")
Back to Journals » International Journal of General Medicine » Volume 16
Genetic Basis of Congenital Central Hypothyroidism in Children: Expanding the Mutational Spectrum of POU1F1 and ATP6V0A4
Authors Fu C , Luo J, Su J, Zhang S, Yang Q, Zhang Y
Received 23 May 2023
Accepted for publication 1 August 2023
Published 8 August 2023 Volume 2023:16 Pages 3355—3362
DOI https://doi.org/10.2147/IJGM.S421382
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Chunyun Fu,1,* Jingsi Luo,2,* Jiasun Su,2 Shujie Zhang,2 Qi Yang,2 Yue Zhang2
1Medical Science Laboratory, Children’s Hospital, Maternal and Child Health Hospital of Guangxi Zhuang Autonomous Region, Nanning, 530003, People’s Republic of China; 2Department of Genetic Metabolism, Children’s Hospital, Maternal and Child Health Hospital of Guangxi Zhuang Autonomous Region, Nanning, 530003, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Chunyun Fu, Email [email protected]
Objective: Congenital central hypothyroidism (CCH) is a rare disorder poorly described in childhood and adolescence. The current knowledge on the genetic bases of CCH is scarce. The purpose of this study was to analyze the clinical characteristics and molecular genetic basis of CCH in children.
Methods: We conducted a thorough evaluation of the clinical features in children diagnosed with CCH. Genomic DNA was extracted from peripheral blood of both children and their parents, and chromosomal microarray analysis and whole-exome sequencing were performed. Candidates for single nucleotide variants were validated using Sanger sequencing and were classified according to the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) guidelines.
Results: Two cases with likely pathogenic variants were detected by whole-exome sequencing. Individual 1 carried a novel homozygous ATP6V0A4 c.1418C>T (p.Ser473Phe) variant and a novel heterozygous POU1F1 c.416G>A. (p.Arg139Gln) variant. Individual 2 had a novel homozygous POU1F1 c.212C>T (p.Ala71Val) variant. The chromosomal microarray detected the presence of a 24 Mb heterozygous deletion (LOH: loss of heterozygosity) in the p12.1p13.13 region of chromosome 2 in individual 3, and the copy number variant was unknown of clinical significance.
Conclusion: Our study employed chromosomal microarray and whole-exome sequencing to investigate central hypothyroidism in seven children, leading to the detection of genetic anomalies in three individuals. The identification of novel variants has contributed to the expanded genetic spectrum of POU1F1 and ATP6V0A4 associated with pediatric central hypothyroidism.
Keywords: congenital central hypothyroidism, genetic, chromosomal microarray, whole-exome sequencing, pathogenic variants
Introduction
Central congenital hypothyroidism (CCH) is a rare disorder that is characterized by a defect in thyroid hormone secretion in an otherwise normal thyroid gland due to insufficient stimulation by thyroid-stimulating hormone (TSH). Estimates of CCH incidence range from about 1:16,000 to 1:80,000 in the general population.1–5 CCH results from the abnormal function of the pituitary gland, the hypothalamus, or both. The disease may occur in isolation, or more frequently in combination with other pituitary hormone deficits.6,7 The diagnosis of central hypothyroidism is based on low circulating levels of free thyroxine (FT4) in the presence of low to normal TSH concentrations. As thyroid hormones are critical for the development of the central nervous system, Individuals with CCH may result in developmental delay due to untreated neonatal hypothyroidism.8 The underlying molecular basis of CCH is poorly understood, although the genetic studies have been reported in a few cases.7–12 At present, genetic defects in only four genes (TSHB, TRHR, IGSF1 and the TBL1X) have been identified in patients with isolated CCH.12 Here, we investigate the genetic mechanisms in seven patients with CCH by chromosomal microarray analysis (CMA) and whole-exome sequencing (WES).
Materials and Methods
Individuals
A total of seven individuals were enrolled who were diagnosed with CCH in our pediatric clinics. This study complies with the Declaration of Helsinki and was approved by the local Medical Ethics Committee of the Maternal and Child Health Hospital of Guangxi Zhuang Autonomous Region. Written informed consent was obtained from the parents of the Individuals.
CMA and WES Analysis and Validation
Peripheral venous blood samples were collected from the Individuals. Genomic DNA was extracted from peripheral blood leukocytes using QIAamp DNA Blood Mini Kit (Qiagen, Germany) according to the manufacturer’s protocol. Genomic profiling was performed using the illumina Human SNP cyto-12 array, which includes over 300 k single nucleotide polymorphisms (SNPs) in the human genome. Exome capture was carried out using Roche NimbleGen SeqCap EZ Exome Library SR platform according to the manufacturer’s protocols. Each captured library then was loaded on illumina Hiseq2000 platform for sequence analysis. Chromosomal microarray analysis (CMA)13 and whole-exome sequencing (WES)14 were performed to detect Copy number variations (CNVs) and Single Nucleotide Variants (SNVs), respectively, as described in detail previously. Sanger sequencing was used to validate mutations identified by WES. The American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) guidelines were used for variant classification.15
Results
Genetic Analysis
Individual 1 was detected with a homozygous ATP6V0A4 c.1418C>T variant, and parents were carriers of this variant (Figure 1A). In addition, Individual 1 was also found with a de novo heterozygous POU1F1 c.416G>A (p. Arg139Gln) variant (Figure 1B). Individual 2 was detected with a homozygous POU1F1 c.212C>T variant, resulting in the encoded amino acid changed from alanine to proline, and parents were carriers of this variant (Figure 2).
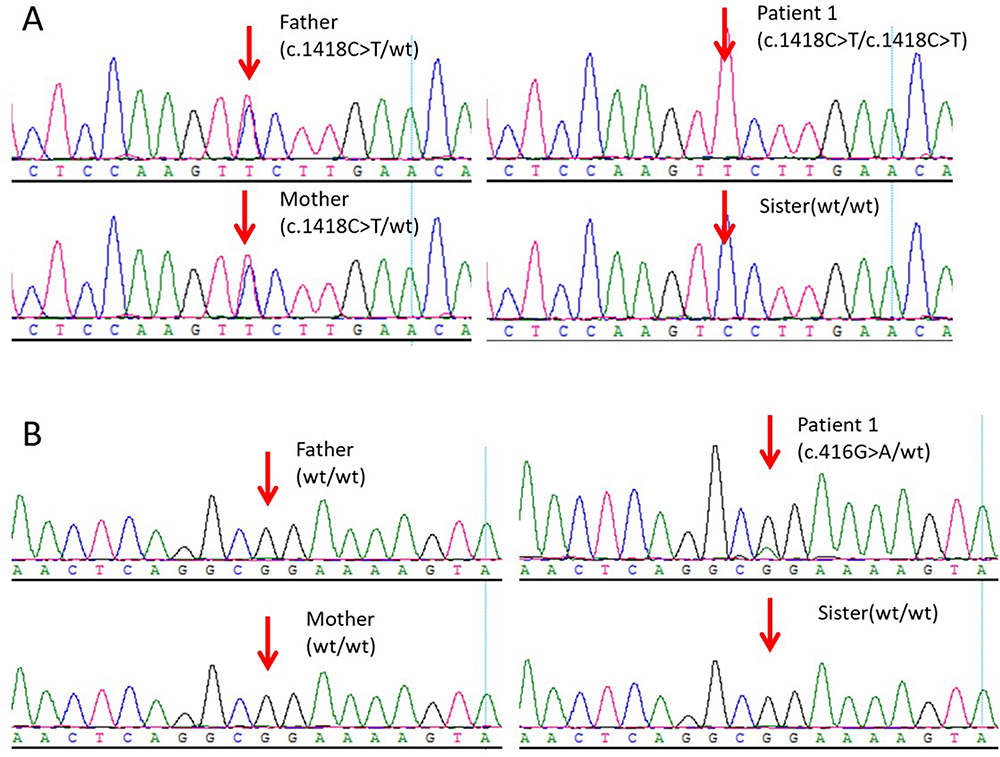 |
Figure 1 Sanger sequencing for validation of the variations detected by the next-generation sequencing platforms. (A) ATP6V0A4 c.1418C>T variant in family members of Individual 1. (B) POU1F1 c.416G>A (p. Arg139Gln) variant in family of Individual 1. |
 |
Figure 2 Sanger sequencing for validation of the variations detected by the next-generation sequencing platforms. POU1F1 c.212C>T variant in family members of Individual 2. |
Of these three variants, the two POU1F1 variants (c.416G>A and c.212C>T) are classified as likely pathogenic according to ACMG/AMP guidelines.15 The remaining ATP6V0A4 variant c.1418C>T was evaluated and classified as variant of unknown significance (VUS) (Table 1).
 |
Table 1 Evaluation and Classification of the Three Genetic Variants Detected by WES |
Individual 3 was detected with a 24 Mb heterozygous deletion (LOH: loss of heterozygosity) in the p12.1p13.13 region of chromosome 2 (Figure 3), and the LOH arr3p12.2q12.2 (81,135,775–100,841,396) x2 was unknown of clinical significance.
 |
Figure 3 CMA testing result of Individual 1. A 24 Mb heterozygous deletion (LOH: loss of heterozygosity) in the 2p12.1p13.13 region was detected. |
Clinical Presentations
The clinical features are summarized in Table 2. All Individuals were born at full-term in non-consanguineous families. Individuals 3 was diagnosed with CCH at newborn screening, and the rest of them were diagnosed during regular visits to a pediatric clinic. All seven Individuals showed normal size and location of thyroid gland. L-T4 replacement therapy was started immediately after the diagnosis of CCH and the dose was adjusted according to the serum TSH, FT4 and FT3 levels. After temporary withdrawal of L-T4 therapy at approximately 3 years of age, Individual 1, Individual 3 and Individual 6 were diagnosed with transient CCH, since measurements of TSH, FT4 and FT3 concentrations were normal, following a temporary withdrawal of L-T4 therapy for 5 weeks. The other four Individuals with a low FT4 level were diagnosed with permanent CCH.
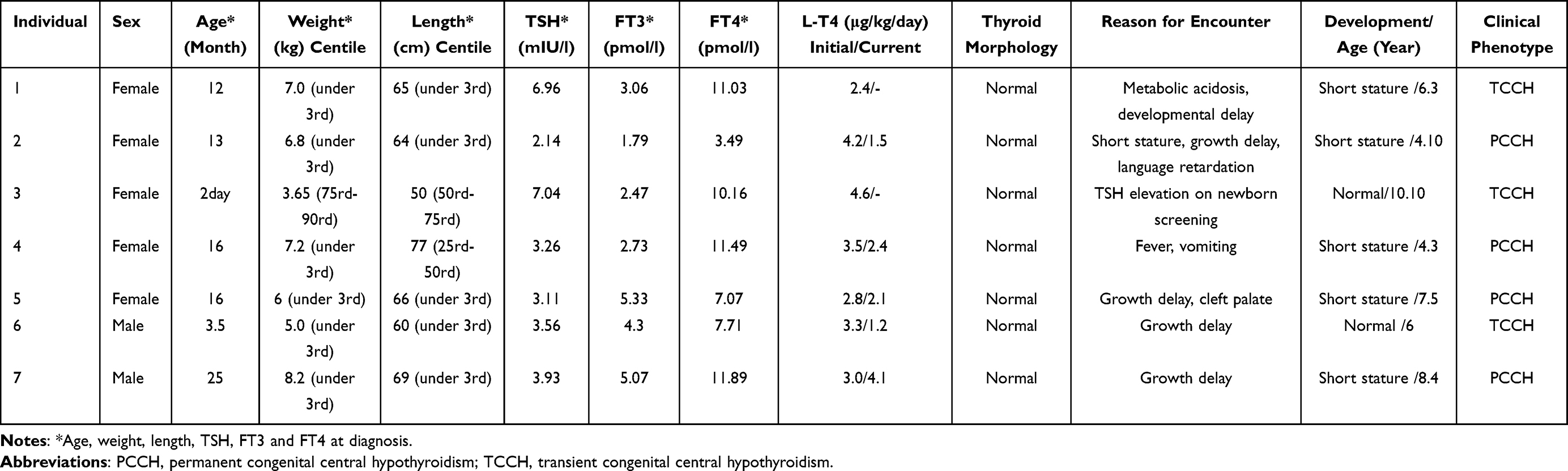 |
Table 2 Clinical Features, Laboratory results in Seven Individuals |
Discussion
A total of 454,240 newborns were screened for CH at the duration from October 2014 to May 2017 at the Newborn Screening Center of Guangxi, China, and seven Individuals (one by newborn screening and six in our pediatric clinic) were diagnosed with CCH, with an incidence of 1:64,891. This incidence is similar to that of USA (1:65,000),16 but lower than that of Netherlands (1:16,000) with a unique screening algorithm based on the combined measurement of TSH, T4 and thyroxine-binding globulin (TBG).4
CCH is associated with either subnormal or inappropriately normal TSH levels, and therefore evades diagnosis in the TSH-based newborn screening for congenital hypothyroidism (CH).17,18 Unfortunately, Individuals may present overt clinical hypothyroidism in later neonatal period. Among the 7 Individuals, only Individual 3 was positive for TSH level in newborn CH screening test. Individual 4, treated with fever and vomiting, was diagnosed with CCH by the thyroid function test. The remaining five CCH Individuals all presented with significant developmental delay with or without other symptoms. After diagnosis, all Individuals were treated with L-T4, the development of most Individuals was still behind that of children at the same age. However, only Individual 3 and Individual 6 developed normally because of timely diagnosis and treatment when they were very young.
The underlying molecular basis of CCH remains elusive, a minority of cases are associated with a known genetic mechanism, and reveals mutations in genes controlling the TSH biosynthetic pathway (TSHB, TRHR, IGSF1) in isolated TSH deficiency,10,19–21 or early (HESX1, LHX3, LHX4, SOX3, OTX2) or late (PROP1, POU1F1) pituitary transcription factors in combined hormone deficits.22,23
In this study, Individual 1 and Individual 2 were found to have variants in one or two CCH-associated genes, and the other five cases remain elusive by WES. This suggests that other unidentified genetic or epigenetic factors may contribute to the etiology of CCH. POU1F1 mutations have been linked to recessive or dominantly inherited complex pituitary hormone deficiency, as characterized by pituitary dysplasia, low hormones associated with pituitary secretion, low central thyroid function, short stature, and facial dysmorphism. ATP6V0A4 mutations, on the other hand, lead to autosomal recessive distal renal tubular acidosis, presenting with high chloride metabolic acidosis, hypokalemia, hypercalcemia, hypocapnia, and urinary enrichment, repeated urinary stone formation, renal calcium deposition and bone demineralization. The mutant phenotypes reported align with the clinical symptoms of Individual 1 and Individual 2 in our study.
The human POU1F1 gene (OMIM 601538), located on chromosome 3p11 and consists of six exons encoding a protein of 291 amino acids with three functional domains: an N-terminal transactivation domain, a POU-specific (POU-S) domain (75 amino acids) and a POU-homeodomain (POU-H) (60 amino acids) connected by a 15-amino-acid flexible linker. The POU-specific (POU-S) domain and the POU homeodomain (POU-H) are critical for the DNA-binding properties of POU1F1. To date, more than 46 POU1F1 mutations in Individuals with combined pituitary hormone deficiency (CPHD) have been described (HGMD; http://www.hgmd.cf.ac.uk), and majority occur within or directly affect the POU-domain DNA-binding module. Both autosomal recessive (AR) and autosomal dominant (AD) modes of inheritance have been described. The AR mode of inheritance appears to be more frequent. Milder phenotypes were found among Individuals with heterozygous mutations than those with homozygous and compound heterozygous mutations. In our study, Individual 1 with POU1F1 heterozygous variant (p.Arg139Gln) was TCCH, while Individual 2 with homozygous variant (p.Ala71Val) turned out to be PCCH.
In summary, the incidence of CCH among newborns in Guangxi Zhuang Autonomous Region, China, was found to be 1:64,891. Our study has shed light on the clinical characteristics and genetic basis of CCH in children, identifying three novel variants that expand the mutational spectrum of POU1F1 and ATP6V0A4. Future function studies on these novel variants will be essential to further elucidate their roles. Additionally, the involvement of other unidentified genetic or epigenetic factors in the etiology of CCH warrants further investigation.
Data Sharing Statement
We will coordinate data sharing and make available both the raw data and analyzed data upon request.
Author Contributions
All authors made significant contributions to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the Guangxi Natural Science Foundation Program (2016GXNSFBA380192).
Disclosure
The authors report no potential conflicts of interest in this work.
References
1. Tajima T, Nakamura A, Morikawa S, et al. Neonatal screening and a new cause of congenital central hypothyroidism. Ann Pediatr Endocrinol Metab. 2014;19(3):117–121. doi:10.6065/apem.2014.19.3.117
2. Adachi M, Soneda A, Asakura Y, et al. Mass screening of newborns for congenital hypothyroidism of central origin by free thyroxine measurement of blood samples on filter paper. Eur J Endocrinol. 2012;166(5):829–838. doi:10.1530/EJE-11-0653
3. Fujiwara F, Fujikura K, Okuhara K, et al. Central congenital hypothyroidism detected by neonatal screening in Sapporo, Japan (2000-2004): it’s Prevalence and clinical Characteristics. Clin Pediatr Endocrinol. 2008;17(3):65–69. doi:10.1297/cpe.17.65
4. Schoenmakers N, Alatzoglou KS, Chatterjee VK, et al. Recent advances in central congenital hypothyroidism. J Endocrinol. 2015;227(3):R51–71. doi:10.1530/JOE-15-0341
5. Lauffer P, Zwaveling-Soonawala N, Naafs JC, et al. Diagnosis and management of central congenital hypothyroidism. Front Endocrinol. 2021;12(686317). doi:10.3389/fendo.2021.686317
6. Boelen A, Van Trotsenburg ASP, Fliers E. Congenital isolated central hypothyroidism: novel mutations and their functional implications. Handb Clin Neurol. 2021;180:161–169.
7. Lauffer P, Bikker H, Boelen A, et al. Mild isolated congenital central hypothyroidism due to a novel homozygous variant in TSHB: a case report. Thyroid. 2022;32(4):472–474. doi:10.1089/thy.2021.0651
8. Turkkahraman D, Karatas Torun N, Randa NC. A case of congenital central hypothyroidism caused by a novel variant (Gln1255Ter) in IGSF1 gene. J Clin Res Pediatr Endocrinol. 2021;13(3):353–357. doi:10.4274/jcrpe.galenos.2020.2020.0149
9. Tajima T, Oguma M. A novel nonsense variant (p.Arg1293Ter) of the immunoglobulin superfamily 1 (IGSF1) associated with congenital hypogonadotropic hypogonadism and central hypothyroidism. Clinical pediatric endocrinology: case reports and clinical investigations. J Clin Res Pediatr Endocrinol. 2022;31(2):98–100.
10. Fourneaux R, Castets S, Godefroy A, et al. Congenital central hypothyroidism caused by a novel IGSF1 variant identified in a French family. Horm Res Paediatr. 2022;95(3):296–303. doi:10.1159/000524233
11. Ebrhim RS, Bruellman RJ, Watanabe Y, et al. Central congenital hypothyroidism caused by a novel mutation, C47W, in the cysteine knot region of TSHbeta. Horm Res Paediatr. 2019;92(6):390–394. doi:10.1159/000504981
12. Garcia M, Gonzalez De Buitrago J, Jimenez-Roses M, et al. Central hypothyroidism due to a TRHR mutation causing impaired ligand affinity and transactivation of Gq. J Clin Endocrinol Metab. 2017;102(7):2433–2442. doi:10.1210/jc.2016-3977
13. Chen R, Li C, Xie B, et al. Clinical and molecular evaluations of siblings with “pure” 11q23.3-qter trisomy or reciprocal monosomy due to a familial translocation t (10;11) (q26; q23.3). Mol Cytogenet. 2014;7(1):101. doi:10.1186/s13039-014-0101-8
14. Fu C, Luo S, Zhang S, et al. Next-generation sequencing analysis of DUOX2 in 192 Chinese subclinical congenital hypothyroidism (SCH) and CH patients. Clin Chim Acta. 2016;458:30–4.
15. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
16. Lafranchi SH. Newborn screening strategies for congenital hypothyroidism: an update. J Inherit Metab Dis. 2010;33(Suppl 2):S225–33. doi:10.1007/s10545-010-9062-1
17. Braslavsky D, Mendez MV, Prieto L, et al. Pilot neonatal screening program for central congenital hypothyroidism: evidence of significant detection. Horm Res Paediatr. 2017;88(3–4):274–280. doi:10.1159/000480293
18. Beck-Peccoz P, Rodari G, Giavoli C, et al. Central hypothyroidism - a neglected thyroid disorder. Nat Rev Endocrinol. 2017;13(10):588–598. doi:10.1038/nrendo.2017.47
19. Elizabeth MSM, Hokken-Koelega A, Visser JA, et al. Case report: a detailed phenotypic description of patients and relatives with combined central hypothyroidism and growth hormone deficiency carrying IGSF1 mutations. Genes. 2022;13(4):623. doi:10.3390/genes13040623
20. Kaplan AI, Luxford C, Clifton-Bligh RJ. Novel TSHB variant (c.217A>C) causing severe central hypothyroidism and pituitary hyperplasia. Endocrinol Diabetes Metab Case Rep. 2022;2022. doi:10.1530/EDM-22-0230
21. Borges MF, Domene HM, Scaglia PA, et al. A recurrent mutation in tshb gene underlying central congenital hypothyroidism undetectable in neonatal screening. Rev Paul Pediatr. 2019;37(4):520–524. doi:10.1590/1984-0462/;2019;37;4;00017
22. Wassner AJ, Cohen LE, Hechter E, et al. Isolated central hypothyroidism in young siblings as a manifestation of PROP1 deficiency: clinical impact of whole exome sequencing. Horm Res Paediatr. 2013;79(6):379–386. doi:10.1159/000350013
23. Chen WY, Niu DM, Chen LZ, et al. Congenital hypopituitarism due to novel compound heterozygous POU1F1 gene mutation: a case report and review of the literature. Mol Genet Metab Rep. 2021;29(100819):100819. doi:10.1016/j.ymgmr.2021.100819
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.