")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 7
Genetic and epigenetic catalysts in early-life programming of adult cardiometabolic disorders
Authors Estampador A, Franks P
Received 5 August 2014
Accepted for publication 22 September 2014
Published 1 December 2014 Volume 2014:7 Pages 575—586
DOI https://doi.org/10.2147/DMSO.S51433
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ming-Hui Zou
Video abstract presented by Paul W Franks
Views: 1248
Angela C Estampador,1,2 Paul W Franks1,3,4
1Department of Clinical Sciences, Genetic and Molecular Epidemiology Unit, Lund University, Skåne University Hospital Malmö, Malmö, Sweden; 2Department of Endocrinology, Rigshospitalet, Copenhagen University Hospital, Copenhagen, Denmark; 3Department of Public Health and Clinical Medicine, Umeå University, Umeå, Sweden; 4Department of Nutrition, Harvard School of Public Health, Boston, MA, USA
Abstract: Evidence has emerged across the past few decades that the lifetime risk of developing morbidities like type 2 diabetes, obesity, and cardiovascular disease may be influenced by exposures that occur in utero and in childhood. Developmental abnormalities are known to occur at various stages in fetal growth. Epidemiological and mechanistic studies have sought to delineate developmental processes and plausible risk factors influencing pregnancy outcomes and later health. Whether these observations reflect causal processes or are confounded by genetic and social factors remains unclear, although animal (and some human) studies suggest that epigenetic programming events may be involved. Regardless of the causal basis to observations of early-life risk factors and later disease risk, the fact that such associations exist and that they are of a fairly large magnitude justifies further research around this topic. Furthermore, additional information is needed to substantiate public health guidelines on lifestyle behaviors during pregnancy to improve infant health outcomes. Indeed, lifestyle intervention clinical trials in pregnancy are now coming online, where materials and data are being collected that should facilitate understanding of the causal nature of intrauterine exposures related with gestational weight gain, such as elevated maternal blood glucose concentrations. In this review, we provide an overview of these concepts.
Keywords: early-life, epigenetic, programming, pregnancy, cardiometabolic, obesity, cardiovascular disease, type 2 diabetes
Background
The global obesity epidemic has occurred in the wake of major recent social, demographic, behavioral, and economic adaptations; of particular relevance are the widespread adoption of sedentary behaviors, reduced physical activity, and ready access to energy-dense, micronutrient-deficient (eg, low iodine, iron, zinc, and vitamin A content) foods.
Adipocyte biogenesis and storage within adipocytes of energy-dense fatty acids (predominantly triglycerides) are natural processes that have enabled humans and other species to survive prolonged periods of food deprivation throughout evolution. Indeed, starvation1,2 and rare diseases associated with very low levels of adipose tissue (eg, partial lipodystrophies, anorexia nervosa, and cachexia) are life-threatening and can render affected women infertile owing to insufficient levels of hormones such as leptin.3,4 Hence, fat accumulation and retention are natural processes that have strong biologic drivers, many encoded by genes. However, energy-dense, nutrient-deficient foods are often easily accessible in most modern industrialized societies. Thus, an unhealthy lifestyle and environment that promote chronic positive energy balance set against a backdrop of genetic susceptibility to protect against leanness likely underlie the high rates of obesity and related cardiometabolic disorders seen in most developed and developing societies today.
Importantly, there is considerable variability in susceptibility to obesity, given exposure to obesogenic environmental factors, with studies in twins5 and family pedigrees6 clearly showing that heritable factors (including genes) contribute to these differences (see Bar-Or et al7 for review of evidence in childhood). Studies designed to identify the specific loci responsible for modifying the effects of environmental exposures on obesity predisposition have been underway for more than 2 decades, yet very little reliable evidence has emerged.8 Nevertheless, there is an emerging body of evidence documenting the molecular changes that underlie these interactions, including those that focus on differences in gene expression and deoxyribonucleic acid (DNA) methylation (epigenetics).
Whilst many of these studies have focused on adult cohorts, risk trajectories for dysmetabolic outcomes, including obesity, type 2 diabetes (T2D), and cardiovascular disease (CVD), are thought to be shaped early in life. The accrual of data in recent years exploring risk factors arising in pregnancy and childhood provides compelling evidence that the risk trajectory for metabolic disease originates in childhood or perhaps even earlier in utero (see Table 1 for a summary of key studies cited in this review). We hypothesize that dysmetabolic programming events occurring within critical windows in development predispose the offspring to CVD and T2D as adults, and that these processes may be mediated by epigenetic processes that give rise to transgenerational inheritance.
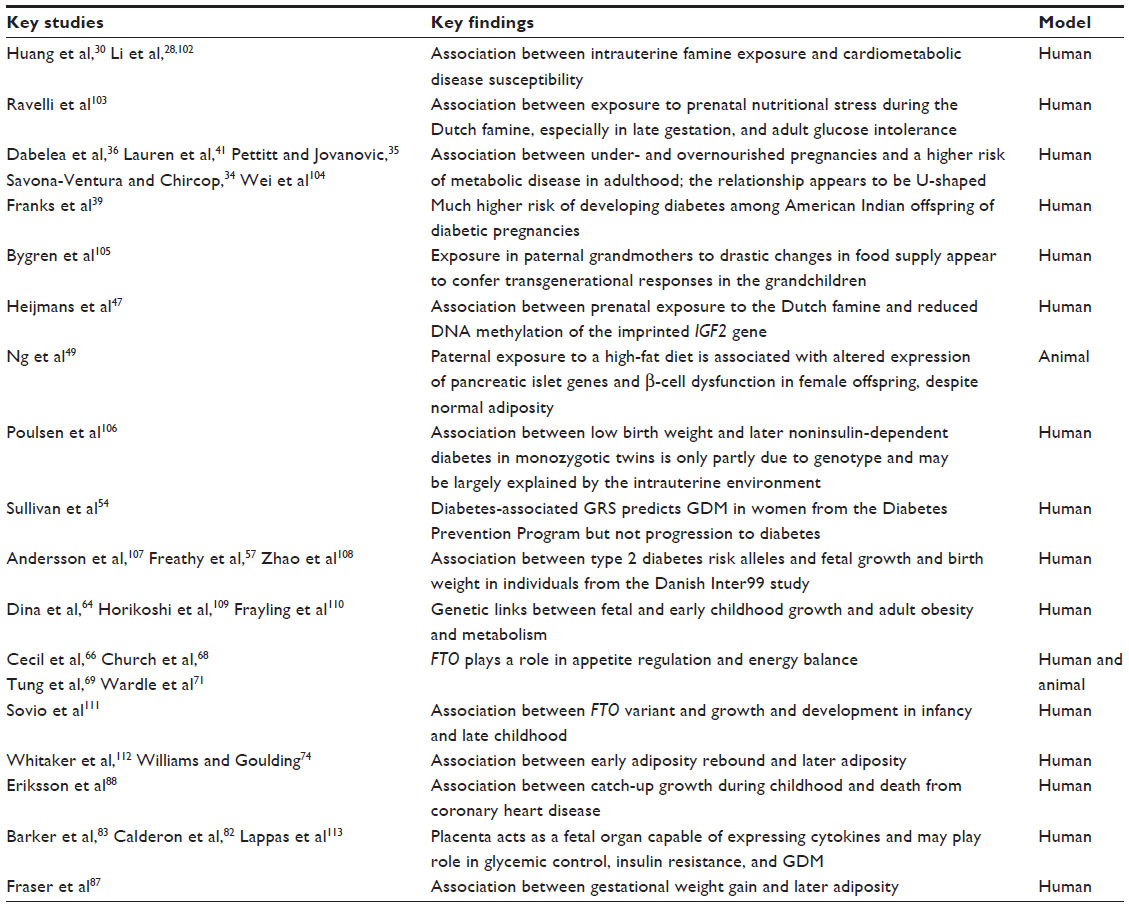 | Table 1 Summary of key studies and their major findings |
The childhood obesity epidemic
Childhood malnutrition has taken on a startling new identity during the past decade, with caloric excess now more common than undernutrition.9–12 The International Association for the Study of Obesity and International Obesity Task Force estimate that at least 200 million school-age children are overweight or obese worldwide.13 Obesity has a long list of comorbidities, including CVD, T2D,14 hypertension, and sleep apnea, as well as emotional/psychological consequences such as social stigmatization, discrimination, and low self-esteem.15,16 Indeed, the emerging epidemic of pediatric T2D is likely to be a direct result of the high prevalence of childhood obesity.17 Together, obesity and its sequelae impose an immense burden at a personal and societal level; thus, childhood obesity represents one of the major threats to global health.
Obesity in childhood, particularly in prepubescent children, is highly predictive of obesity in adulthood, especially when one (or both) parents is obese.18 Childhood body mass index (BMI), a strong correlate of adiposity in children, predicts premature death from endogenous causes, as do a number of other cardiometabolic risk factors (eg, glucose tolerance and hypertension).19–23 In a study of American Indian children, extreme BMI (the highest quartile compared with the lowest quartile) doubled the hazards of premature death.19 The relationship between childhood obesity and mortality may be mediated, in part at least, by dysregulated glucose and by hypertension, with abdominal obesity as one of the earliest manifestations of metabolic dysregulation. Notably, abdominal obesity occurring early in childhood (age <10 years) is a very strong risk factor for T2D later in life relative to other clinical risk factors, and appears to precede the development of glucose intolerance (an important precursor to diabetes) and dyslipidemia (a driver of insulin resistance);24 indeed, elevated BMI, glucose intolerance, and hypertension, when present in childhood, all raise the hazards of premature death.19
Given that childhood obesity is a major determinant of cardiometabolic risk later in life, understanding the modifiable risk factors for childhood obesity is well justified from a public health perspective. Defining early-life risk factors is a necessary step for designing interventions that tackle adult metabolic disease at its roots.
Metabolic and fetal programming
Pregnancy represents a critical period of development for fetal organs involved in maintaining energy homeostasis, not least for the pancreas and adipocytes.25 The fetal insulin hypothesis describes a scenario where poor fetal nutrition induces adaptations that are advantageous to the short-term survival of the fetus but have lasting detrimental effects on the offspring’s metabolic function that, in later life, cause metabolic disease.26 Both under- and overnutrition appear to convey similar effects on disease risk, although the proposed mechanisms differ. In undernourished pregnancies, the offspring’s metabolism is hypothesized to be optimized for extrauterine environments that mirror the intrauterine experience; accordingly, the obesogenic environments that offspring of undernourished pregnancies experience often appear to trigger cardiometabolic disease in these individuals.
In one of the earliest studies documenting the link between impaired fetal growth and CVD, investigators sought to explain observable differences in CVD risk seen across social gradients in England.27 Data recorded in 5,654 boys born in 1911 in the county of Hertfordshire showed a 3-fold difference in death rates from ischemic heart disease in adulthood between those with the lowest and highest birth weights. Data from epidemiologic studies of cohorts born during or soon after more recent manmade famines in the People’s Republic of China28–30 and Europe31 have provided some of the most striking examples of intrauterine malnutrition and cardiometabolic disease susceptibility later in life. Those studies indicate that the intrauterine growth of these children is restricted, as indicated by low birth weights for gestational age, smaller placentas, and shorter stature than are seen in children born outside the periods of famine; correspondingly, susceptibility to cardiometabolic diseases is generally higher in the former than in the latter. One of the most famous examples of such is the Dutch winter famine, which occurred toward the end of World War II (1944 and 1945) as a consequence of an embargo imposed by German forces on food supplies to the western parts of the Netherlands. Although the embargo was lifted in November of 1944, extreme winter conditions made it impossible for food supplies to reach urban areas to the west of the country.31 With food supplies dangerously low, rationing was enforced such that daily caloric intake was typically around 1,000 calories/day, dropping to 400 calories/day at the height of the famine. Thus, the many children born at or around this time were severely malnourished during critical periods of growth and development. The tragedy of the Dutch famine has since been used by epidemiologists to shed light on the role of early-life malnutrition on metabolic programming. Those studies have shown that individuals exposed to prenatal undernutrition, especially in late gestation, developed glucose intolerance as adults.32
Whilst undernourished pregnancies have been the model for much of the seminal work on the developmental origins of adult disease, a more widespread problem in contemporary societies is that of gestational obesity and fetal overnourishment. Offspring of both under- and overnourished pregnancies are at higher risk of metabolic disease in adulthood compared with moderately nourished offspring; thus, the relationship between birth weight and these diseases is U-shaped.33–37
Although maternal obesity in pregnancy is strongly associated with an adverse metabolic profile in the progeny, obesity per se may not cause deleterious programming events in the offspring; rather, factors that coalesce with maternal obesity, such as hyperglycemia, dyslipidemia, hypertension, or inflammation, are likely to be the causal factors.38 High intrauterine glucose concentrations, for example, drive the pancreatic β-cells to secrete excessive quantities of insulin. This process stresses the developing pancreas and, because insulin is a growth hormone, stimulates growth, as reflected in the heavier birth weights of the offspring.
Extensive epidemiological data demonstrate that the offspring of diabetic pregnancies are at much higher risk of developing diabetes themselves later in life; in a study of 911 nondiabetic mothers and 1,436 of their children, we showed that the relationship between a mother’s third trimester glucose tolerance (for every standard deviation [SD] higher glucose [~1.3 mmol/L]) equates to a 1.6-fold (95% confidence interval 1.3–2.0, P<0.0001) higher risk of diabetes in her offspring. This relationship was approximately dose dependent and was reflected by a 56 g higher offspring birth weight per SD unit maternal glucose (P=0.0002).39 In offspring of mothers with manifest diabetes before pregnancy, the risk of diabetes was higher still, with roughly half of the children born to these women having developed T2D themselves by the age of 25 years.39
Elsewhere, in a subcohort of pregnant women with and without gestational diabetes mellitus (GDM) from the National Collaborative Perinatal Project and their offspring, offspring of diabetic mothers had heavier birth weights compared with the offspring of mothers without GDM.40 Furthermore, at age 7 years, the same offspring born to GDM mothers had more adipose tissue compared with their unexposed counterparts, even after adjusting for birth weight, thereby indicating that the relationship between GDM exposure and excessive growth of the offspring extends beyond the perinatal period. Consequently, the diabetic pregnancy is viewed by many to be pivotal in adversely impacting the trajectory for T2D later in life, independently of genetic factors.41 It may also be that exposure to diabetes in utero has wider impacts on health, such that offspring of diabetic pregnancies are more predisposed to CVD42 and tend to die younger.19 The mechanisms for how intrauterine exposures influence the fetus’s cardiometabolic risk trajectories are not well understood in humans, as almost all data are observational, but in animals suggest that alterations in the transcriptional capacity of genes occur, which may involve “epigenetic” processes such as DNA methylation, histone modification, chromatin unraveling, and the actions of small (micro)ribonucleic acids (RNAs).43
Epigenetics
Unlike germline genetic variation that remains unchanging in all cells of the body, epigenetic variation is dynamic and varies across tissues in response to a wide spectrum of environmental stimuli, including those guiding tissue differentiation during growth and development, as well as harmful exposures that elicit an adaptive response from cells. Epigenetic processes are hypothesized to modulate the effects of almost all environmental exposures on the phenotypes of health and disease via transcriptional regulation brought about by, for example, methylation of CpG islands in nuclear DNA; post-translational modification of histone proteins such as acetylation, methylation, phosphorylation, and ubiquitination; and RNA interference. Epigenetic modifications are an important aspect of biology, as they impart to cells the capacity for cellular plasticity and may also act, in response to intrauterine triggers, to program cells in preparation for the extrauterine environment, essentially priming the genome of the developing fetus for the world outside the womb. In this way, epigenetic alterations might potentiate a survival advantage through differential regulation of the genes encoding proteins involved in energy metabolism and adipogenesis. The conservation across generations of cellular memory through epigenetic processes has been widely speculated on, with a number of intriguing transgenerational observational studies suggesting this to be true,44–46 but with no compelling evidence to back this up in humans. Thus, the impact of environmental factors on cellular programming and the extent to which these are conserved across generations represents an intriguing concept but requires further investigation.
One study by Heijmans et al47 examined patterns of DNA methylation at IGF2, an imprinted locus, in people who endured exposure to famine in utero during the Dutch Hunger Winter. The study indicated that there are critical periods of fetal development when the IGF2 promoter is susceptible to hypomethylation. The IGF2 locus was hypomethylated in the offspring who were conceived during the famine, whereas there were no changes in DNA methylation in the offspring exposed to famine in late gestation.47
The impact of genomic imprinting (the parental origin of specific effects of inherited alleles) on disease has been well studied in mouse models and some human studies (see Peters48 for a review of evidence on genomic imprinting); evidence from these studies shows that parent-specific imprinted genes have a significant range of effects on biological processes extending into adulthood. Until recently, transgenerational effects of paternal exposures on subsequent generation(s) were rarely considered within the context of the transgenerational cycle of disease. Ng et al49 investigated the effects of paternal diet on adult rat offspring. The authors observed that glucose tolerance was significantly impaired in the female but not the male offspring.49 In a subsequent study, the authors sought to investigate further the effects of paternal exposure on the female offspring. They found that paternal exposure to a high-fat diet induced impaired glucose tolerance and diminished insulin secretion early in life, despite normal adiposity and growth rates in the female offspring. The mechanisms that underlie these observations appear to involve impaired β-cell replication and epigenetic modification at Il13ra2.49 Another murine study showed that in utero caloric restriction is associated with loci-specific hypomethylation in the male germline at differentially methylated regions that can partially survive reprogramming processes in early embryo development, with altered gene expression at discrete sites.
Epigenetic programming and gene–environment interactions appear to be important factors to consider when unraveling the dynamic (epi)genetic architecture of complex diseases across lineages. Although it seems highly plausible that transgenerational epigenetic processes may affect disease predisposition, it is difficult to exclude cultural and societal factors serving concurrently, or alternatively, as a vehicle for disease transmission across generations. The integration of nonbiological (socioeconomic, cultural, and lifestyle) and biological (genetic and epigenetic) information into disease prediction models is an increasingly important area of research that may have relevance when considering early interventions against adult cardiometabolic diseases.50
Genetics
A major challenge in examining the impact of environmental components in pregnancy and early life on later disease risk is that these relationships are often confounded by genetic factors, which are also shared by the mother and her offspring. Most complex metabolic traits are probably the consequence of interactions between genetic and environmental (especially lifestyle) factors. Studying such interactions is challenging, owing to the relatively small magnitude of the anticipated interaction effects and the difficulty in accurately and precisely quantifying lifestyle behaviors. Early attempts to address this challenge have involved the use of discordant monozygotic twins to disentangle the effects of the intrauterine milieu from those primarily attributable to the offspring’s genotype.51
There is increasing information regarding the genetic basis of T2D and obesity, two metabolic disorders long since determined to be heritable.52 Genome-wide association studies have been the mainstay of population genetics research for much of the past decade and have been used to uncover a plethora of novel loci associated with complex traits. To date, more than 50 established genomic regions harboring variants associated with T2D have been discovered and confirmed.53 However, heritability studies that seek to partition the genetic from nongenetic sources of variance in a trait indicate that whilst T2D has a large genetic component, the role of environmental risk factors is likely to be larger still.51
The role of genetic factors in gestational diabetes is less well studied. However, in a recent analysis of the Diabetes Prevention Program (DPP), a randomized controlled trial (RCT) of more than 3,000 adults randomized to receive a program of intensive lifestyle modification, metformin treatment, or placebo control intervention, we examined the association of 34 established T2D loci with history of GDM, response to intervention, and indices of β-cell function.54 We modeled the genetic loci in aggregate using a genetic risk score. Women with a history of GDM had diminished β-cell function, and the genetic risk score was associated with GDM history but not with response to the DPP interventions. These data suggest that the genetic defects that lead to T2D are likely to overlap with those involved in the pathogenesis of GDM.
Owing to the role of fetal growth and development in T2D, genes that encode these phenotypes may be plausible candidates for T2D. This hypothesis has been tested in a number of candidate gene and genome-wide association studies analyses, with several birth weight-associated loci also being implicated in T2D and other metabolic traits:55–57 eg, common variants at the ADCY5 and CDKAL1 with T2D, fasting glucose, and fasting insulin concentrations, with the allele associated with lower birth weight also being associated with high diabetes risk55–57 and levels of glucose and insulin.58–60 Similarly, the birth weight-associated allele at ADRB1 has been associated with adult blood pressure.61 Moreover, when 47 established T2D-associated variants were tested in unison for association with birth weight, the diabetes risk-raising alleles were statistically associated with lower birth weight than would be expected by chance.62 Although these data support a causal relationship between fetal growth and later risk of cardiometabolic disease, some genes will have pleiotropic effects on growth, development, and adult disease, which may confound associations between birth weight and later disease.
As indicated, interactions between genetic variants and environmental risk factors may also determine the risk of obesity and diabetes in childhood and adult life. One such locus is proximal to the FTO gene, which harbors the strongest single nucleotide variant (rs9960939) for polygenic obesity in children and adults.63–65 FTO may play a role in appetite regulation and energy balance.66–69 For example, in a study of Scottish school children, Cecil et al66 reported a higher ratio of energy intake and energy expenditure in carriers of the minor (A) allele compared with those carrying the lower-risk T allele; interestingly, energy expenditure, albeit insufficient to offset energy intake, was higher than in the T allele carriers, a finding that was subsequently confirmed in adults.70 As well, Wardle et al71 showed that the T allele was associated with lower food intake, suggesting that the T allele promotes sensitivity to satiety. Elsewhere, Church et al68 reported an interaction of the rs9960939 variant with age on children’s weight gain trajectories, and found that the A allele was associated with lower BMI in infancy but a higher BMI in later childhood. Furthermore, adiposity rebound occurred earlier in developmental age in children carrying the minor A allele compared with noncarriers.72 This observation may be important, as early onset of adiposity rebound has been shown to predict later overweight and obesity.73,74 Hence, it is possible that FTO influences developmental processes in distinct ways throughout infancy and later childhood, although underlying mechanisms remain unknown. A prospective population-based study of children residing in the Brazilian Amazon examined the effects of the FTO rs9939609 minor risk allele on weight gain in childhood of predominantly mixed race/ethnicity and observed that those associations were modified by vitamin D levels, which have been shown to be negatively correlated with measures of child adiposity.75 Common FTO variants also appear to be associated with rate of childhood weight gain, which may be augmented in children with lower levels of vitamin D.76
The role of the placenta
Maternal nutrition and placental insufficiency77 are especially relevant in fetal growth and development, as these factors have a direct effect on determining the amount and quality of nutrient supply to the embryo and fetus. A mother’s lifestyle behaviors in pregnancy regulate energy input and output (ie, diet/nutrition and physical activity) and may position the fetus’s energy set point to affect obesity predisposition later in life. The placenta not only acts as a buffer to regulate the transfer of small molecules such as fatty acids to the fetal compartment but also acts as a sink for excess fetal glucose. However, when the placenta’s capacity to do so is exceeded, and the fetus is exposed to high glucose and protein levels, fetal macrosomia can ensue.78 Together, maternal behaviors and the ability of the placenta to function appropriately determine the availability of necessary nutrients to the fetus and also the ability to maintain a homeostatic environment and to avoid exposure to excessive levels of glucose and proteins, some of which are harmful to the fetus.
The placenta is a fetal organ capable of expressing cytokines that may play a role in glycemic control, insulin resistance, and GDM.79 Placenta tissues, for example, secrete leptin during hyperglycemia.80 Leptin and insulin are hormones that play a coordinated role in sympathetic nerve activity38 and in the programming of hypothalamic circuitry involved in appetite regulation.81 Additionally, placenta may undergo morphological adaptations in response to intrauterine exposures, such as levels of glycemia,82 which may result in long-term deleterious effects on metabolic programming. For example, high placenta weight, particularly in lower birth weight infants, is associated with elevated blood pressure in adulthood.83
Obesity and weight gain trajectories
In 2009, the Institute of Medicine (IOM) established new guidelines for limiting maternal weight gain during pregnancy by trimester based on the mother’s prepregnancy BMI. With the release of these new recommendations, the IOM also highlighted the need for more research investigating the impact of these new guidelines on pediatric health outcomes.84 In response to this solicitation, the association of weight gain in pregnancy and childhood overweight/obesity at age 2–5 years was assessed.85 The study showed that children of mothers who exceeded the recommended weight gain were larger at birth and had a greater odds of overweight/obesity in childhood, after adjusting for potential confounders, compared with the offspring of mothers whose weight gain was within the recommended range; the study also showed that prepregnancy BMI may modify the association of gestational weight gain and infant risk. Elsewhere, relationships between maternal prepregnancy BMI, gestational weight gain, and maternal and child health outcomes at birth were assessed in 33,973 Chinese mother–infant dyads.86 The study showed that mothers who were overweight or obese prior to pregnancy, compared with those who were normal weight, were at higher risk of GDM despite gestational weight gain being within the range defined as adequate by the IOM.
In a subsample of the Avon Longitudinal Study of Parents and Children, a prospective cohort study of pregnant women and their offspring, investigators examined the impact of gestational weight gain by trimesters and found a positive association between gestational weight gain in early pregnancy and infant adiposity at age 9 years; the magnitude of this relationship was even greater in mothers who gained more than 500 g/wk. The same relationship persisted in midpregnancy only in mothers who gained more than 500 g/wk.87
Eriksson et al88 found correlations between growth trajectories in childhood and coronary heart disease. Most affected were children who experienced compensatory “catch-up growth” or accelerated weight gain in childhood. These children exhibited a 5-fold elevated risk of death from coronary heart disease.
High-risk populations
Indigenous populations such as Alaskan Natives, American Indians, Inuits, and Australian Aborigines are perceived by many to be genetically prone to obesity and diabetes, largely because of their high degree of susceptibility to these diseases following abrupt unhealthful shifts in lifestyle over the past few decades, yet the specific risk loci are largely unknown. Analysis of data from Pima Indians residing in southern Arizona has contributed to a number of landmark studies focused on intrauterine exposure to diabetes and later T2D risk. Those studies have shown, for example, that Pima Indian children tend to be overweight and hyperinsulinemic.89 In this population, a diabetic intrauterine environment conveys increased risk for childhood hyperglycemia and systolic hypertension, independently of adiposity.90 As adults, Pimas born to diabetic pregnancies are at far higher risk of developing early onset T2D due to lower insulin secretory rates.91 With hyperglycemia and overt T2D occurring at even younger ages (ie, within reproductive age) than in most other populations, the intrauterine risks associated with gestational diabetes are readily transmitted from one generation to the next, thus perpetuating the transgenerational cycle of T2D and obesity within these populations (Figure 1).
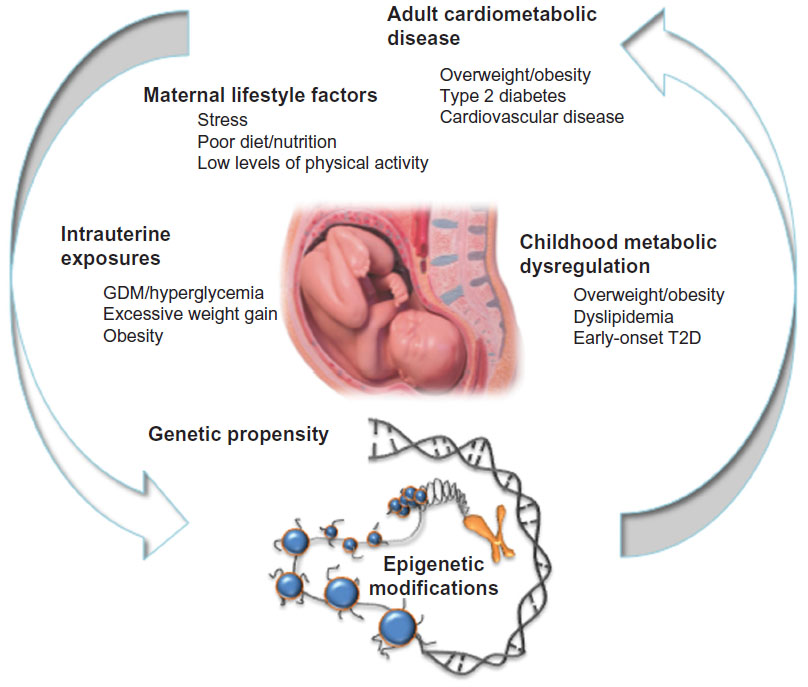 | Figure 1 The vicious cycle of cardiometabolic disease. |
Although mainland indigenous minority populations are often at high risk of obesity and diabetes, indigenous island populations appear even more so, with seven out of the top ten countries/territories with the highest prevalence of age-adjusted diabetes worldwide being those that reside on small islands.92 Populations of small islands are especially susceptible to this phenomenon, probably as a result of strong genetic founder effects that interact with the recent transition from subsistence farming to Westernized lifestyles, where island food supplies now often depend heavily on imported foods that are energy dense and nutritionally deficient. This process is thought to have created a “mismatch” between the fetus’s epigenome, which has been programmed to expect a thrifty extrauterine environment, and the extrauterine reality of caloric excess.93
Evidence from RCTs
While substantial observational evidence appears to support hypotheses that cardiometabolic risk trajectories are established as a result of events in early life, far fewer supportive data are available from RCTs. To date, there is relatively limited information examining whether interventions to prevent and/or reduce harmful intrauterine exposures are effective in improving health outcomes in both the mother and child.94 Large-scale, consortium-wide pregnancy RCTs as well as smaller regional RCTs have been implemented in the past several years to investigate the impact of lifestyle interventions during pregnancy and early life. However, intervention strategies and the IOM recommendations84 are careful to focus on weight gain in pregnancy rather than weight loss, as safety considerations surrounding weight loss in pregnancy have yet to be adequately addressed.95 The Treatment of Obese Pregnant Women (TOP) study was an RCT in Denmark that recruited obese (BMI ≥30 kg/m2) pregnant women from 2009 to 2012.96 Four hundred and twenty-five women were randomly allocated to one of three treatment groups: 1) physical activity and diet, 2) physical activity only, or 3) no intervention, standard care. The authors of the study concluded that physical activity measured using a pedometer, regardless of treatment group, resulted in a reduction in gestational weight gain in these women. As with similar pregnancy intervention trials, the relatively small sample size may have yielded insufficient statistical power to detect differences in infant health outcomes. Currently, larger RCTs are being implemented to assess lifestyle effects on pregnancy outcomes. The LIMIT study,97 a more recent RCT in Australia, is designed to investigate whether limiting gestational weight gain in overweight and obese pregnant women through comprehensive dietary and lifestyle recommendations will reduce the number of infants who are excessive birth weight and associated maternal and infant morbidities. Similarly, in 2012, the US National Institutes of Health (NIH) established the Lifestyle Interventions for Expectant Moms (LIFE-Moms) Consortium consisting of seven sites across the US.98 The aim of the consortium is to identify effective lifestyle interventions that favorably impact gestational weight gain and other measures of cardiometabolic health in the mother and infant. An additional ongoing RCT from the UK (UPBEAT99) is recruiting 1,546 obese pregnant women, who are subsequently randomized to receive a program of lifestyle intervention or control. The primary outcomes of the trial are GDM and large for gestational age infants. The findings of these studies are expected to resolve some of the ambiguity surrounding pregnancy and early lifestyle interventions and their effectiveness as public health strategies for improving maternal, neonatal, and infant cardiometabolic health.
Future clinical and public health implications based on current evidence
The majority of today’s guidelines and intervention strategies against obesity-related diseases often do not account for the possibility that cardiometabolic risk trajectories may be set in the very early stages of life, when system-wide injuries affecting energy homeostasis begin to occur. However, guidelines for improving cardiometabolic health in the US population have been issued by the NIH Heart, Lung, and Blood Institute,84 which focus on screening of childhood obesity and limiting weight gain in pregnancy, respectively. Targeting children within the population who are at greatest risk for cardiometabolic complications in adulthood requires being able to accurately identify those with adverse lipid profiles, excess body fat, glucose dysregulation, high blood pressure, and other indicators. The National Heart, Lung, and Blood Institute calls for universal lipid screening between the ages of 9 and 11 years;84 however, the feasibility and economic implications of these recommendations are not known. In addition, lifestyle interventions in childhood may be perceived as extreme, and general screening may be unable to detect underlying pathophysiological mechanisms that may not fully manifest until later in life.
Although lifestyle interventions (physical activity and diet) are important public health strategies against obesity-related diseases because they beneficially affect multiple systems in the body, this approach alone is unlikely to curb the global obesity epidemic. Optimizing detection and preventative strategies will entail exploration in early life, from preconception to early childhood, to identify novel biomarkers that indicate aberrant metabolic programming events early on. More targeted strategies for reducing T2D and CVD may require risk stratification approaches based on intermediate risk factors such as molecular (eg, micro-RNA and [epi]genetic), anthropometric, and biological (eg, microbiota) markers, rather than overt phenotypes, like diabetes status, coronary heart disease, stroke, or metabolic syndrome, that are detectable in early stages of development. For example, altered or impaired epigenetic dynamics in preadipocyte differentiation may lead to permanent morphological and functional changes in fat tissue100 – a major player in the endocrine system orchestrating a cascade of transcription factors, including the peroxisome proliferator-activated receptor γ involved in signaling networks of glucose regulation and energy balance.101 Furthermore, assessing the validity of epigenetic inheritance, in other words the transmission of epigenetic regulatory features acquired as an adaptive response to an environmental signal affecting one generation to subsequent generation(s) that has not been exposed directly to the environmental “trigger(s)”, will have major implications in forecasting the societal burden of chronic disease. In this way, investigating the primordial defects associated with the cluster of traits defining metabolic syndrome may provide important insight into potential targets for future preventative strategies.
Conclusion
Maximizing strategies for preventing or modifying excessive morbidity and mortality associated with complex diseases requires a more complete understanding of its earliest modifiable risk factors and consideration of this knowledge in public health recommendations. Epidemiological studies have already made major contributions to our understanding of how adverse exposures in utero and during childhood impact disease risk trajectories, which now require experimental validation. Numerous clinical trials are now underway or completed to assess the effects of intervening during pregnancy and childhood to improve prognosis in those considered at high risk, usually determined on the basis of the mother’s early pregnancy weight or glucose tolerance. As the body of evidence from clinical trials grows, the extent to which the hypotheses set forth through observational studies are valid will become clear, and solid evidence-based guidelines designed to prevent obesity, T2D, and CVD, which account for early-life exposures, will likely take form.
Acknowledgments
ACE is supported in part by a doctoral fellowship from the Danish Diabetes Academy supported through the Novo Nordisk Foundation. PWF is supported in part by the Novo Nordisk Foundation, Swedish Research Council, and EXODIAB.
Disclosure
The authors have no disclosures relevant to this manuscript.
References
Sims EA, Horton ES. Endocrine and metabolic adaptation to obesity and starvation. Am J Clin Nutr. 1968;21(12):1455–1470. | |
Sparen P, Vagero D, Shestov DB, et al. Long term mortality after severe starvation during the siege of Leningrad: prospective cohort study. BMJ. 2004;328(7430):11. | |
Chan JL, Mantzoros CS. Role of leptin in energy-deprivation states: normal human physiology and clinical implications for hypothalamic amenorrhoea and anorexia nervosa. Lancet. 2005;366(9479):74–85. | |
Bluher S, Mantzoros CS. Leptin in reproduction. Curr Opin Endocrinol Diabetes Obes. 2007;14(6):458–464. | |
Bouchard C, Tremblay A, Despres JP, et al. The response to long-term overfeeding in identical twins. N Engl J Med. 1990;322(21):1477–1482. | |
Wilmore JH, Despres JP, Stanforth PR, et al. Alterations in body weight and composition consequent to 20 wk of endurance training: the HERITAGE Family Study. Am J Clin Nutr. 1999;70(3):346–352. | |
Bar-Or O, Foreyt J, Bouchard C, et al. Physical activity, genetic, and nutritional considerations in childhood weight management. Med Sci Sports Exerc. 1998;30(1):2–10. | |
Ahmad S, Varga TV, Franks PW. Gene x environment interactions in obesity: the state of the evidence. Hum Hered. 2013;75(2–4):106–115. | |
Wang Y, Monteiro C, Popkin BM. Trends of obesity and underweight in older children and adolescents in the United States, Brazil, China, and Russia. Am J Clin Nutr. 2002;75(6):971–977. | |
Popkin BM, Gordon-Larsen P. The nutrition transition: worldwide obesity dynamics and their determinants. Int J Obes Relat Metab Disord. 2004;28 Suppl 3:S2–S9. | |
Popkin BM, Adair LS, Ng SW. Global nutrition transition and the pandemic of obesity in developing countries. Nutr Rev. 2012;70(1):3–21. | |
Swinburn B, Gill T, Kumanyika S. Obesity prevention: a proposed framework for translating evidence into action. Obes Rev. 2005;6(1):23–33. | |
International Obesity Taskforce. Obesity the global epidemic. 2011. Available at: http://www.worldobesity.org/iotf/obesity/obesitytheglobalepidemic/. Accessed August 3, 2014. | |
Freedman DS, Mei Z, Srinivasan SR, Berenson GS, Dietz WH. Cardiovascular risk factors and excess adiposity among overweight children and adolescents: the Bogalusa Heart Study. J Pediatr. 2007;150(1):12–17. | |
Koplan JP, Liverman CT, Kraak VI. Preventing childhood obesity: health in the balance: executive summary. J Am Diet Assoc. 2005; 105(1):131–138. | |
Lobstein T, Baur L, Uauy R. Obesity in children and young people: a crisis in public health. Obes Rev. 2004;5 Suppl 1:4–104. | |
Dabelea D, Mayer-Davis EJ, Saydah S, et al. Prevalence of type 1 and type 2 diabetes among children and adolescents from 2001 to 2009. JAMA. 2014;311(17):1778–1786. | |
Whitaker RC, Wright JA, Pepe MS, Seidel KD, Dietz WH. Predicting obesity in young adulthood from childhood and parental obesity. N Engl J Med. 1997;337(13):869–873. | |
Franks PW, Hanson RL, Knowler WC, Sievers ML, Bennett PH, Looker HC. Childhood obesity, other cardiovascular risk factors, and premature death. N Engl J Med. 2010;362(6):485–493. | |
Must A, Jacques PF, Dallal GE, Bajema CJ, Dietz WH. Long-term morbidity and mortality of overweight adolescents. A follow-up of the Harvard Growth Study of 1922 to 1935. N Engl J Med. 1992;327(19):1350–1355. | |
Gunnell DJ, Frankel SJ, Nanchahal K, Peters TJ, Davey Smith G. Childhood obesity and adult cardiovascular mortality: a 57-y follow-up study based on the Boyd Orr cohort. Am J Clin Nutr. 1998;67(6):1111–1118. | |
DiPietro L, Mossberg HO, Stunkard AJ. A 40-year history of overweight children in Stockholm: life-time overweight, morbidity, and mortality. Int J Obes Relat Metab Disord. 1994;18(9):585–590. | |
Engeland A, Bjorge T, Sogaard AJ, Tverdal A. Body mass index in adolescence in relation to total mortality: 32-year follow-up of 227,000 Norwegian boys and girls. Am J Epidemiol. 2003;157(6):517–523. | |
Franks PW, Hanson RL, Knowler WC, et al. Childhood predictors of young-onset type 2 diabetes. Diabetes. 2007;56(12):2964–2972. | |
Hellerstrom C, Andersson A, Groth CG, et al. Experimental pancreatic transplantation in diabetes. Diabetes Care. 1988;11 Suppl 1:45–53. | |
Hales CN, Barker DJ. The thrifty phenotype hypothesis. Br Med Bull. 2001;60:5–20. | |
Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet. 1989;2(8663):577–580. | |
Li Y, He Y, Qi L, et al. Exposure to the Chinese famine in early life and the risk of hyperglycemia and type 2 diabetes in adulthood. Diabetes. 2010;59(10):2400–2406. | |
Li Y, Jaddoe VW, Qi L, et al. Exposure to the chinese famine in early life and the risk of metabolic syndrome in adulthood. Diabetes Care. 2011;34(4):1014–1018. | |
Huang C, Li Z, Wang M, Martorell R. Early life exposure to the 1959–1961 Chinese famine has long-term health consequences. J Nutr. 2010;140(10):1874–1878. | |
Lumey LH, Stein AD, Kahn HS, et al. Cohort profile: the Dutch Hunger Winter families study. Int J Epidemiol. 2007;36(6):1196–1204. | |
Ravelli AC, van der Meulen JH, Michels RP, et al. Glucose tolerance in adults after prenatal exposure to famine. Lancet. 1998;351(9097):173–177. | |
Lauren L, Jarvelin M-R, Elliott P, et al. Relationship between birthweight and blood lipid concentrations in later life: evidence from the existing literature. Int J Epidemiol. 2003;32(5):862–876. | |
Savona-Ventura C, Chircop M. Birth weight influence on the subsequent development of gestational diabetes mellitus. Acta Diabetol. 2003;40(2):101–104. | |
Pettitt DJ, Jovanovic L. Low birth weight as a risk factor for gestational diabetes, diabetes, and impaired glucose tolerance during pregnancy. Diabetes Care. 2007;30 Suppl 2:S147–S149. | |
Dabelea D, Pettitt DJ, Hanson RL, Imperatore G, Bennett PH, Knowler WC. Birth weight, type 2 diabetes, and insulin resistance in Pima Indian children and young adults. Diabetes Care. 1999;22(6):944–950. | |
Wei J-N, Sung F-C, Li C-Y, et al. Low birth weight and high birth weight infants are both at an increased risk to have type 2 diabetes among schoolchildren in Taiwan. Diabetes Care. 2003;26(2):343–348. | |
Henry SL, Barzel B, Wood-Bradley RJ, Burke SL, Head GA, Armitage JA. Developmental origins of obesity-related hypertension. Clin Exp Pharmacol Physiol. 2012;39(9):799–806. | |
Franks PW, Looker HC, Kobes S, et al. Gestational glucose tolerance and risk of type 2 diabetes in young Pima Indian offspring. Diabetes. 2006;55(2):460–465. | |
Baptiste-Roberts K, Nicholson WK, Wang N-Y, Brancati FL. Gestational diabetes and subsequent growth patterns of offspring: the National Collaborative Perinatal Project. Matern Child Health J. 2012;16(1):125–132. | |
Lauren L, Jarvelin M-R, Elliott P, et al. Relationship between birthweight and blood lipid concentrations in later life: evidence from the existing literature. Int J Epidemiol. 2003;32(5):862–876. | |
Manderson JG, Mullan B, Patterson CC, Hadden DR, Traub AI, McCance DR. Cardiovascular and metabolic abnormalities in the offspring of diabetic pregnancy. Diabetologia. 2002;45(7):991–996. | |
Chuang JC, Jones PA. Epigenetics and MicroRNAs. Pediatr Res. 61(5 Part 2):24R–29R. | |
Bygren LO, Tinghog P, Carstensen J, et al. Change in paternal grandmothers’ early food supply influenced cardiovascular mortality of the female grandchildren. BMC Genet. 2014;15:12. | |
Kaati G, Bygren LO, Edvinsson S. Cardiovascular and diabetes mortality determined by nutrition during parents’ and grandparents’ slow growth period. Eur J Hum Genet. 2002;10(11):682–688. | |
Kaati G, Bygren LO, Pembrey M, Sjostrom M. Transgenerational response to nutrition, early life circumstances and longevity. Eur J Hum Genet. 2007;15(7):784–790. | |
Heijmans BT, Tobi EW, Stein AD, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105(44):17046–17049. | |
Peters J. The role of genomic imprinting in biology and disease: an expanding view. Nat Rev Genet. 2014;15(8):517–530. | |
Ng S-F, Lin RCY, Laybutt DR, Barres R, Owens JA, Morris MJ. Chronic high-fat diet in fathers programs beta-cell dysfunction in female rat offspring. Nature. 2010;467(7318):963–966. | |
Pembrey M, Saffery R, Bygren LO. Human transgenerational responses to early-life experience: potential impact on development, health and biomedical research. J Med Genet. 2014;51(9):563–572. | |
Poulsen P, Vaag AA, Kyvik KO, Moller Jensen D, Beck-Nielsen H. Low birth weight is associated with NIDDM in discordant monozygotic and dizygotic twin pairs. Diabetologia. 1997;40(4):439–446. | |
Drong AW, Lindgren CM, McCarthy MI. The genetic and epigenetic basis of type 2 diabetes and obesity. Clin Pharmacol Ther. 2012;92(6):707–715. | |
Morris AP, Voight BF, Teslovich TM, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44(9):981–990. | |
Sullivan SD, Jablonski KA, Florez JC, et al. Genetic risk of progression to type 2 diabetes and response to intensive lifestyle or metformin in prediabetic women with and without a history of gestational diabetes mellitus. Diabetes Care. 2014;37(4):909–911. | |
Zhao J, Li M, Bradfield JP, et al. Examination of type 2 diabetes loci implicates CDKAL1 as a birth weight gene. Diabetes. 2009;58(10):2414–2418. | |
Andersson EA, Pilgaard K, Pisinger C, et al. Type 2 diabetes risk alleles near ADCY5, CDKAL1 and HHEX-IDE are associated with reduced birthweight. Diabetologia. 2010;53(9):1908–1916. | |
Freathy RM, Mook-Kanamori DO, Sovio U, et al. Variants in ADCY5 and near CCNL1 are associated with fetal growth and birth weight. Nat Genet. 2010;42(5):430–435. | |
Dupuis J, Langenberg C, Prokopenko I, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42(2):105–116. | |
Saxena R, Hivert M-F, Langenberg C, et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet. 2010;42(2):142–148. | |
Steinthorsdottir V, Thorleifsson G, Reynisdottir I, et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet. 2007;39(6):770–775. | |
Johnson AD, Newton-Cheh C, Chasman DI, et al. Association of hypertension drug target genes with blood pressure and hypertension in 86,588 individuals. Hypertension. 2011;57(5):903–910. | |
Horikoshi M, Yaghootkar H, Mook-Kanamori DO, et al. New loci associated with birth weight identify genetic links between intrauterine growth and adult height and metabolism. Nat Genet. 2013;45(1):76–82. | |
Frayling TM, Timpson NJ, Weedon MN, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316(5826):889–894. | |
Dina C, Meyre D, Gallina S, et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. 2007;39(6):724–726. | |
Scott LJ, Mohlke KL, Bonnycastle LL, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316(5829):1341–1345. | |
Cecil JE, Tavendale R, Watt P, Hetherington MM, Palmer CNA. An obesity-associated FTO gene variant and increased energy intake in children. N Engl J Med. 2008;359(24):2558–2566. | |
Wardle J, Llewellyn C, Sanderson S, Plomin R. The FTO gene and measured food intake in children. Int J Obes (Lond). 2009;33(1):42–45. | |
Church C, Lee S, Bagg EAL, et al. A mouse model for the metabolic effects of the human fat mass and obesity associated FTO gene. PLoS Genet. 2009;5(8):e1000599. | |
Tung Y-CL, Ayuso E, Shan X, et al. Hypothalamic-specific manipulation of Fto, the ortholog of the human obesity gene FTO, affects food intake in rats. PLoS One. 2010;5(1):e8771. | |
Jonsson A, Franks PW. Obesity, FTO gene variant, and energy intake in children. N Engl J Med. 2009;360(15):1571–1572. | |
Wardle J, Llewellyn C, Sanderson S, Plomin R. The FTO gene and measured food intake in children. Int J Obes (Lond). 2009;33(1):42–45. | |
Sovio U, Mook-Kanamori DO, Warrington NM, et al. Association between common variation at the FTO locus and changes in body mass index from infancy to late childhood: the complex nature of genetic association through growth and development. PLoS Genet. 2011; 7(2):e1001307. | |
Whitaker RC, Pepe MS, Wright JA, Seidel KD, Dietz WH. Early adiposity rebound and the risk of adult obesity. Pediatrics. 1998;101(3):E5. | |
Williams SM, Goulding A. Early adiposity rebound is an important predictor of later obesity. Obesity (Silver Spring). 2009;17(7):1310. | |
Lourenco BH, Qi L, Willett WC, Cardoso MA. FTO genotype, vitamin D status, and weight gain during childhood. Diabetes. 2014;63(2):808–814. | |
Turer CB, Lin H, Flores G. Prevalence of vitamin D deficiency among overweight and obese US children. Pediatrics. 2013;131(1): e152–e161. | |
Henriksen T, Clausen T. The fetal origins hypothesis: placental insufficiency and inheritance versus maternal malnutrition in well-nourished populations. Acta Obstet Gynecol Scand. 2002;81(2):112–114. | |
Desoye G, Korgun ET, Ghaffari-Tabrizi N, Hahn T. Is fetal macrosomia in adequately controlled diabetic women the result of a placental defect? A hypothesis. J Matern Fetal Neonatal Med. 2002;11(4):258–261. | |
Desoye G, Hauguel-de Mouzon S. The human placenta in gestational diabetes mellitus. The insulin and cytokine network. Diabetes Care. 2007;30 Suppl 2:S120–S126. | |
Lappas M, Yee K, Permezel M, Rice GE. Release and regulation of leptin, resistin and adiponectin from human placenta, fetal membranes, and maternal adipose tissue and skeletal muscle from normal and gestational diabetes mellitus-complicated pregnancies. J Endocrinol. 2005;186(3):457–465. | |
Plagemann A. Perinatal programming and functional teratogenesis: impact on body weight regulation and obesity. Physiol Behav. 2005;86(5):661–668. | |
Calderon IMP, Damasceno DC, Amorin RL, Costa RAA, Brasil MAM, Rudge MVC. Morphometric study of placental villi and vessels in women with mild hyperglycemia or gestational or overt diabetes. Diabetes Res Clin Pract. 2007;78(1):65–71. | |
Barker DJ, Bull AR, Osmond C, Simmonds SJ. Fetal and placental size and risk of hypertension in adult life. BMJ. 1990;301(6746):259–262. | |
Institute of Medicine (US) and National Research Council (US) Committee to Reexamine IOM Pregnancy Weight Guidelines, Rasmussen KM, Yaktine AL. Weight Gain During Pregnancy: Reexamining the Guidelines. Washington, DC: National Academies Press; 2009. | |
Sridhar SB, Darbinian J, Ehrlich SF, et al. Maternal gestational weight gain and offspring risk for childhood overweight or obesity. Am J Obstet Gynecol. 2014;211(3):259. e1–e8. | |
Liu L, Li M, Yang L, et al. Measuring coverage in MNCH: a validation study linking population survey derived coverage to maternal, newborn, and child health care records in rural China. PLoS One. 2013; 8(5):e60762. | |
Fraser A, Tilling K, Macdonald-Wallis C, et al. Associations of gestational weight gain with maternal body mass index, waist circumference, and blood pressure measured 16 y after pregnancy: the Avon Longitudinal Study of Parents and Children (ALSPAC). Am J Clin Nutr. 2011;93(6):1285–1292. | |
Eriksson JG, Forsen T, Tuomilehto J, Winter PD, Osmond C, Barker DJ. Catch-up growth in childhood and death from coronary heart disease: longitudinal study. BMJ. 1999;318(7181):427–431. | |
Salbe AD, Weyer C, Lindsay RS, Ravussin E, Tataranni PA. Assessing risk factors for obesity between childhood and adolescence: I. Birth weight, childhood adiposity, parental obesity, insulin, and leptin. Pediatrics. 2002;110(2 Pt 1):299–306. | |
Bunt JC, Tataranni PA, Salbe AD. Intrauterine exposure to diabetes is a determinant of hemoglobin A(1)c and systolic blood pressure in pima Indian children. J Clin Endocrinol Metab. 2005;90(6):3225–3229. | |
Gautier JF, Wilson C, Weyer C, et al. Low acute insulin secretory responses in adult offspring of people with early onset type 2 diabetes. Diabetes. 2001;50(8):1828–1833. | |
Aguiree F, Brown A, Cho NH, Dahlquist G, Dodd S. IDF Diabetes Atlas. International Diabetes Foundation; 2013. Available from: http://www.idf.org/sites/default/files/EN_6E_Atlas_Full_0.pdf. Accessed September 23, 2014. | |
Godfrey KM, Lillycrop KA, Burdge GC, Gluckman PD, Hanson MA. Epigenetic mechanisms and the mismatch concept of the developmental origins of health and disease. Pediatr Res. 2007;61(5 Pt 2):5R–10R. | |
Dodd JM, Crowther CA, Robinson JS. Dietary and lifestyle interventions to limit weight gain during pregnancy for obese or overweight women: a systematic review. Acta Obstet Gynecol Scand. 2008;87(7):702–706. | |
Furber CM, McGowan L, Bower P, Kontopantelis E, Quenby S, Lavender T. Antenatal interventions for reducing weight in obese women for improving pregnancy outcome. Cochrane Database Syst Rev. 2013;1:CD009334. | |
Renault KM, Norgaard K, Nilas L, et al. The Treatment of Obese Pregnant Women (TOP) study: a randomized controlled trial of the effect of physical activity intervention assessed by pedometer with or without dietary intervention in obese pregnant women. Am J Obstet Gynecol. 2014;210(2):134. e1–e9. | |
Dodd JM, Turnbull DA, McPhee AJ, Wittert G, Crowther CA, Robinson JS. Limiting weight gain in overweight and obese women during pregnancy to improve health outcomes: the LIMIT randomised controlled trial. BMC Pregnancy Childbirth. 2011;11:79. | |
Lifestyle Interventions for Expectant Moms: LIFE-Moms [homepage on the Internet]. Available from: https://lifemoms.bsc.gwu.edu/web/lifemoms. Accessed September 23, 2014. | |
Briley AL, Barr S, Badger S, et al. A complex intervention to improve pregnancy outcome in obese women; the UPBEAT randomised controlled trial. BMC Pregnancy Childbirth. 2014;14:74. | |
Schultz NS, Broholm C, Gillberg L, et al. Impaired leptin gene expression and release in cultured preadipocytes isolated from individuals born with low birth weight. Diabetes. 2014;63(1):111–121. | |
Rosen ED, Hsu C-H, Wang X, et al. C/EBPalpha induces adipogenesis through PPARgamma: a unified pathway. Genes Dev. 2002;16(1):22–26. | |
Li Y, Jaddoe VW, Qi L, et al. Exposure to the chinese famine in early life and the risk of metabolic syndrome in adulthood. Diabetes Care. 2011;34(4):1014–1018. | |
Ravelli AC, van der Meulen JH, Michels RP, et al. Glucose tolerance in adults after prenatal exposure to famine. Lancet. 1998;351(9097):173–177. | |
Wei J-N, Sung F-C, Li C-Y, et al. Low birth weight and high birth weight infants are both at an increased risk to have type 2 diabetes among schoolchildren in taiwan. Diabetes Care. 2003;26(2):343–348. | |
Bygren LO, Tinghog P, Carstensen J, et al. Change in paternal grandmothers’ early food supply influenced cardiovascular mortality of the female grandchildren. BMC Genet. 2014;15:12. | |
Poulsen P, Vaag AA, Kyvik KO, Jensen DM, Beck-Nielsen H. [Low birth weight is associated with non-insulin-dependent diabetes mellitus in discordant monozygotic and dizygotic twins]. Ugeskr Laeger. 1998;160(16):2382–2387. Danish. | |
Andersson EA, Pilgaard K, Pisinger C, et al. Type 2 diabetes risk alleles near ADCY5, CDKAL1 and HHEX-IDE are associated with reduced birthweight. Diabetologia. 2010;53(9):1908–1916. | |
Zhao J, Li M, Bradfield JP, et al. Examination of type 2 diabetes loci implicates CDKAL1 as a birth weight gene. Diabetes. 2009;58(10):2414–2418. | |
Horikoshi M, Yaghootkar H, Mook-Kanamori DO, et al. New loci associated with birth weight identify genetic links between intrauterine growth and adult height and metabolism. Nat Genet. 2013;45(1):76–82. | |
Frayling TM, Timpson NJ, Weedon MN, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316(5826):889–894. | |
Sovio U, Mook-Kanamori DO, Warrington NM, et al. Association between common variation at the FTO locus and changes in body mass index from infancy to late childhood: the complex nature of genetic association through growth and development. PLoS Genet. 2011;7(2):e1001307. | |
Whitaker RC, Pepe MS, Wright JA, Seidel KD, Dietz WH. Early adiposity rebound and the risk of adult obesity. Pediatrics. 1998; 101(3):E5. | |
Lappas M, Yee K, Permezel M, Rice GE. Release and regulation of leptin, resistin and adiponectin from human placenta, fetal membranes, and maternal adipose tissue and skeletal muscle from normal and gestational diabetes mellitus-complicated pregnancies. J Endocrinol. 2005;186(3):457–465. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.