49. Ichida F. Left ventricular noncompaction – risk stratification and genetic consideration. J Cardiol. 2020;75(1):1–9. doi:10.1016/j.jjcc.2019.09.011
50. Rupp S, Felimban M, Schänzer A, et al. Genetic basis of hypertrophic cardiomyopathy in children. Clin Res Cardiol. 2019;108(3):282–289. doi:10.1007/s00392-018-1354-8
51. Brion M, Allegue C, Santori M, et al. Sarcomeric gene mutations in sudden infant death syndrome (SIDS). Forensic Sci Int. 2012;219(1–3):278–281. doi:10.1016/j.forsciint.2012.01.018
52. Tester DJ, Wong LCH, Chanana P, et al. Cardiac genetic predisposition in sudden infant death syndrome. J Am Coll Cardiol. 2018;71(11):1217–1227. doi:10.1016/j.jacc.2018.01.030
53. Lin Y, Williams N, Wang D, et al. Applying high-resolution variant classification to cardiac arrhythmogenic gene testing in a demographically diverse cohort of sudden unexplained deaths. Circ Cardiovasc Genet. 2017;10(6):e001839. doi:10.1161/CIRCGENETICS.117.001839
54. Richards S, Aziz N, Bale S, et al. ACMG laboratory quality assurance committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
55. Subbotina E, Williams N, Sampson BA, Tang Y, Coetzee WA. Functional characterization of TRPM4 variants identified in sudden unexpected natural death. Forensic Sci Int. 2018;293:37–46. doi:10.1016/j.forsciint.2018.10.006
56. Subbotina E, Yang HQ, Gando I, et al. Functional characterization of ABCC9 variants identified in sudden unexpected natural death. Forensic Sci Int. 2019;298:80–87. doi:10.1016/j.forsciint.2019.02.035
57. Stanley KE, Giordano J, Thorsten V, et al. Causal genetic variants in stillbirth. N Engl J Med. 2020;383(12):1107–1116. doi:10.1056/NEJMoa1908753
58. Schechtman VL, Lee MY, Wilson AJ, Harper RM. Dynamics of respiratory patterning in normal infants and infants who subsequently died of the sudden infant death syndrome. Pediatr Res. 1996;40(4):571–577. doi:10.1203/00006450-199610000-00010
59. Sridhar R, Thach BT, Kelly DH, Henslee JA. Characterization of successful and failed autoresuscitation in human infants, including those dying of SIDS. Pediatr Pulmonol. 2003;36(2):113–122. doi:10.1002/ppul.10287
60. Kahn A, Groswasser J, Rebuffat E, et al. Sleep and cardiorespiratory characteristics of infant victims of sudden death: a prospective case-control study. Sleep. 1992;15(4):287–292. doi:10.1093/sleep/15.4.287
61. Kato I, Franco P, Groswasser J, et al. Incomplete arousal processes in infants who were victims of sudden death. Am J Respir Crit Care Med. 2003;168(11):1298–1303. doi:10.1164/rccm.200301-134OC
62. Paterson DS, Trachtenberg FL, Thompson EG, et al. Multiple serotonergic brainstem abnormalities in sudden infant death syndrome. JAMA. 2006;296(17):2124–2132. doi:10.1001/jama.296.17.2124
63. Duncan JR, Paterson DS, Hoffman JM, et al. Brainstem serotonergic deficiency in sudden infant death syndrome. JAMA. 2010;303(5):430–437. doi:10.1001/jama.2010.45
64. Haynes RL, Frelinger AL, Giles EK, et al. High serum serotonin in sudden infant death syndrome. Proc Natl Acad Sci U S A. 2017;114(29):7695–7700. doi:10.1073/pnas.1617374114
65. Dosumu-Johnson RT, Cocoran AE, Chang Y, Nattie E, Dymecki SM. Acute perturbation of Pet1-neuron activity in neonatal mice impairs cardiorespiratory homeostatic recovery. Elife. 2018;7:e37857. doi:10.7554/eLife.37857
66. Paterson DS. Serotonin gene variants are unlikely to play a significant role in the pathogenesis of the sudden infant death syndrome. Respir Physiol Neurobiol. 2013;189(2):301–314. doi:10.1016/j.resp.2013.07.001
67. Blakely RD, DeFelice LJ, Galli A. Biogenic amine neurotransmitter transporters: just when you thought you knew them. Physiology (Bethesda). 2005;20:225–231. doi:10.1152/physiol.00013.2005
68. Heils A, Mössner R, Lesch KP. The human serotonin transporter gene polymorphism-basic research and clinical implications. J Neural Transm (Vienna). 1997;104(10):1005–1014. doi:10.1007/BF01273314
69. Ogilvie AD, Battersby S, Bubb VJ, et al. Polymorphism in serotonin transporter gene associated with susceptibility to major depression. Lancet. 1996;347(9003):731–733. doi:10.1016/S0140-6736(96)90079-3
70. Cases O, Seif I, Grimsby J, et al. Aggressive behavior and altered amounts of brain serotonin and norepinephrine in mice lacking MAOA. Science. 1995;268(5218):1763–1766. doi:10.1126/science.7792602
71. Sabol SZ, Hu S, Hamer D. A functional polymorphism in the monoamine oxidase A gene promoter. Hum Genet. 1998;103(3):273–279. doi:10.1007/s004390050816
72. Sunderland R, Emery JL. Febrile convulsions and cot death. Lancet. 1981;2(8239):176–178. doi:10.1016/S0140-6736(81)90359-7
73. Vestergaard M, Pedersen MG, Ostergaard JR, Pedersen CB, Olsen J, Christensen J. Death in children with febrile seizures: a population-based cohort study. Lancet. 2008;372(9637):457–463. doi:10.1016/S0140-6736(08)61198-8
74. Kinney HC, Poduri AH, Cryan JB, et al. Hippocampal formation maldevelopment and sudden unexpected death across the pediatric age spectrum. J Neuropathol Exp Neurol. 2016;75(10):981–997. doi:10.1093/jnen/nlw075
75. Kinney HC, Cryan JB, Haynes RL, et al. Dentate gyrus abnormalities in sudden unexplained death in infants: morphological marker of underlying brain vulnerability. Acta Neuropathol. 2015;129(1):65–80. doi:10.1007/s00401-014-1357-0
76. Houser CR. Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain Res. 1990;535(2):195–204. doi:10.1016/0006-8993(90)91601-C
77. Chan S, Bello JA, Shinnar S, et al. Hippocampal malrotation is associated with prolonged febrile seizures: results of the FEBSTAT study. AJR Am J Roentgenol. 2015;205:1068–1074. doi:10.2214/AJR.14.13330
78. Saetre E, Abdelnoor M. Incidence rate of sudden death in epilepsy: a systematic review and meta-analysis. Epilepsy Behav. 2018;86:193–199. doi:10.1016/j.yebeh.2018.06.037
79. Verducci C, Hussain F, Donner E, et al. SUDEP in the North American SUDEP Registry: the full spectrum of epilepsies. Neurology. 2019;93(3):e227–e236. doi:10.1212/WNL.0000000000007778
80. Kinney HC, McDonald AG, Minter ME, Berry GT, Poduri A, Goldstein RD. Witnessed sleep-related seizure and sudden unexpected death in infancy: a case report. Forensic Sci Med Pathol. 2013;9(3):418–421. doi:10.1007/s12024-013-9448-0
81. Hefti MM, Kinney HC, Cryan JB, et al. Sudden unexpected death in early childhood: general observations in a series of 151 cases: part 1 of the investigations of the San Diego SUDC research project. Forensic Sci Med Pathol. 2016;12(1):4–13. doi:10.1007/s12024-015-9724-2
82. Hefti MM, Cryan JB, Haas EA, et al. Hippocampal malformation associated with sudden death in early childhood: a neuropathologic study: part 2 of the investigations of The San Diego SUDC research project. Forensic Sci Med Pathol. 2016;12(1):14–25. doi:10.1007/s12024-015-9731-3
83. Kon FC, Vázquez RZ, Lang A, Cohen MC. Hippocampal abnormalities and seizures: a 16-year single center review of sudden unexpected death in childhood, sudden unexpected death in epilepsy and SIDS. Forensic Sci Med Pathol. 2020;16(3):423–434. doi:10.1007/s12024-020-00268-7
84. Brownstein CA, Goldstein RD, Thompson CH, et al. SCN1A variants associated with sudden infant death syndrome. Epilepsia. 2018;59(4):e56–e62. doi:10.1111/epi.14055
85. Frasier CR, Zhang H, Offord J, et al. Channelopathy as a SUDEP biomarker in dravet syndrome patient-derived cardiac myocytes. Stem Cell Reports. 2018;11(3):626–634. doi:10.1016/j.stemcr.2018.07.012
86. Halvorsen M, Petrovski S, Shellhaas R, et al. Mosaic mutations in early-onset genetic diseases. Genet Med. 2016;18(7):746–749. doi:10.1038/gim.2015.155
87. Catterall WA. Forty years of sodium channels: structure, function, pharmacology, and epilepsy. Neurochem Res. 2017;42(9):2495–2504. doi:10.1007/s11064-017-2314-9
88. Gando I, Williams N, Fishman GI, Sampson BA, Tang Y, Coetzee WA. Functional characterization of SCN10A variants in several cases of sudden unexplained death. Forensic Sci Int. 2019;301:289–298. doi:10.1016/j.forsciint.2019.05.042
89. Holm H, Gudbjartsson DF, Arnar DO, et al. Several common variants modulate heart rate, PR interval and QRS duration. Nat Genet. 2010;42(2):117–122. doi:10.1038/ng.511
90. Zhang L, Zhou F, Huang L, et al. Association of common and rare variants of SCN10A gene with sudden unexplained nocturnal death syndrome in Chinese Han population. Int J Legal Med. 2017;131(1):53–60. doi:10.1007/s00414-016-1397-1
91. Swenson BR, Louie T, Lin HJ, et al. GWAS of QRS duration identifies new loci specific to Hispanic/Latino populations. PLoS One. 2019;14(6):e0217796. doi:10.1371/journal.pone.0217796
92. Männikkö R, Wong L, Tester DJ, et al. Dysfunction of NaV1.4, a skeletal muscle voltage-gated sodium channel, in sudden infant death syndrome: a case-control study. Lancet. 2018;391(10129):1483–1492. doi:10.1016/S0140-6736(18)30021-7
93. Lion-Francois L, Mignot C, Vicart S, et al. Severe neonatal episodic laryngospasm due to de novo SCN4A mutations: a new treatable disorder. Neurology. 2010;75(7):641–645. doi:10.1212/WNL.0b013e3181ed9e96
94. Türkdoğan D, Matthews E, Usluer S, et al. Possible role of SCN4A skeletal muscle mutation in apnea during seizure. Epilepsia Open. 2019;4(3):498–503. doi:10.1002/epi4.12347
95. Nakase K, Kollmar R, Lazar J, et al. Laryngospasm, central and obstructive apnea during seizures: defining pathophysiology for sudden death in a rat model. Epilepsy Res. 2016;128:126–139. doi:10.1016/j.eplepsyres.2016.08.004
96. Hawkins NA, Calhoun JD, Huffman AM, Kearney JA. Gene expression profiling in a mouse model of Dravet syndrome. Exp Neurol. 2019;311:247–256. doi:10.1016/j.expneurol.2018.10.010
97. Goldman AM, Glasscock E, Yoo J, Chen TT, Klassen TL, Noebels JL. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl Med. 2009;1(2):2ra6. doi:10.1126/scitranslmed.3000289
98. Johnson JN, Hofman N, Haglund CM, Cascino GD, Wilde AA, Ackerman MJ. Identification of a possible pathogenic link between congenital long QT syndrome and epilepsy. Neurology. 2009;72(3):224–231. doi:10.1212/01.wnl.0000335760.02995.ca
99. Tu E, Bagnall RD, Duflou J, Semsarian C. Post-mortem review and genetic analysis of sudden unexpected death in epilepsy (SUDEP) cases. Brain Pathol. 2011;21(2):201–208. doi:10.1111/j.1750-3639.2010.00438.x
100. Toczek MT, Carson RE, Lang L, et al. PET imaging of 5-HT1A receptor binding in patients with temporal lobe epilepsy. Neurology. 2003;60(5):749–756. doi:10.1212/01.WNL.0000049930.93113.20
101. Savic I, Lindström P, Gulyás B, Halldin C, Andrée B, Farde L. Limbic reductions of 5-HT1A receptor binding in human temporal lobe epilepsy. Neurology. 2004;62(8):1343–1351. doi:10.1212/01.WNL.0000123696.98166.AF
102. Merlet I, Ostrowsky K, Costes N, et al. 5-HT1A receptor binding and intracerebral activity in temporal lobe epilepsy: an [18F]MPPF-PET study. Brain. 2004;127(4):900–913. doi:10.1093/brain/awh109
103. Statnick MA, Maring-Smith ML, Clough RW, et al. Effect of 5,7-dihydroxytryptamine on audiogenic seizures in genetically epilepsy-prone rats. Life Sci. 1996;59(21):1763–1771. doi:10.1016/0024-3205(96)00519-X
104. Richerson GB, Buchanan GF. The serotonin axis: shared mechanisms in seizures, depression, and SUDEP. Epilepsia. 2011;52(1):28–38. doi:10.1111/j.1528-1167.2010.02908.x
105. Kim Y, Bravo E, Thirnbeck CK, et al. Severe peri-ictal respiratory dysfunction is common in Dravet syndrome. J Clin Invest. 2018;128(3):1141–1153. doi:10.1172/JCI94999
106. Opdal SH. Cytokines, Infection, and Immunity. In: Duncan JR, Byard RW, editors. SIDS Sudden Infant and Early Childhood Death: The Past, the Present and the Future. The University of Adelaide: University of Adelaide Press; 2018:689–710.
107. Platt MW, Blair PS, Fleming PJ, et al. A clinical comparison of SIDS and explained sudden infant deaths: how healthy and how normal? CESDI SUDI Research Group. Confidential inquiry into stillbirths and deaths in infancy study. Arch Dis Child. 2000;82(2):98–106. doi:10.1136/adc.82.2.98
108. Fard D, Läer K, Rothämel T, et al. Candidate gene variants of the immune system and sudden infant death syndrome. Int J Legal Med. 2016;130(4):1025–1033. doi:10.1007/s00414-016-1347-y
109. Opdal SH, Rognum TO. The IL6-174G/C polymorphism and sudden infant death syndrome. Hum Immunol. 2007;68(6):541–543. doi:10.1016/j.humimm.2007.02.008
110. Moscovis SM, Gordon AE, Al Madani OM, et al. Interleukin-10 and sudden infant death syndrome. FEMS Immunol Med Microbiol. 2004;42(1):130–138. doi:10.1016/j.femsim.2004.06.020
111. Courts C, Madea B. No association of IL-10 promoter SNP −592 and −1082 and SIDS. Forensic Sci Int. 2011;204(1–3):179–181. doi:10.1016/j.forsciint.2010.06.001
112. Moscovis SM, Gordon AE, Al Madani OM, et al. Genetic and environmental factors affecting TNF-α responses in relation to sudden infant death syndrome. Front Immunol. 2015;6:374. doi:10.3389/fimmu.2015.00374
113. Perskvist N, Skoglund K, Edston E, Bäckström G, Lodestad I, Palm U. TNF-alpha and IL-10 gene polymorphisms versus cardioimmunological responses in sudden infant death. Fetal Pediatr Pathol. 2008;27(3):149–165. doi:10.1080/15513810802077651
114. Ferrante L, Opdal SH, Vege A, Rognum TO. IL-1 gene cluster polymorphisms and sudden infant death syndrome. Hum Immunol. 2010;71(4):402–406. doi:10.1016/j.humimm.2010.01.011
115. Kawaguchi Y, Tochimoto A, Hara M, et al. Contribution of single nucleotide polymorphisms of the IL1A gene to the cleavage of precursor IL-1alpha and its transcription activity. Immunogenetics. 2007;59(6):441–448. doi:10.1007/s00251-007-0213-y
116. Highet AR, Berry AM, Goldwater PN. Distribution of interleukin-1 receptor antagonist genotypes in sudden unexpected death in infancy (SUDI); unexplained SUDI have a higher frequency of allele 2. Ann Med. 2010;42(1):64–69. doi:10.3109/07853890903325360
117. Hurme M, Santtila S. IL-1 receptor antagonist (IL-1Ra) plasma levels are co-ordinately regulated by both IL-1Ra and IL-1beta genes. Eur J Immunol. 1998;28(8):2598–2602. doi:10.1002/(SICI)1521-4141(199808)28:08<2598::AID-IMMU2598>3.0.CO;2-K
118. Ferrante L, Opdal SH, Vege A, Rognum TO. TNF-alpha promoter polymorphisms in sudden infant death. Hum Immunol. 2008;69(6):368–373. doi:10.1016/j.humimm.2008.04.006
119. Hafke A, Schürmann P, Rothämel T, Dörk T, Klintschar M. Evidence for an association of interferon gene variants with sudden infant death syndrome. Int J Legal Med. 2019;133(3):863–869. doi:10.1007/s00414-018-1974-6
120. Puffenberger EG, Hu-Lince D, Parod JM, et al. Mapping of sudden infant death with dysgenesis of the testes syndrome (SIDDT) by a SNP genome scan and identification of TSPYL loss of function. Proc Natl Acad Sci USA. 2004;101(32):11689–11694. doi:10.1073/pnas.0401194101
121. Slater B, Glinton K, Dai H, et al. Sudden infant death with dysgenesis of the testes syndrome in a non-Amish infant: a case report. Am J Med Genet. 2020;182(11):2751–2754. doi:10.1002/ajmg.a.61842
122. Conomy JP, Levinsohn M, Fanaroff A. Familial infantile myasthenia gravis: a cause of sudden death in young children. J Pediatr. 1975;87(3):428–430. doi:10.1016/S0022-3476(75)80651-2
123. Robertson WC, Chun RW, Kornguth SE. Familial infantile myasthenia. Arch Neurol. 1980;37(2):117–119. doi:10.1001/archneur.1980.00500510075018
124. Finsterer J. Congenital myasthenic syndromes. Orphanet J Rare Dis. 2019;14(1):57. doi:10.1186/s13023-019-1025-5
125. Byring RF, Pihko H, Tsujino A, et al. Congenital myasthenic syndrome associated with episodic apnea and sudden infant death. Neuromuscul Disord. 2002;12(6):548–553. doi:10.1016/S0960-8966(01)00336-4
126. Bachetti T, Ceccherini I. Causative and common PHOX2B variants define a broad phenotypic spectrum. Clin Genet. 2020;97(1):103–113. doi:10.1111/cge.13633
127. Amiel J, Salomon R, Attié T, et al. Mutations of the RET-GDNF signaling pathway in Ondine’s curse. Am J Hum Genet. 1998;62(3):715–717. doi:10.1086/301759
128. de Pontual L, Népote V, Attié-Bitach T, et al. Noradrenergic neuronal development is impaired by mutation of the proneural HASH-1 gene in congenital central hypoventilation syndrome (Ondine’s curse). Hum Mol Genet. 2003;12(23):3173–3180. doi:10.1093/hmg/ddg339
129. Bolk S, Angrist M, Xie J, et al. Endothelin-3 frameshift mutation in congenital central hypoventilation syndrome. Nat Genet. 1996;13(4):395–396. doi:10.1038/ng0896-395
130. Belligni EF, Palmer RW, Hennekam RC. MECP2 duplication in a patient with congenital central hypoventilation. Am J Med Genet. 2010;152A(6):1591–1593. doi:10.1002/ajmg.a.33311
131. Guilleminault C, McQuitty J, Ariagno RL, Challamel MJ, Korobkin R, McClead RE. Congenital central alveolar hypoventilation syndrome in six infants. Pediatrics. 1982;70(5):684–694.
132. Pattyn A, Morin X, Cremer H, Goridis C, Brunet J-F. The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature. 1999;399(6734):366–370. doi:10.1038/20700
133. Stornetta RL, Moreira TS, Takakura AC, et al. Expression of Phox2b by brainstem neurons involved in chemosensory integration in the adult rat. J Neurosci. 2006;26(40):10305–10314. doi:10.1523/JNEUROSCI.2917-06.2006
134. Berry-Kravis EM, Zhou L, Rand CM, Weese-Mayer DE. Congenital central hypoventilation syndrome: PHOX2B mutations and phenotype. Am J Respir Crit Care Med. 2006;174(10):1139–1144. doi:10.1164/rccm.200602-305OC
135. Liebrechts-Akkerman G, Liu F, Lao O, et al. PHOX2B polyalanine repeat length is associated with sudden infant death syndrome and unclassified sudden infant death in the Dutch population. Int J Legal Med. 2014;128(4):621–629. doi:10.1007/s00414-013-0962-0
136. Ventura F, Barranco R, Bachetti T, et al. Medico-legal investigation in an explicable case of congenital central hypoventilation syndrome due to a rare variant of the PHOX2B gene. J Forensic Leg Med. 2018;58:1–5. doi:10.1016/j.jflm.2018.04.009
137. Di Zanni E, Adamo A, Belligni E, et al. Common PHOX2B poly-alanine contractions impair RET gene transcription, predisposing to Hirschsprung disease. Biochim Biophys Acta Mol Basis Dis. 2017;1863(7):1770–1777. doi:10.1016/j.bbadis.2017.04.017
138. Fernández RM, Mathieu Y, Luzón-Toro B, et al. Contributions of PHOX2B in the pathogenesis of Hirschsprung disease. PLoS One. 2013;8(1):e54043. doi:10.1371/journal.pone.0054043
139. Ruiz-Ferrer M, Fernández RM, Antiñolo G, López-Alonso M, Eng C, Borrego S. A complex additive model of inheritance for Hirschsprung disease is supported by both RET mutations and predisposing RET haplotypes. Genet Med. 2006;8(11):704–710. doi:10.1097/01.gim.0000245632.06064.f1
140. Wallace DC. Mitochondrial genetic medicine. Nat Genet. 2018;50(12):1642–1649. doi:10.1038/s41588-018-0264-z
141. Läer K, Vennemann M, Rothämel T, Klintschar M. Mitochondrial deoxyribonucleic acid may play a role in a subset of sudden infant death syndrome cases. Acta Paediatr. 2014;103(7):775–779. doi:10.1111/apa.12609
142. Opdal SH, Rognum TO, Torgersen H, Vege A. Mitochondrial DNA point mutations detected in four cases of sudden infant death syndrome. Acta Paediatr. 1999;88(9):957–960. doi:10.1111/j.1651-2227.1999.tb00189.x
143. Ogle RF, Christodoulou J, Fagan E, et al. Mitochondrial myopathy with tRNA(Leu(UUR)) mutation and complex I deficiency responsive to riboflavin. J Pediatr. 1997;130(1):138–145. doi:10.1016/S0022-3476(97)70323-8
144. Silvestri G, Santorelli FM, Shanske S, et al. A new mtDNA mutation in the tRNA(Leu(UUR)) gene associated with maternally inherited cardiomyopathy. Hum Mutat. 1994;3(1):37–43. doi:10.1002/humu.1380030107
145. Tester DJ, Wong LCH, Chanana P, et al. Exome-wide rare variant analyses in sudden infant death syndrome. J Pediatr. 2018;203:423–428.e11. doi:10.1016/j.jpeds.2018.08.011
146. Toruner GA, Kurvathi R, Sugalski R, et al. Copy number variations in three children with sudden infant death. Clin Genet. 2009;76(1):63–68. doi:10.1111/j.1399-0004.2009.01161.x
147. Manning M, Hudgins L; Professional Practice and Guidelines Committee. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet Med. 2010;12(11):742–745. doi:10.1097/GIM.0b013e3181f8baad
148. Lee JS, Hwang H, Kim SY, et al. Chromosomal microarray with clinical diagnostic utility in children with developmental delay or intellectual disability. Ann Lab Med. 2018;38(5):473–480. doi:10.3343/alm.2018.38.5.473
149. Allen NM, Conroy J, Shahwan A, et al. Chromosomal microarray in unexplained severe early onset epilepsy - A single centre cohort. Eur J Paediatr Neurol. 2015;19(4):390–394. doi:10.1016/j.ejpn.2015.03.010
150. Truty R, Paul J, Kennemer M, et al. Prevalence and properties of intragenic copy-number variation in Mendelian disease genes. Genet Med. 2019;21(1):114–123. doi:10.1038/s41436-018-0033-5
151. Middleton O, Baxter S, Demo E, et al. National association of medical examiners position paper: retaining postmortem samples for genetic testing. Acad Forensic Pathol. 2013;3(2):191–194. doi:10.23907/2013.024
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.