Back to Journals » Drug Design, Development and Therapy » Volume 15
Gene Therapy for Rdh12-Associated Retinal Diseases Helps to Delay Retinal Degeneration and Vision Loss
Authors Bian J, Chen H, Sun J ![]() , Cao Y, An J, Pan Q, Qi M
, Cao Y, An J, Pan Q, Qi M
Received 20 May 2021
Accepted for publication 4 August 2021
Published 17 August 2021 Volume 2021:15 Pages 3581—3591
DOI https://doi.org/10.2147/DDDT.S305378
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Georgios Panos
Jiaxin Bian,1,2 Hongyu Chen,1,2 Junhui Sun,1,2 Yuqing Cao,3 Jianhong An,3 Qing Pan,4 Ming Qi1,2,5– 8
1Department of Cell Biology and Medical Genetics, School of Medicine Zhejiang University, Hangzhou, 310000, People’s Republic of China; 2Center for Precision Medicine, Zhejiang-California International NanoSystems Institute, Hangzhou, 310000, People’s Republic of China; 3School of Optometry and Ophthalmology Wenzhou Medical College, Wenzhou, People’s Republic of China; 4Department of Ophthalmology, Zhejiang University Medical School First Affiliated Hospital, Hangzhou, 310000, People’s Republic of China; 5Assisted Reproduction Unit, Department of Obstetrics and Gynecology, Department of Laboratory Medicine, Sir Run Run Shaw Hospital, Zhejiang University School of Medicine, Key Laboratory of Reproductive Dysfunction Management of Zhejiang Province, Hangzhou, 310000, People’s Republic of China; 6DIAN Diagnostics, Hangzhou, 310000, People’s Republic of China; 7Department of Pathology and Laboratory of Medicine, University of Rochester Medical Centre, Rochester, NY, 14609, USA; 8HVP-China, Hangzhou, 310000, People’s Republic of China
Correspondence: Qing Pan
Department of Ophthalmology, Zhejiang University Medical School First Affiliated Hospital, 79 Qingchun Road, Hangzhou, People’s Republic of China
Tel +86 13515814709
Email [email protected]
Ming Qi
Department of Cell Biology and Medical Genetics, School of Medicine Zhejiang University, Research Building A713, Yuhangtang Road 866, Hangzhou, People’s Republic of China
Tel +86 13666686943
Fax +86 57188208274
Email [email protected]; [email protected]
Purpose: The aim of study was to establish Rdh12-associated inherited retinal disease (Rdh12-IRD) mouse model and to identify the best timepoint for gene therapy.
Methods: We induced retinal degeneration in Rdh12−/− mice using a bright light. We clarified the establishment of Rdh12-IRD mouse model by analyzing the thickness of retinal layers and electroretinography (ERG). Rdh12-IRD mice received a subretinal injection of adeno-associated virus 2/8-packaged Rdh12 cDNA for treatment. We evaluated the visual function and retinal structure in the treated and untreated eyes to identify the best timepoint for gene therapy.
Results: Rdh12-IRD mice showed significant differences in ERG amplitudes and photoreceptor survival compared to Rdh12+/+ mice. Preventive gene therapy not only maintained normal visual function but also prevented photoreceptor loss. Salvage gene therapy could not reverse the retinal degeneration phenotype of Rdh12-IRD mice, but it could slow down the loss of visual function.
Conclusion: The light-induced retinal degeneration in our Rdh12−/− mice indicated that a defect in Rdh12 alone was sufficient to cause visual dysfunction and photoreceptor degeneration, which reproduced the phenotypes observed in RDH12-IRD patients. This model is suitable for gene therapy studies. Early treatment of the primary Rdh12 defect helps to delay the later onset of photoreceptor degeneration and maintains visual function in Rdh12-IRD mice.
Keywords: Rdh12, retinal diseases, mouse model, gene therapy
Introduction
Retinol dehydrogenase 12 (RDH12) is a nicotinamide adenine dinucleotide phosphate-dependent retinal reductase, which specifically localizes in the inner segments of rod and cone photoreceptor cells.1 RDH12 protects photoreceptors from intense illumination-induced cytotoxicity by reducing free all-trans-retinal.2–4 Recessive RDH12 mutations are associated with inherited retinal diseases (IRDs), diagnosed as Leber congenital amaurosis type 13 or early-onset severe retinal dystrophy, which accounts for approximately 3.4–10.5% of IRD cases.5,6 RDH12-IRD is a complex disease in which vision loss is caused by two pathological mechanisms: visual dysfunction and photoreceptor degeneration.7,8 With the success of retinal pigment epithelium-specific protein 65 kDa (RPE65) gene therapy, there is new hope for other retinal diseases.9
The most common retinal phenotypes in patients with RDH12-IRD are progressive rod-cone dystrophy and severe macular atrophy.7,8 The clinical signs of RDH12-IRD include universal reduction in rod and cone electroretinography (ERG) responses10 and abnormal macular structure with an almost undetectable outer nuclear layer (ONL) within a thinned foveal center by spectral domain optical coherence tomography (SD-OCT).8 The degree of retinal degeneration in RDH12-IRD is more serious than those of other types of IRD.11 However, Rdh12 knockout mice (Rdh12−/− mice) lack the rapid retinal degeneration phenotypes observed in humans.2,12,13 Without the retinal disease phenotypes of visual function and retinal structure, it is difficult to judge the effect of gene therapy. The establishment of Rdh12-IRD mouse model is essential for gene therapy research.
Studies have shown that C57BL/6 mice with RPE65 (Met450Leu) variant are more susceptible to light damage.14 Rdh12−/− C57BL/6 mice with RPE65 (Met450Leu) variant are also susceptible to light-induced photoreceptor apoptosis, and bright light could induce retinal degeneration.12 This phenotype is similar to that of human IRD, but it may be due to a joint influence of Rdh12 and Rpe65. Moreover, the changes in visual function and fundus phenotype induced by light are unknown. In our study, Rdh12−/− C57BL/6 with RPE65 (Met-450) was used as an Rdh12-IRD mouse model, which expresses RPE65 at a significantly low level.14 Since we want to study the effect of Rdh12 gene therapy, it is appropriate to study Rdh12 defect-only mice.
Adeno-associated virus 2/5 (AAV2/5) gene therapy has been performed in Rdh12−/− BALB/cJ with RPE65 (Leu-450), which prevents light-induced visual dysfunction in mice, but the changes of retinal structure remain unclear.6 Presumably, the correction of potential cellular dysfunction will promote neuronal survival in human recessive retinal degeneration.15 Rdh12-IRD gene therapy should not only focus on the improvement of visual function, but also on slowing the progress of photoreceptor degeneration. Furthermore, the effect after the degenerative phase of IRD for gene therapy is also important.
In our study, we explored the effects of gene therapy before (preventive gene therapy) and after (salvage gene therapy) the onset of IRD. We aimed to determine (1) whether bright light could induce human IRD phenotypes of dysfunction and degeneration of photoreceptors in Rdh12 defect-only mice, (2) whether gene therapy had similar therapeutic effects in Rdh12−/− C57BL/6 mice as in Rdh12−/− BALB/cJ mice, (3) whether preventive gene therapy was effective for visual function and whether it had an effect on retinal degeneration, and (4) whether salvage gene therapy was effective.
Materials and Methods
Experimental Animals
The Rdh12−/− mice were generated by Biocytogen (Beijing, China) (license number: SCXK [Su] 2016-0004) using CRISPR/Cas9– based EGE system. Briefly, two single guide RNAs (sgRNAs) were designed to target the upstream of exon 1 and the downstream of exon 5 in the non-conservative region of Rdh12. The sgRNAs plasmid with Cas9 mRNA was co-injected into C57BL/6 with RPE65 (Met-450) mouse zygotes, and surviving zygotes were transferred into KM albino pseudo-pregnant mice. All treatment and care of animals were performed according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Animal Care and Ethics Committee at Zhejiang Provincial People’s Hospital (Hangzhou, China).
Virus Preparation
The cDNA sequence (951 bp) of mRdh12 (NM_030017) was cloned into AAV2/8 plasmid that yielded AAV2/8-CMV-mRdh12 and the control AAV2/8-CMV-GFP. After sequence verification, amplicons were packaged using a three-plasmid transient transfection of human embryonic kidney 293T cells (ATCC, Manassas, VA). The transfection and purification were performed by using a protocol as published16,17 (Supplementary Methods).
Rdh12-IRD Mouse Model Construction
Light damage was induced as previously published.12,14,18 6–8-week-old mice were dark-adapted overnight and exposed to 3 Klux diffuse white fluorescent light (lights on at 11:00 a.m.) for 48 h after pupil dilation with 0.01% atropine eye gel. After light exposure and dark adaptation for 24 h, retinal structure was evaluated by analyzing the thickness of retinal layers. Moreover, visual function and fundus phenotype were detected by ERG recording and fundus imaging 7 days after exposure.
Subretinal Injections and Gene Therapy
Subretinal injections were performed as reported6 with slight modifications. Xylazine (7 mg/kg) and ketamine (70 mg/kg) were used as general anesthetics, and 2.5% phenylephrine hydrochloride with 1% tropicamide was administered for pupil dilation. The tip of a 29-gauge hypodermic needle was used to make a small incision through the cornea adjacent to the limbus under an ophthalmic surgical microscope. After penetration, the needle orientation was changed to be parallel to the anterior surface of the lens, which would avoid possible injuries to the lens. A Hamilton syringe with 33-gauge blunt needle was inserted through the incision, avoiding the lens to go deeper into the vitreous cavity, and pushed through the retina. Mice received a subretinal injection of 1 µL each to produce bullous retinal detachment in the inferior or superior hemisphere within the nasal quadrant, covering approximately one-third of the retina. After 5 days, OCT imaging detected the disappearance of bullous retinal detachment and the restoration of retinal structure, which can be considered a successful injection. The successfully injected mice were used for further experiments.
The experiment of gene therapy was divided 6–8-week-old mice into four groups, Rdh12+/+ negative light-induced retinal degeneration (Rdh12+/+-LD) (negative control), Rdh12−/−-LD (positive control), preventive gene therapy (Rdh12−/− mice-preventive), and salvage gene therapy (Rdh12−/− mice-salvage) (n=8–10 each). The preventive and salvage groups received the subretinal injection of AAV2/8-mRdh12 (4.5×1010 vg) in one eye and no treatment in the contralateral eye. In the preventive group, gene therapy was performed first, and LD was performed after the expression of Rdh12 detected. In the salvage group, LD was performed first and then followed by gene therapy. Young adult mice were randomly grouped, and both sexes in equal numbers were used for injections. After 3 and 6 months of gene therapy, the effects of gene therapy were assessed by retinal structure and visual function.
Hematoxylin-Eosin Staining and Photography
Eye samples were enucleated and fixed in 75% ethanol: H2O: methanol: acetic acid (10:7:2:1) for 48 h. The tissues were embedded in paraffin, sectioned vertically through the optic nerve, and then stained with hematoxylin-eosin (HE). Images were taken using a microscope (Olympus BX61, Japan) equipped with a digital camera (Olympus DF72, Japan). The ce11Sens 1.14 software was used to measure the thicknesses of each retinal layer and total retina in the inferior and superior retina as per standard convention.
There were two measurement methods.19,20 First, differences of each layer thickness in mice retina were measured at 1000 µm from the optic nerve head to determine the degree of light-induced photoreceptor degeneration. Second, Rdh12 located in the inner segment (IS) layer. The thicknesses of IS+OS and ONL were measured to determine the effects of gene therapy in the location: 400 µm (S-3), 800 µm (S-2), and 1200 µm (S-1) superior and 400 µm (I-3), 800 µm (I-2), and 1200 µm (I-1) inferior to the optic disc.
Electroretinography Recordings
ERG was recorded as previously published.21 Five intensities for scotopic flash ERG (−3.699, −2.201, −0.699, 0.301, and 0.799 log cd·s/m2) and photopic stimuli (−0.699, −0.201, 0.301, 0.799, and 1.301 log cd·s/m2) were observed. Photopic ERGs were recorded after 10 min of light adaptation with a rod-saturating background (1.398 log cd/m2).
The ERG protocol of gene therapy consisted of recording scotopic responses to LED stimuli (−2.201 log cd·s/m2 for rod isolated B-wave, 0.799 log cd·s/m2 for rod-cone combined response). Photopic ERGs were in response to 0.799 log cd·s/m2 intensity flashes. All mice were dark-adapted overnight before the experiments. Mice were anesthetized by a mixture of ketamine/xylazine as described above.
Two-Choice Cued Water Maze Task
A Morris water maze was used to detect cone visual function. Morris water maze was performed according to the previously published protocol.22 First, mice were trained to habituate to the water and one stable platform. During the first day of the experiment, mice were trained to associate red with a stable platform. During the second day of the experiment, mice had to discriminate between two visible platforms, red (stable and correct) and green (unstable and incorrect). The reflectance of the red-colored cue was well within the spectral window of the wild-type mouse M-opsin, not green. Mice with normal vision could distinguish between red and green.23,24 The number of correct choices, time to stable platform and travelled distance were used for evaluating cone visual function of mice.
Statistical Analyses
Statistical analyses were performed using a statistical software (Statistical Package for the Social Sciences software, version 21.0, NY, USA). The data was analyzed using the Mann–Whitney U-test and Kruskal–Wallis test. Statistical difference was determined by the Mann–Whitney U-test between the two groups. The Kruskal–Wallis test was used for multiple groups. Results were presented as median (min - max). P values < 0.05 were considered significant.
Results
Bright Light Induced IRD in Rdh12−/− Mice
We aimed to detect differences in visual function and retinal structure before and after LD in mice. The abolished expression of Rdh12 was confirmed (Supplementary Figure 1). The differences between overall retinal function in Rdh12−/− and Rdh12+/+ mice were evaluated by ERG. There were no significant differences in the amplitudes of a- or b-waves between Rdh12−/− and Rdh12+/+ mice at 6–8 weeks (Figure 1A and B). After 48 h of illumination, there was a significant reduction in a- and b-waves in Rdh12−/− mice (Figure 1C and D), indicating that Rdh12−/− mice were more susceptible to LD than Rdh12+/+ mice.
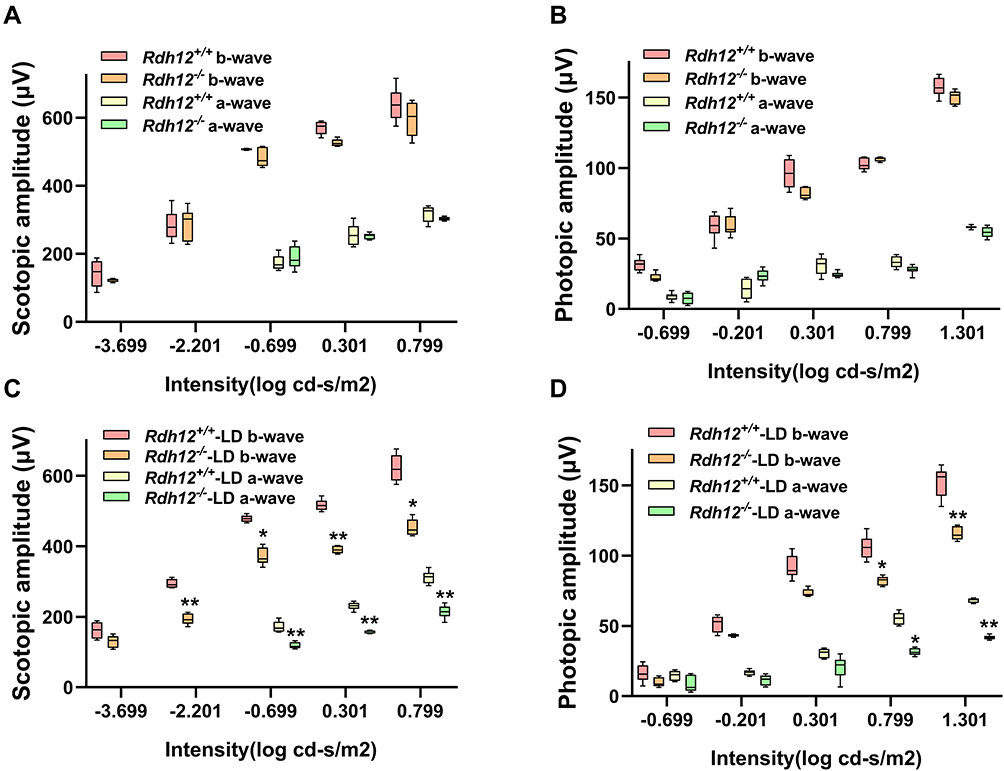 |
Figure 1 Visual dysfunction was induced by bright light in Rdh12−/− mice. (A) A-wave, b-wave amplitudes of scotopic ERG in Rdh12+/+ and Rdh12−/− mice. (B) A-wave, b-wave amplitudes of photopic ERG in Rdh12+/+ and Rdh12−/− mice. (C) The scotopic ERG of Rdh12+/+-LD and Rdh12−/−-LD mice. (D) The photopic ERG of Rdh12+/+-LD and Rdh12−/−-LD mice. Results were presented as median (min - max). n=8–10 per group. *P < 0.05; **P < 0.01 vs Rdh12+/+-LD group. |
We examined the histological differences between Rdh12+/+ and Rdh12−/− mice under natural or bright-light conditions. No apparent differences were noted in the retinal structure of Rdh12+/+ or Rdh12−/− mice over the course of 1 year (Figure 2A and B). After 48 h of illumination, the ONL of Rdh12−/− mice around the central area was reduced; no changes were observed in the same area in Rdh12+/+ mice. No significant changes in the peripheral retina were detected in either genotype. Other retinal layers in Rdh12−/−-LD mice were decreased to some extent (Figure 2C and D). Numerous yellow-white punctate retinal flecks were detected in the fundus of Rdh12−/−-LD mice (Figure 2E), but not in Rdh12+/+-LD mice.
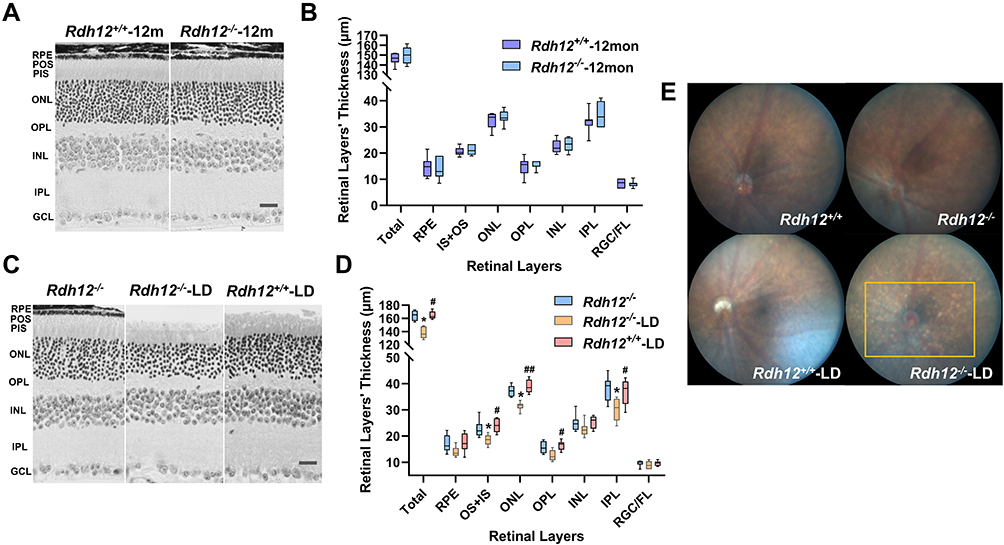 |
Figure 2 Photoreceptor degeneration was induced by bright light in Rdh12−/− mice. (A) Montage of cross-sections of the retina from Rdh12−/− and Rdh12+/+ mice that raised in 12h dark/12h light conditions for 1 year. (B) Quantify the thickness of different layers in Rdh12−/− and Rdh12+/+ mice retina, which were measured at 1000 µm from the optic nerve head. (C) Montage of cross-sections of the retina from Rdh12−/−, Rdh12−/−-LD and Rdh12+/+-LD mice, which exposed to light for 48 h and then dark-adapted for 24h. The ages of mice were 6–8 weeks. (D) Quantify the thickness of different layers in Rdh12−/−, Rdh12−/−-LD and Rdh12+/+-LD mice retinas, which were measured at 1000 µm from the optic nerve head. (E) Fundus camera detected fundus appearance. Results were presented as median (min - max). Scale bar, 20 µm. n=8–12 per group. *P < 0.05 vs Rdh12−/− group. #P < 0.05; ##P < 0.01 vs Rdh12−/−-LD group. Abbreviations: RGC/FL, retinal ganglion cell/fiber layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; IS, inner segment; OS, outer segment; RPE, retinal pigment epithelium. |
Rdh12−/− mice had normal photopic and scotopic ERG kinetics, indicating that these mice have normal phototransduction signaling in the natural state in rod and cone photoreceptors. Bright light induced visual dysfunction and photoreceptor degeneration in Rdh12−/− mice. Yellow-white punctate retinal flecks in the fundus of Rdh12−/− mice revealed retinal related diseases.
Earlier Treatment Was More Beneficial in Rdh12-IRD Mice Retinas
Gene therapy was performed in Rdh12−/− mice before and after the onset of LD-IRD to study the effects of preventive and salvage treatments related to Rdh12 defects on visual dysfunction and photoreceptor degeneration. The Rdh12 mRNA level of treated Rdh12−/− mice reached 70–80% of that in Rdh12+/+ mice after gene therapy (Supplementary Figures 2 and 3). Moreover, the restoration of retinal structure was confirmed (Supplementary Figure 4). Mice underwent ERG analysis of rod- and cone-mediated light responses 3 (Figure 3A–D) and 6 months (Figure 3E–H) post-injection. The amplitudes of rod- and cone-mediated responses in eyes of the preventive group were significantly greater than those in the untreated Rdh12−/−-LD and salvage group, both in photopic and scotopic ERG. In addition, the eyes of the salvage group showed minor differences in activity compared to Rdh12−/−-LD (positive control). The high level of significance obtained in pairwise comparisons of rod and cone function in the preventive and salvage group provides strong evidence for the importance of treatment timepoint. Furthermore, the percentage of cone B-wave and combined rod-cone B-wave remaining in eyes of the preventive and salvage groups was significantly greater than that remaining in Rdh12−/−-LD mice (Figure 3I–L). The remaining was calculated from the ratio of 6- and 3-month ERG level.
 |
Figure 3 Rod- and cone-mediated light responses after gene therapy in four groups. Scotopic (rod-isolated and combined rod-cone) and photopic (cone-isolated) responses were quantified. A-wave, b-wave amplitudes were calculated for injected and noninjected eyes. Results of 3 months (A–D) and 6 months (E–H) post-injection, and 3 months of comparison ERG remaining (I–L) were showed. Results were presented as median (min - max). n=8–10 per group. *P <0.05; **P <0.01 vs Rdh12+/+-LD group. #P < 0.05; ##P <0.01 vs Rdh12−/−-LD group. ΔP < 0.05; ΔΔP <0.01 vs preventive group. |
We hypothesized that the preventive group had greater rod and cone activity because of a decreased degree of retinal degeneration. To confirm this, histological differences between the four groups were examined. The measurement position was shown below (Figure 4A). The results of HE staining showed that the retinal thickness of the preventive group was evidently thicker than that in the salvage group (Figure 4B and C). As the IS layer was significantly thin to measure by itself, we evaluated the combined thickness of IS+OS layers, where the cones and rods were located (Figure 5A and B). The thickness of the ONL was significantly different after LD (Figure 5C and D). These results supported the conclusion that preventive gene therapy can slow down photoreceptor loss. There were no significant differences between the salvage group and Rdh12−/−-LD mice.
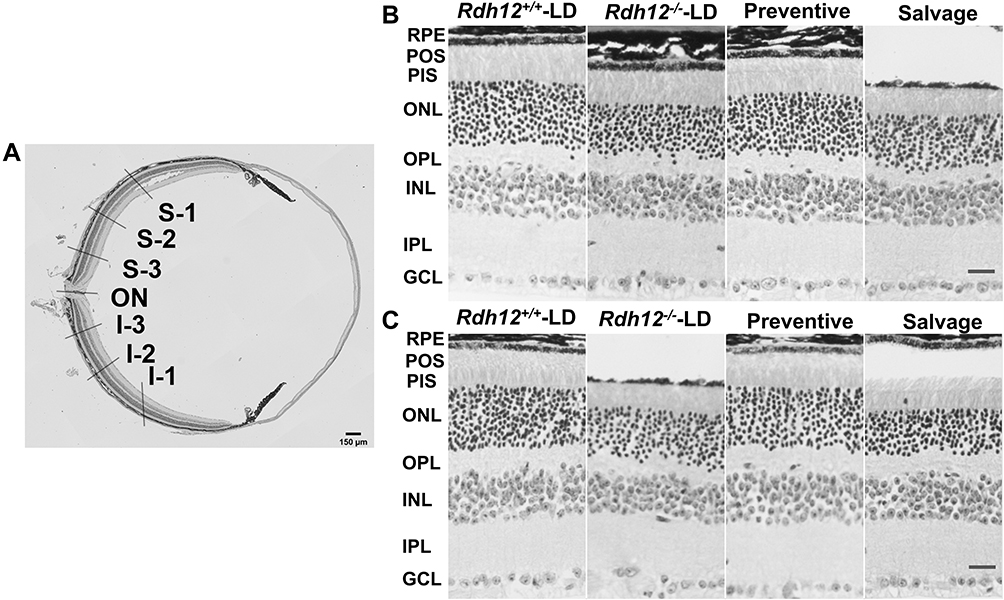 |
Figure 4 Histological differences after gene therapy in four groups. (A) HE staining of mouse retina in the low magnification micrograph. (B) (3 months)/ (C) (6 months): HE staining in S-2 of four groups. Scale bar, 20 µm. |
 |
Figure 5 Quantitative analysis of histological differences after gene therapy in four groups. The thicknesses of the combined IS and OS, ONL were measured every 400 µm across both the superior (S1-3) and inferior hemispheres (I1-3).(A) (3 months)/ (B) (6 months): quantification of the thickness of OS+IS of the retina from four groups. (C) (3 months)/ (D) (6 months): quantification of the thickness of ONL of the retina from four groups. Results were presented as median (min - max). n=8–12 per group. *P <0.05; **P <0.01 vs Rdh12+/+-LD group. #P < 0.05 vs Rdh12−/−-LD group. ΔP < 0.05 vs preventive group. |
Cone-Guided Behavior of Rdh12−/− Mice in the Two-Choice Cued Water Maze
Two-choice cued water maze experiments are highly dependent on cone-mediated vision under photopic conditions. All groups of mice were able to differentiate between the two platforms based on the visual cues. However, the correct rate (Figure 6A), time to platform (Figure 6B), and travelled distance (Figure 6C and D) were significantly different. The preventive group mice performed significantly better than the salvage group and were similar to Rdh12+/+-LD mice. In addition, the salvage group showed minor differences compared to Rdh12−/−-LD (positive control). This confirmed that preventive gene therapy was sufficient to restore cone-mediated visual behavior.
 |
Figure 6 Preventive gene therapy improved cone-mediated visual processing in Rdh12−/−-LD mice. (A) The correct choices for six trials during the discrimination test. (B) Time to platform. (C) Distance travelled. (D) Representative swim paths of four groups. Results were presented as median (min - max). n=10–12 per group. *P <0.05; **P <0.01 vs Rdh12+/+-LD group. ΔP < 0.05 vs preventive group. |
Discussion
The Rdh12−/− mice do not recapitulate the severe phenotypes observed in human patients, such as decreased scotopic and photopic ERG responses or retinal degeneration,3,12 thus presenting several limitations for the study of RDH12 related disease mechanisms and gene therapy. We found that light-induced visual dysfunction and retinal degeneration could establish a better IRD model in Rdh12−/− C57BL/6 RPE65 (Met-450) mice. Our results from a light-induced Rdh12-IRD model showed significant differences in ERG amplitudes and photoreceptor survival compared to Rdh12+/+ mice, indicating that a defect in the Rdh12 gene alone was sufficient to cause visual dysfunction and photoreceptor degeneration. In addition, yellow-white punctate retinal flecks were detected, which is a characteristic fundus phenotype of retinal degeneration,25,26 indicating cone cell27 or RPE cell28 dysfunction. The above phenotypes were consistent with the characteristics of human RDH12-IRD patients.7,8,10
Studies have shown that AAV2/5-hRDH12 gene therapy can maintain visual function under the light exposure that caused significant retinal damage in Rdh12−/− BALB/cJ mice, with no toxic effect found in 54 weeks of testing.6 Our study confirmed and extended previous gene therapy studies. We used AAV2/8-mRdh12 gene therapy in Rdh12−/− C57BL/6 mice before and after the onset of IRD. It has been reported29 that AAV2/8 has higher transduction efficiency than AAV2/5 in the treatment of severe retinal degeneration animal models. C57BL/6 mice have a black fundus, which is similar to the human retina. In addition, it is important to note that retinal degeneration is a typical feature of RDH12-IRD, which has not been involved in previous gene therapy studies.
For the past 10 years, subretinal drug delivery has been widely used in gene therapy, which has been considered to be safe and effective. Nevertheless, there can be complications as with any surgery, including macular holes, choroidal effusions, cataracts, and rarely, permanent vision loss.30 At present, OCT has been used clinically to visually observe the ocular surface passages to reduce the risk of complications during the operation.31,32 In our study, we used OCT imaging to detect mice successfully injected into the subretinal space for gene therapy research. We evaluated the visual function and retinal structure of treated and untreated eyes in Rdh12-IRD mice by ERG and HE staining. ERG permits the quantification of retinal function, and histology is used to define the degree of retinal degeneration.
Preventive gene therapy could significantly improve visual function and slow down photoreceptor degeneration. However, when photoreceptor cell degeneration occurred in mice, salvage gene therapy could neither reverse nor slow down the natural process of degeneration. This means that preventive gene therapy of the primary Rdh12 defect contributes to delayed luminescent photoreceptor degeneration in Rdh12-IRD mice. Gene therapy applied after the onset of retinal degeneration did not seem to improve visual function or increase photoreceptor survival, but it helped slow down the loss of visual function, which mirrored the effects of gene augmentation therapy for some RPE65-LCA patients.9 Clinical research has shown9 that patients with less-advanced retinal disease would have the most improvement by gene augmentation therapy. The practical implication of our results is that early intervention for patients with RDH12-IRD is of great significance to the treatment of the disease. Salvage gene therapy might be improved by refined delivery of agents to increase the activities of cone and rod cells, sequentially or simultaneously with a more advanced gene therapy, such as pro-survival, or antiapoptotic factors.33–35
Given both the bright light-induced Rdh12-IRD mouse model and the progressive development of retinal degeneration in humans with RDH12-IRD, our findings suggest that light may promote the development of RDH12-IRD, and conscious avoidance of light may have a positive effect on delaying the development of retinal degeneration in patients with RDH12 mutations. Adolescence may be a period of relatively rapid progression,5,36 and retained photoreceptors have been found within the central retina in all mutant-RDH12 patients examined,8 which makes it possible for gene therapy to prevent degeneration and improve vision. The future therapeutic strategies should combine gene testing with gene therapy, which emphasizes the preservation of potentially vision and highlights the window of opportunity toward young children.
Conclusion
We found that a defect in Rdh12 alone mice was sufficient to cause visual dysfunction and photoreceptor degeneration, which reproduced the phenotypes seen in RDH12-IRD patients. Preventive gene therapy of the Rdh12 defect helps to delay the later onset of photoreceptor degeneration and maintains visual function in Rdh12-IRD mice, which provides theoretical support for the clinical application of RDH12 gene therapy in the future.
Abbreviations
IRDs, inherited retinal diseases; LD, light-induced retinal degeneration; ERG, electroretinography; RDH12, retinol dehydrogenase 12; LCA13, Leber congenital amaurosis type 13; ONL, outer nuclear layer.
Acknowledgments
The authors thank Xiangtian Zhou from Wenzhou Medical University for providing the support on our electroretinogram detection.
Funding
The study was supported by grants from the Science and Technology Project of Zhejiang Province (2018C37121), Natural Science Foundation of Zhejiang Province (LZ14C060001) and National Natural Science Foundation of China (31371271).
Disclosure
Hongyu Chen’s current affiliation is at National Clinical Research Center for Child Health, The Children’s Hospital of Zhejiang University School of Medicine, Hangzhou 310000, China. Junhui Sun’s current affiliation is at Reproductive Medicine Center, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou 325000, China. The authors report no conflicts of interest in this work.
References
1. Belyaeva OV, Korkina OV, Stetsenko AV, Kim T, Nelson PS, Kedishvili NY. Biochemical properties of purified human retinol dehydrogenase 12 (RDH12): catalytic Efficiency toward retinoids and C9 aldehydes and effects of cellular retinol-binding protein type I (CRBPI) and cellular retinaldehyde-binding protein (CRALBP) on the oxidation and reduction of retinoids. Biochemistry. 2005;44(18):7035–7047. doi:10.1021/bi050226k
2. Kurth I, Thompson DA, Ruther K, et al. Targeted disruption of the murine retinal dehydrogenase gene Rdh12 does not limit visual cycle function. Research support, N.I.H., Extramural. Research support, Non-U.S. Gov’t. Mol Cell Biol. 2007;27(4):1370–1379. doi:10.1128/MCB.01486-06
3. Chen C, Thompson DA, Koutalos Y. Reduction of all-trans-retinal in vertebrate rod photoreceptors requires the combined action of RDH8 and RDH12. Research Support, N.I.H., Extramural. Research Support, Non-U.S. Gov’t. J Biol Chem. 2012;287(29):24662–24670. doi:10.1074/jbc.M112.354514
4. Hofmann L, Tsybovsky Y, Alexander NS, et al. Structural insights into the drosophila melanogaster retinol dehydrogenase, a member of the short-chain dehydrogenase/reductase family. Biochemistry. 2016;55(47):6545–6557. doi:10.1021/acs.biochem.6b00907
5. Fahim AT, Bouzia Z, Branham KH, et al. Detailed clinical characterisation, unique features and natural history of autosomal recessive RDH12-associated retinal degeneration. Br J Ophthalmol. 2019;103(12):1789. doi:10.1136/bjophthalmol-2018-313580
6. Feathers KL, Jia L, Perera ND, et al. Development of a gene therapy vector for RDH12-associated retinal dystrophy. Hum Gene Ther. 2019;30(11):1325–1335. doi:10.1089/hum.2019.017
7. Garg A, Lee W, Sengillo JD, Allikmets R, Garg K, Tsang SH. Peripapillary sparing in RDH12-associated Leber congenital amaurosis. Ophthalmic Genet. 2017;38(6):575–579. doi:10.1080/13816810.2017.1323339
8. Aleman TS, Uyhazi KE, Serrano LW, et al. RDH12 mutations cause a severe retinal degeneration with relatively spared rod function. Invest Ophthalmol Vis Sci. 2018;59(12):5225–5236. doi:10.1167/iovs.18-24708
9. Bainbridge JWB, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. New Eng J Med. 2008;358(21):2231–2239. doi:10.1056/NEJMoa0802268
10. Fahim AT, Thompson DA. Natural history and genotype-phenotype correlations in RDH12-associated retinal degeneration. Review. Adv Exp Med Biol. 2019;1185:209–213. doi:10.1007/978-3-030-27378-1_34
11. Kumaran N, Moore AT, Weleber RG, Michaelides M. Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. Br J Ophthalmol. 2017;101(9):1147–1154. doi:10.1136/bjophthalmol-2016-309975
12. Maeda A, Maeda T, Imanishi Y, et al. Retinol dehydrogenase (RDH12) protects photoreceptors from light-induced degeneration in mice. Research support, N.I.H., Extramural research support, Non-U.S. Gov’t. J Biol Chem. 2006;281(49):37697–37704. doi:10.1074/jbc.M608375200
13. Chrispell JD, Feathers KL, Kane MA, et al. Rdh12 activity and effects on retinoid processing in the murine retina. Research support, N.I.H., Extramural research support, non-U.S. Gov’t. J Biol Chem. 2009;284(32):21468–21477. doi:10.1074/jbc.M109.020966
14. Wenzel A, Reme CE, Williams TP, Hafezi F, Grimm C. The Rpe65 Leu450Met variation increases retinal resistance against light-induced degeneration by slowing rhodopsin regeneration. J Neurosci. 2001;21(1):53–58. doi:10.1523/jneurosci.21-01-00053.2001
15. Cideciyan AV, Jacobson SG, Beltran WA, et al. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t. Proc Natl Acad Sci U S A. 2013;110(6):E517–25. doi:10.1073/pnas.1218933110
16. Nishiguchi KM, Carvalho LS, Rizzi M, et al. Gene therapy restores vision in rd1 mice after removal of a confounding mutation in Gpr179. Nat Commun. 2015;6:6006. doi:10.1038/ncomms7006
17. Georgiadis A, Duran Y, Ribeiro J, et al. Development of an optimized AAV2/5 gene therapy vector for Leber congenital amaurosis owing to defects in RPE65. Gene Ther. 2016;23(12):857–862. doi:10.1038/gt.2016.66
18. Wenzel A, Grimm C, Marti A, et al. c-fos controls the “private pathway” of light-induced apoptosis of retinal photoreceptors. J Neurosci. 2000;20(1):81–88. doi:10.1523/jneurosci.20-01-00081.2000
19. Ueno S, Nishiguchi KM, Tanioka H, et al. Degeneration of retinal on bipolar cells induced by serum including autoantibody against TRPM1 in mouse model of paraneoplastic retinopathy. PLoS One. 2013;8(11):e81507–e81507. doi:10.1371/journal.pone.0081507
20. Poon AWH, Ma EXH, Vadivel A, et al. Impact of bronchopulmonary dysplasia on brain and retina. Biol Open. 2016;5(4):475–483. doi:10.1242/bio.017665
21. Huang F, Zhang L, Wang Q, et al. Dopamine D1 receptors contribute critically to the apomorphine-induced inhibition of form-deprivation myopia in mice. Invest Ophthalmol Vis Sci. 2018;59(6):2623–2634. doi:10.1167/iovs.17-22578
22. Michalakis S, Mühlfriedel R, Tanimoto N, et al. Restoration of cone vision in the CNGA3-/- mouse model of congenital complete lack of cone photoreceptor function. Mol Ther. 2010;18(12):2057–2063. doi:10.1038/mt.2010.149
23. Jacobs GH, Fenwick JC, Calderone JB, Deeb SS. Human cone pigment expressed in transgenic mice yields altered vision. J Neurosci. 1999;19(8):3258–3265. doi:10.1523/jneurosci.19-08-03258.1999
24. Jacobs GH, Williams GA, Cahill H, Nathans J. Emergence of novel color vision in mice engineered to express a human cone photopigment. Science. 2007;315(5819):1723. doi:10.1126/science.1138838
25. Müller PL, Birtel J, Herrmann P, Holz FG, Charbel Issa P, Gliem M. Functional relevance and structural correlates of near infrared and short wavelength fundus autofluorescence imaging in ABCA4-related retinopathy. Transl Vis Sci Technol. 2019;8(6):46. doi:10.1167/tvst.8.6.46
26. Pichi F, Abboud EB, Ghazi NG, Khan AO. Fundus autofluorescence imaging in hereditary retinal diseases. Acta Ophthalmol. 2018;96(5):e549–e561. doi:10.1111/aos.13602
27. Makiyama Y, Ooto S, Hangai M, et al. Cone abnormalities in fundus albipunctatus associated with RDH5 mutations assessed using adaptive optics scanning laser ophthalmoscopy. Am J Ophthalmol. 2014;157(3):558–570.e4. doi:10.1016/j.ajo.2013.10.021
28. Cicinelli MV, Battista M, Starace V, Battaglia Parodi M, Bandello F. Monitoring and management of the patient with Stargardt disease. Clin Ophthalmol. 2019;11:151–165. doi:10.2147/opto.s226595
29. Natkunarajah M, Trittibach P, McIntosh J, et al. Assessment of ocular transduction using single-stranded and self-complementary recombinant adeno-associated virus serotype 2/8. Gene Ther. 2008;15(6):463–467. doi:10.1038/sj.gt.3303074
30. Darrow JJ. Luxturna: FDA documents reveal the value of a costly gene therapy. Drug Discov Today. 2019;24(4):949–954. doi:10.1016/j.drudis.2019.01.019
31. Galantuomo MS, Fossarello M, Cuccu A, et al. Rebound macular edema following oral acetazolamide therapy for juvenile X-linked retinoschisis in an Italian family. Clin Ophthalmol. 2016;10:2377–2382. doi:10.2147/opth.s114568
32. Napoli PE, Nioi M, Mangoni L, et al. Fourier-domain OCT imaging of the ocular surface and tear film dynamics: a review of the state of the art and an integrative model of the tear behavior during the inter-blink period and visual fixation. J Clin Med. 2020;9(3):668. doi:10.3390/jcm9030668
33. Gorbatyuk MS, Knox T, Lavail MM, et al. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc Natl Acad Sci U S A. 2010;107(13):5961–5966. doi:10.1073/pnas.0911991107
34. Dalkara D, Kolstad KD, Guerin KI, et al. AAV mediated GDNF secretion from retinal glia slows down retinal degeneration in a rat model of retinitis pigmentosa. Mol Ther. 2011;19(9):1602–1608. doi:10.1038/mt.2011.62
35. Ohnaka M, Miki K, Gong YY, et al. Long-term expression of glial cell line-derived neurotrophic factor slows, but does not stop retinal degeneration in a model of retinitis pigmentosa. J Neurochem. 2012;122(5):1047–1053. doi:10.1111/j.1471-4159.2012.07842.x
36. Ba-Abbad R, Arno G, Robson AG, Bouras K, Michaelides M. Macula-predominant retinopathy associated with biallelic variants in RDH12. Ophthalmic Genet. 2020;41:612–615.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.