")
Back to Journals » International Journal of Nanomedicine » Volume 16
Gene Therapy for Drug-Resistant Glioblastoma via Lipid-Polymer Hybrid Nanoparticles Combined with Focused Ultrasound
Authors Yang Q , Zhou Y, Chen J, Huang N, Wang Z , Cheng Y
Received 14 October 2020
Accepted for publication 19 November 2020
Published 8 January 2021 Volume 2021:16 Pages 185—199
DOI https://doi.org/10.2147/IJN.S286221
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Israel (Rudi) Rubinstein
Qiang Yang,1 Yanghao Zhou,1 Jin Chen,1 Ning Huang,1 Zhigang Wang,2 Yuan Cheng1
1Department of Neurosurgery, The Second Affiliated Hospital of Chongqing Medical University, Chongqing 400010, People’s Republic of China; 2Institute of Ultrasound Imaging, The Second Affiliated Hospital of Chongqing Medical University, Chongqing 400010, People’s Republic of China
Correspondence: Yuan Cheng
Department of Neurosurgery, The Second Affiliated Hospital of Chongqing Medical University, Chongqing 400010, People’s Republic of China
Tel +86 023 63693540
Email [email protected]
Background: Therapy for glioblastoma (GBM) has always been very challenging, not only because of the presence of the blood-brain barrier (BBB) but also due to susceptibility to drug resistance. Recently, the clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein 9 (CRISPR/Cas9) has revolutionized gene editing technology and is capable of treating a variety of genetic diseases, including human tumors, but there is a lack of safe and effective targeting delivery systems in vivo, especially in the central nervous system (CNS).
Methods: Lipid-polymer hybrid nanoparticles (LPHNs-cRGD) were constructed for efficient and targeting delivery of CRISPR/Cas9 plasmids targeting O6-methylguanine-DNA methyltransferase (MGMT), a drug-resistance gene to temozolomide (TMZ). Focused ultrasound (FUS)-microbubbles (MBs) were used to non-invasively and locally open the BBB to further facilitate gene delivery into glioblastoma in vivo. The gene editing efficiency and drug sensitivity changes were evaluated both in vitro and in vivo.
Results: The gene-loaded LPHNs-cRGD were successfully synthesized and could protect pCas9/MGMT from enzyme degradation. LPHNs-cRGD could target GBM cells and mediate the transfection of pCas9/MGMT to downregulate the expression of MGMT, resulting in an increased sensitivity of GBM cells to TMZ. MBs-LPHNs-cRGD complexes could safely and locally increase the permeability of the BBB with FUS irradiation in vivo and facilitated the accumulation of nanoparticles at the tumor region in orthotopic tumor-bearing mice. Furthermore, the FUS-assisted MBs-LPHNspCas9/MGMT-cRGD enhanced the therapeutic effects of TMZ in glioblastoma, inhibited tumor growth, and prolonged survival of tumor-bearing mice, with a high level of biosafety.
Conclusion: In this work, we constructed LPHNs-cRGD for targeting delivery of the CRISPR/Cas9 system, in combination with FUS-MBs to open the BBB. The MBs-LPHNs-cRGD delivery system could be a potential alternative for efficient targeting gene delivery for the treatment of glioblastoma.
Keywords: CRISPR/Cas9, LPHNs, FUS, microbubbles, glioblastoma
Introduction
Glioblastoma (GBM) is the most common and lethal primary brain tumor in adults. The mean survival of patients with glioblastoma is only 12–14 months, even after treatment with comprehensive therapy including surgery, chemotherapy, and/or radiotherapy.1,2 Temozolomide (TMZ), an alkylating agent, is the main drug therapy for patients with glioblastoma because TMZ can easily cross the blood-brain barrier (BBB) and inhibit GBM cell proliferation, with low levels of systematic toxicity.3 However, the therapeutic efficacy of TMZ is often limited by O6-methylguanine-DNA methyltransferase (MGMT), which can repair the O6-methylguanine lesions caused by TMZ, inducing TMZ resistance in GBM cells.4,5 Downregulating the expression of MGMT can reduce chemoresistance and increase the therapeutic effects of TMZ in GBM cells.6,7
The clustered regularly interspaced short palindromic repeat (CRISPR)-associated protein 9 (CRISPR/Cas9) has been considered one of the most significant gene editing technologies. In this system, the Cas9 endonuclease, under the direction of guide RNA (gRNA), can recognize and cleave specific DNA sequences, generating double-stranded breaks (DSBs), leading to insertions, deletions or mutations at target genomic locations.8,9 With high efficiency, specificity, and simplicity, the CRISPR/Cas9 system shows tremendous potential for various cancer treatments via genome editing in cells and animals.10,11 However, current delivery systems for CRISPR/Cas9 are mostly viral vectors such as lentiviruses (LVs), adenoviruses (AVs), which can be immunogenic and carcinogenic, especially in vivo.12,13 Thus, non-viral vectors have emerged as important alternatives for gene delivery with the advantages of good biocompatibility, low immunogenicity, and easy modification.14
Nanoparticles, such as lipid- and polymer-based systems, have made significant progress in targeting drug delivery over the past decades. Lipids, the traditional vectors, have many favorable properties such as high biological compatibility, low toxicity, and ease of surface modification, but their instability, poor bioavailability, and low drug-loading hinder their clinical use.15 Alternatively, polymeric nanoparticles (such as PLGAs) have high stability in systemic circulation, a controlled-release profile, and high drug-loading efficiency. However, the biocompatibility of synthetic polymers remains a significant concern for therapy.16–18 Therefore, lipid-polymer hybrid nanoparticles (LPHNs) were investigated in an effort to combine the complementary advantages of lipids and polymer nanoparticles.19,20 LPHNs are typically composed of three different functional components: a polymer core encapsulating therapeutic agents; an inner lipid layer surrounding the polymer core, which can confer biocompatibility and retain therapeutic agents inside the polymer core; and an outer lipid-PEG layer coating the lipid layer to extend the blood circulation time and enhance steric stabilization.21 Additionally, the PEG chains can be further modified with various bioactive molecules to meet different needs, such as the use of antibodies for active targeting therapy for tumors.22 As a new generation delivery vector, use of LPHNs has been expanded to deliver various diagnosis and treatment agents, such as Dox/ICG,23 siRNA for gene therapy,24 diagnostic imaging agents.25
However, the blood-brain barrier (BBB) presents a major obstacle for glioblastoma therapy.26 The BBB blocks more than 98% of material to prevent toxic substances from entering the central nervous system (CNS)and maintain brain homeostasis. However, this can also prevent the diffusion of effective therapeutic agents from entering the brain.27 Increasing permeability of the BBB is an important method to enhance drug delivery. Focused ultrasound (FUS) shows tremendous potential in this field, especially when combined with microbubbles (MBs).28 Compared with other strategies, such as hypertonic solutions and direct injection, FUS in the presence of MBs, through the cavitation effect, can non-invasively, reversibly, and site-specifically increase the permeability of the BBB without causing damage to the surrounding brain tissue.29,30 Thus, FUS combined with MBs is an important alternative strategy to permeabilize the BBB and facilitate the delivery of therapeutic agents into the brain.
In this study, we developed LPHNs nanocarriers loaded with CRISPR/Cas9 plasmids targeting the MGMT gene and modified with the cRGD peptide which targets the overexpressed integrin αvβ3 receptors in tumor cells (LPHNspCas9/MGMT-cRGD) and evaluated the gene editing efficiency in vitro. Subsequently, we constructed MBs-LPHNspCas9/MGMT-cRGD complexes for BBB-opening and targeting gene delivery in vivo under FUS irradiation. The results showed that the complexes can execute effective gene delivery and restore the sensitivity of GBM cells to TMZ, with a high level of biosafety and biocompatibility both in vitro and in vivo. This work provides a promising strategy to construct a safe, efficient and multi-functional complex for the targeted delivery of CRISPR/Cas9 and the treatment of TMZ-resistant glioblastoma.
Materials and Methods
Materials and Cells
PLGA (lactide: glycolide = 50:50, Molecular Weight: 12,000) was purchased from Jinan Daigang Biological Material Company (Shandong, China). 1.2-Dipalmitoyl-sn-glycero-3-phosphatidylcholine (DPPC), 3beta [N-(N’,N’-dimethylaminoethane)-carbamoyl] cholesterol (DC-Chol), and cholesterol were provided by Avanti Polar Lipids, Inc. (Alabaster, AL, USA). 1.2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] (DSPE-PEG2000), DSPE-PEG2000-cRGD and DSPE-PEG2000-Biotin were obtained from Xi’an Ruixi Biological Technology Co., Ltd. (Xi’an, China). Lipofectamine 3000 was purchased from Invitrogen (Carlsbad, CA, USA). Streptavidin was bought from Solarbio (Beijing, China). DiI and DiO were purchased from Beyotime Biotechnology Company (Shanghai, China). DAPI and TMZ were purchased from Mengbo Biotechnology Co., Ltd. (Chongqing, China). DiR was obtained from Sigma-Aldrich Chemical Co. (St Louis, MO, USA). The cell Counting Kit-8 (CCK-8) was purchased from Dojindo Laboratories (Kumamoto, Japan). All reagents and solvents were of analytical grade.
The human glioblastoma cell line T98G was obtained from the American type culture collection (ATCC) and maintained in MEM (Hyclone, USA) supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% penicillin-streptomycin. Cells were incubated in a 37°C incubator under 5% CO2 atmosphere.
Three pairs of candidate dual-gRNA/Cas9 plasmids targeting MGMT gene (pCas9/MGMT, containing an EGFP cassette) were purchased from GenePharma (Shanghai, China). A blank plasmid was used as control (pctrl). The editing effects of the plasmids were tested via PCR and Sanger sequencing in 293T cells (Figure S1–3 and Table S1). Plasmids with effective editing efficiency were selected for the subsequent experiments.
Preparation and Characterization of Lipid-Polymer-Hybrid PEG Nanoparticles (LPHNs-cRGD)
LPHNs-cRGD were prepared via nanoprecipitation method.31,32 Briefly, 5 mg of PLGA was dissolved in 1 mL of acetone as an organic phase. Lecithin, DSPE-PEG-cRGD, and DSPE-PEG-biotin (at a molar ratio of 7:1.5:1.5) were distributed in 20 mL of 4% ethanol solution as an aqueous phase. The organic solution was then added dropwise to the aqueous solution under vigorous stirring. The nanoparticles self-assembled and the acetone evaporated after 2–3 hours with slow and continuous stirring at room temperature. The LPHNs-cRGD were then washed three times in phosphate-buffered saline (PBS) to remove remaining organic solvents using Amicon tubes (Molecular weight cut off: 10 kDa, Millipore), diluted in 1 mL of PBS, and stored at 4°C. For gene-loaded LPHNs-cRGD, 300 μg of plasmids and 1mg DC-chol were co-incubated for 30 min previously in acetone containing PLGA. The same method was used to synthesize non-targeting LPHNs using DSPE-PEG.
The particle size, zeta potential and polydispersity index (PDI) were determined using a Zetasizer Nano ZS instrument (Malvern Instruments, UK). The encapsulation efficiency of the pCas9/MGMT in LPHNs-cRGD was determined from the ratio of the amount of encapsulated pCas9/MGMT to the amount of pCas9/MGMT initially added using PicoGreen dsDNA Quantitation Kit (Thermo Fisher, USA) according to the manufacturer’s instruction. The structure of the plasmid in nanoparticles was observed through agarose electrophoresis after extracted from nanoparticles. Fluorescence microscope (Nikon, Japan) was used to observe the DiI-labeled LPHNs-cRGD. The morphology and structure of the LPHNs-cRGD were observed via transmission electron microscopy (TEM, Hitachi H-7600, Japan). Stability of nanoparticles was examined in MEM with 10% FBS (v/v) at 37°C, and the size of nanoparticles was monitored as above at different time intervals.
Release of pCas9/MGMT in vitro
In vitro release of pCas9/MGMT from nanoparticles was carried out in PBS at 37°C. 5 mg of pCas9/MGMT-loaded nanoparticles was suspended in 1 mL PBS and shaken slowly at 37°C. At a predetermined time, the solution was centrifuged and the supernatants were collected to measure the released pCas9/MGMT using the Picogreen assay as mentioned above. Nanoparticles were then immediately resuspended in fresh PBS and shaken in the incubator at 37°C.
Protection from DNase I
DNase I was used to investigate whether LPHNs-cRGD can protect pCas9/MGMT from enzymatic digestion. In brief, 100 μL nanoparticles were suspended in reaction buffer containing 1U of DNase I (Beyotime, Shanghai, China) per microgram of DNA. The solution was incubated at 37°C for different time. And then nanoparticles were separated by high-speed centrifugation, dissolved in 1 mL of chloroform followed by adding 500 μL TE buffer. The mixture was rapidly agitated using a magnetic stirrer for 2 h at room temperature to extract pCas9/MGMT from nanoparticles. After centrifugation, the supernatant containing extracted pCas9/MGMT was collected. As a control, 50 μL naked pCas9/MGMT was incubated with DNase I at the above concentration for 3 min at 37°C. Then, samples were added with DNA loading buffer before applied to a 0.8% agarose gel in 0.5 x TAE buffer containing gold-view for 40 min at 70 mV, and images were observed using Bio-Rad imaging system (Hercules, CA, USA).
Cytotoxicity and Targeting Ability of LPHNs-cRGD in vitro
5 × 103 T98G cells per well were seeded in 96-well plates and incubated overnight, then cells were added with different concentrations of LPHNs or LPHNs-cRGD and further incubated for 48 h. Untreated cells in culture medium were used as the negative control groups. After incubation, the cells were added with 10μL of CCK-8 solution and further incubated for 1–2 h. Then the absorbance of solution in each well was measured at 450 nm using a microplate reader (Synergy HT, Bio-Tek, USA). The cell viability (%) was calculated according to the instructions of the CCK-8 assay.
The targeting ability of the LPHNs-cRGD toward T98G cells was observed using both Laser scanning confocal microscope (LSCM) and flow cytometry (FCM). T98G cells were seeded into confocal culture dishes with a density of 1.0 × 105 cells per dish and incubated overnight. The cells were then divided into three groups: a control group (PBS), an LPHNs group and an LPHNs-cRGD group. After 24 h of incubation, the LPHNs group was incubated with 10 μL DiI-labeled LPHNs emulsions, the LPHNs-cRGD group was mixed with 10 μL DiI-labeled LPHNs-cRGD emulsions. After 4 h of coincubation, the cells in the dishes were rinsed 3 times with PBS and fixed with 4% paraformaldehyde for 15 min at room temperature, then stained with DAPI for 5 min. After rinsing again, the fixed cells were imaged using LSCM. For FCM, T98G cells were seeded into 6-well plates with a density of 2.0 × 105 cells per dish and incubated overnight. The cells were then mixed with different nanoparticles as previously described. After 4 h of co-incubation, the cells were washed with PBS, followed by digestion with trypsin, centrifugation, and suspension in 500 μL PBS. Finally, the fluorescence intensity of the cellular uptake was detected via FCM.
Transfection Efficiency and Gene Editing with LPHNs-cRGD in vitro
Fluorescence microscopy and FCM were used to evaluate the transfection efficiency. T98G cells were seeded into 6-well plates and incubated overnight, respectively. The cells were then treated with naked pCas9/MGMT, LPHNspCas9/MGMT, and LPHNspCas9/MGMT -cRDG, respectively. Lip3000 was used as a positive control according to the product manual. The amount of pCas9/MGMT in all of the groups was 5 μg. After incubation for 48 h, EGFP expression was observed with a fluorescence microscope, and then cells were collected for FCM as described above.
Western blotting was carried out to test the expression of MGMT protein after transfection with different nanoparticles. T98G cells were seeded into 6-well plates and incubated overnight. LPHNspCas9/MGMT and LPHNspCas9/MGMT-cRGD (5 μg of pCas9/MGMT) were then added to the medium and incubated for 48 h. Untreated cells, PBS and LPHNspctrl-cRGD were used as negative controls. Whole-cell protein was isolated using RIPA lysis buffer (Beyotime, Shanghai, China) with phenylmethylsulfonyl fluoride and quantified by a BCA assay. Isolated protein was separated on SDS-PAGE gel and transferred to polyvinylidene fluoride membranes. The membranes were incubated with the appropriate antibodies against MGMT (Abcam, USA) and subsequently incubated with secondary anti-rabbit IgG-HRP antibodies (Beyotime, China). Images were obtained using the Bio-Rad imaging system.
CCK-8 and Apoptosis Assays for Chemosensitivity Testing
The sensitivity of the GBM cells to TMZ after treatment with LPHNspCas9/MGMT in vitro was evaluated using CCK-8 and apoptosis assays. The T98G cells were seeded into 96-well plates and incubated overnight. The cells were randomly divided into five groups: a negative control group, an LPHNspCas9/MGMT-cRGD group, a TMZ group, an LPHNspCas9/MGMT + TMZ group, and an LPHNspCas9/MGMT-cRGD + TMZ group. In the LPHNspCas9/MGMT-cRGD and TMZ groups, cells were treated with LPHNspCas9/MGMT-cRGD or TMZ alone. In the LPHNspCas9/MGMT + TMZ and LPHNspCas9/MGMT-cRGD + TMZ groups, the cells were first transfected with LPHNspCas9/MGMT or LPHNspCas9/MGMT-cRGD for 48 h, and then the culture medium was replaced with fresh medium containing TMZ (150 μM) and incubated continuously for 36 h. The amount of pCas9/MGMT in all of the groups was 0.2 μg. Finally, cell viability was evaluated using the CCK-8 assay as previously described.
For apoptosis assay, T98G cells were seeded into 6-well plates, divided into five groups, and treated as described above. The amount of pCas9/MGMT in all of the groups was 5 μg. After co-incubation, the cells were washed three times with PBS, digested, centrifuged, and resuspended in 500 μL of PBS. Finally, cells were subjected to flow cytometry analysis.
Preparation of Microbubbles (MBs) and MBs-LPHNs-cRGD
Microbubbles were prepared via the lipid film hydration method. Briefly, DPPC, DSPE-PEG-biotin, and cholesterol (at a mass ratio of 5:2:1) were dissolved in 5 mL of chloroform in a round-bottomed flask and processed in a rotatory evaporator in a water bath at 50°C to form lipid film. The dried lipid film was rehydrated with 0.5 mL of PBS and glycerine solution (at a volume ratio of 9:1) followed by addition of perfluoropropane (C3F8). The mixture was rapidly mechanically vibrated for 50 s. Finally, the biotinylated MBs were purified by centrifugation and stored at 4°C for further use.
The MBs and LPHNs-cRGD were bound together via the biotin-avidin linkage. Briefly, 10 μL of MBs was diluted in 1 mL PBS and were added with excessive avidin. After 30 min of incubation at room temperature, the mixture solution was washed to remove unreacted agents. The biotin-avidin MBs were added with LPHNs-cRGD emulsions and incubated for 1 h to obtain MBs-LPHNs-cRGD. The MBs-LPHNs were prepared using the same method. The particle size and zeta potential were determined as above. MBs-LPHNs-cRGD fabricated by DiO-labeled LPHNs-cRGD and DiI-labeled MBs were observed by fluorescence microscope to confirm the connection of the two nanoparticles.
Opening the Blood Brain Barrier
NOD-SCID mice (4–6 weeks) were purchased from Beijing HFK Bioscience (Beijing, China) and raised in a suitable environment in the Experimental Animal Center of Chongqing Medical University. The animals received care in accordance with the Guide for the Care and Use of Laboratory Animals. The procedures were approved by the Ethics Committee of The Second Affiliated Hospital of Chongqing Medical University.
Mice were anesthetized with pentobarbital and immobilized on a stereotaxic device. The hair on top of the heads was shaved. The focused ultrasound probe was placed above the heads of mice and vertically fixed on the stereotactic frame to ensure the focus position was 2 mm to the right laterally and 0.5 mm anterior to bregma and was 2.5 mm under the dura. The mice were randomly divided into different groups according to the irradiation times and amounts of MBs-LPHNs-cRGD. After injected with MBs-LPHNs-cRGD through the tail vein, the mice were exposed to FUS irradiation immediately followed by injected with 2% Evans blue dye (EB). 1 h later, the mice were sacrificed and perfused transcardially with saline, and the brain tissue was removed and coronally incised for digital photography and H&E staining.
Orthotopic Xenograft Model
NOD-SCID mice were anesthetized with pentobarbital and immobilized on a stereotaxic device. The T98G cells (2 × 105) were intracranially injected into the striatum of the mice using the stereotaxic device (2 mm right laterally, 0.5 mm anterior to the bregma, and 2.5 mm depth from the dura). Tumor growth was monitored by a 7.0T MRI scanner (Biospec70/20USR, Bruke, Germany) seven days after implantation.
Targeting Ability, Distribution and Biosafety in vivo
Orthotopic xenograft NOD-SCID mice were randomly divided into 4 groups. Group 1 was treated with DiR-labeled LPHNs-cRGD. Group 2 was treated with DiR-labeled MBs-LPHNs-cRGD. Group 3 was treated with DiR-labeled MBs-LPHNs followed by FUS irradiation. Group 4 was treated with DiR-labeled MBs-LPHNs-cRGD and immediately exposed to FUS irradiation. 2 h and 24 h later, images of mice were acquired using a fluorescence imaging system (Berthold Night OWL LB 983, Germany) after hair removal. Then, brains and major organs were collected for ex vivo imaging. The fluorescence intensity at the tumor sites and major organs was calculated by the imaging system.
Mice were divided into two groups (a control and an MBs-LPHNs-cRGD group). Saline and MBs-LPHNs-cRGD emulsions were intravenously injected into the mice, respectively. The mice were sacrificed seven days after injection. The major organs (liver, spleen, kidneys, heart, and lungs) were harvested and fixed via polyoxymethylene for H&E staining.
Therapeutic Effect in vivo
Mice with orthotopic T98G glioblastoma were divided into five groups. Group 1 (control) was treated with saline. Group 2 was treated with TMZ alone. Group 3 was treated with MBs-LPHNspCas9/MGMT-cRGD and TMZ. Group 4 was treated with MBs-LPHNspCas9/MGMT followed by FUS irradiation and TMZ. Group 5 was treated with MBs-LPHNspCas9/MGMT-cRGD followed by FUS exposure and TMZ. Nanocomplexes (MBs-LPHNspCas9/MGMT-cRGD or MBs-LPHNspCas9/MGMT) were intravenously injected and then the mice were exposed to FUS irradiation immediately or not. Two days later, TMZ (50 mg/kg) was administered via daily intraperitoneal injection for 5 consecutive days. Three mice in each group were scanned with a 7.0T MRI to monitor tumor volume every seven days and then all mice were administered with the next cycle of treatment. The mice were sacrificed when they demonstrated severe neurological symptoms.33 Tumor volume was calculated as follows: volume = [length (max layer) × width (max layer)] × layers × 0.8/2 (0.8 mean the slices interval). The survival of each mouse was recorded and the median survival for each group was calculated.
Statistical Analysis
All experiments were performed in triplicate. Statistical analysis was performed using SPSS software (SPSS version 23.0) and GraphPad software (GraphPad Prism 8.0). Data are presented as the mean ± standard deviation (SD). p < 0.05 were considered to be statistically significant.
Results
Formulation and Characterization of the LPHNs-cRGD
The diameters and zeta potentials of the LPHNs-cRGD were 179.6 ± 44.82 nm and 29.6 ± 4.33 mv, respectively, with a small PDI (0.048) (Figure 1A and B). And the encapsulation efficiency was 76.5 ± 7.2%. During the preparation process, the structure of the pCas9/MGMT was unaffected and remained intact (Figure S4). After staining by DiI, the LPHNs-cRGD exhibited uniform red fluorescence signal (Figure 1C). TEM showed that the LPHNs-cRGD had uniform spherical shape and typical core-shell structures (Figure 1D and E). The inner, darker region with high electron density was indicative of the polymer core, and the outer, brighter section indicated the lipid layer. In addition, the nanoparticle size in MEM containing 10% FBS did not significantly increase, indicating the excellent colloidal stability of nanoparticles under physiological conditions (Figure S5).
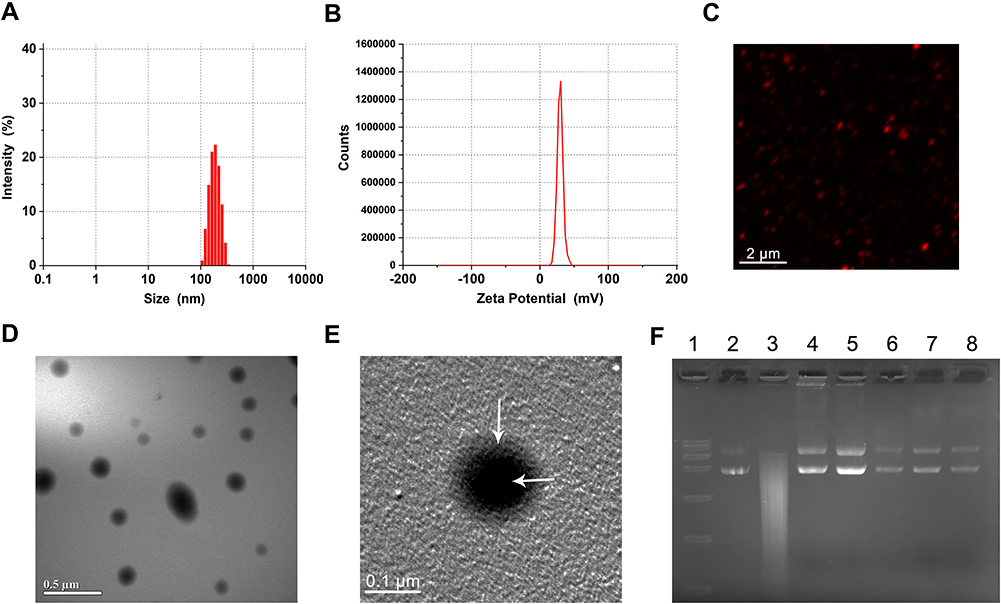 |
Figure 1 Characterization of nanoparticles. Abbreviations: LPHNs, lipid–polymer hybrid nanoparticles; cRGD, cyclic arginine-glycine-aspartic acid; DiI, 1.1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate; TEM, transmission electron microscopy; pCas9/MGMT, Cas9/MGMT plasmid; DNase I, Deoxyribonuclease I. Notes: (A) Particle size and (B) zeta potential of LPHNs-cRGD. (C) Fluorescent image of DiI-labeled LPHNs-cRGD. (D) and (E) TEM image of LPHNs- cRGD, White arrow: lipid layer and polymer core of nanoparticles, respectively. (F) Protection assay of LPHNs-cRGD from DNaseI. Lane 1, DNA maker; Lane 2, untreated pCas9/MGMT; Lane 3, naked pCas9/MGMT after incubation with DNase I for 3min; Lanes 4–8 correspond to the extracted pCas9/MGMT from LPHNs-cRGD after incubation with DNase I for 3 min, 15 min, 30 min, 1 h and 2 h, respectively. |
Release of Plasmids in vitro and Protection from DNase I
A release study of pCas9/MGMT-loading LPHNs and LPHNs-cRGD was carried out in a PBS solution. As shown in Figure S6, the nanoparticles exhibited a sustained release profile with a short fast release period in the first hours, followed by a long-term sustained release period. This may be related to that most of the pCas9/MGMT was encapsulated within the nanoparticles, which must diffuse from the PLGA matrix into the aqueous medium.
To test whether the LPHNs-cRGD could protect the pCas9/MGMT from enzymatic digestion, the nanoparticles were exposed to DNase I for varying lengths of time; then, pCas9/MGMT was extracted for agarose electrophoresis. As (Figure 1F) shows, the naked pCas9/MGMT gradually degraded within 3 min of incubation with the DNase I. In contrast, pCas9/MGMT in the LPHNs-cRGD remained intact throughout various lengths of incubation with DNase I. The increased ratio of circular plasmids might be related to the long duration and rapidity of stirring when extracting the pCas9/MGMT from nanoparticles. This demonstrated that the LPHNs-cRGD could protect pCas9/MGMT from nuclease digestion.
Targeting Ability and Biosafety Study in vitro
The targeting ability of LPHNs-cRGD to T98G cells was evaluated via LSCM and FCM (Figure 2A and Figure S7). LSCM images showed strong red fluorescence in the T98G cells treated with the LPHNs-cRGD, which was much stronger than that in the LPHNs group. The results of the FCM analysis also showed that the fluorescence intensity in the LPHNs-cRGD group was higher than that in the LPHNs group. Those suggested that the nanoparticles with cRGD peptides were more readily taken up than the nanoparticles without cRGD.
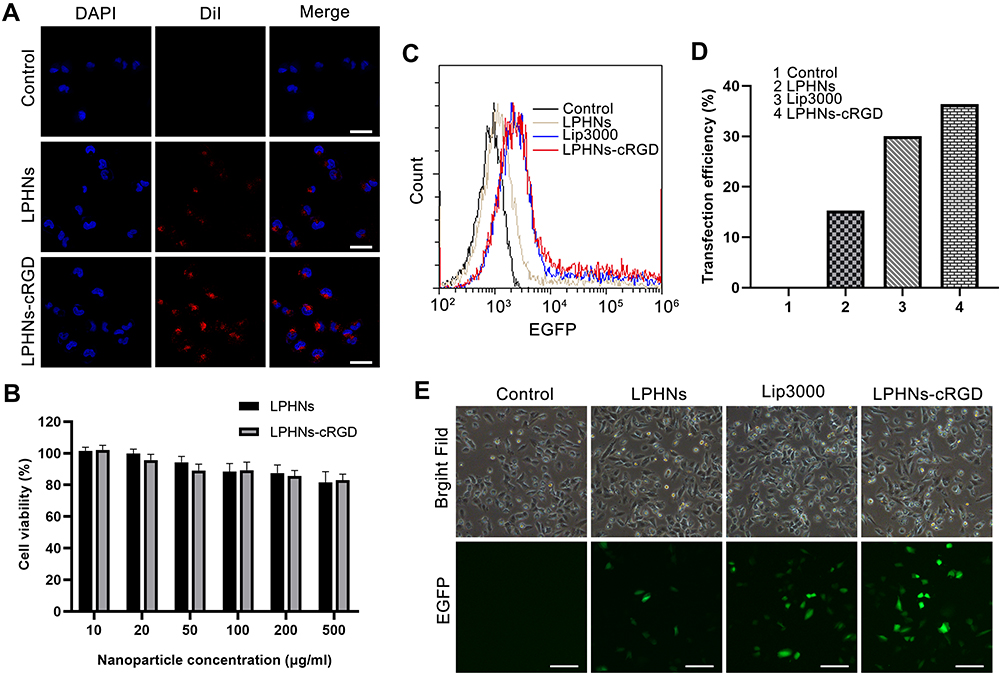 |
Figure 2 Targeting ability, biosafety, and transfection efficiency of LPHNs-cRGD in vitro. Abbreviations: LPHNs, lipid–polymer hybrid nanoparticles; cRGD, cyclic arginine-glycine-aspartic acid; LSCM, laser scanning confocal microscopy; DiI, 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate; FCM, flow cytometry; EGFP, enhanced green fluorescent protein; pCas9/MGMT, Cas9/MGMT plasmid. Notes: (A) The LSCM images of T98G cells with different treatments (Control, DiI-labeled LPHNs, DiI-labeled LPHNs-cRGD). The scale bar is 20 μm. (B) Cell viability at various concentrations of LPHNs and LPHNs-cRGD in T98G cells after 48 h (n=3). (C) and (D) FCM and statistical analysis of EGFP-positive T98G cells after transfected with different pCas9/MGMT formulations for 48 h. (E) Microscopy images of EGFP expression in T98G cells after transfected with different pCas9/MGMT formulations for 48h. The scale bar is 50 μm. |
The in vitro biosafety of the nanoparticles was assessed using a CCK-8 assay to verify that the nanoparticles were biocompatible and non-toxic to cells. As shown in Figure 2B, the results of this assay demonstrated that the cell viability was >80% after exposure to different concentrations of nanoparticles, indicating that nanoparticles were safe for cells.
Transfection Efficiency
To examine whether the pCas9/MGMT in the LPHNs-cRGD could be successfully transfected into GBM cells, FCM and fluorescence microscopy were used to observe the expression of EGFP. As shown in Figure 2C and D, FCM results showed no EGFP fluorescence signal in T98G cells treated with pCas9/MGMT alone, since naked DNA could not pass through the cell membrane. EGFP fluorescence signal was detected in the group transfected with the LPHNs, LPHNs-cRGD, and Lip3000. After an additional 48 h, the transfection efficiency of the LPHNs-cRGD group was 36.39%, which was slightly higher than that in the Lip3000 group (30.09%), and much higher than that in the LPHNs group (15.27%). The relatively low expression of EGFP might due to the large size of the pCas9/MGMT.
We then observed EGFP expression using fluorescence microscopy at 48 h after transfection (Figure 2E). The cells treated with LPHNs-cRGD and Lip3000 showed significant EGFP signal, while dim green fluorescence was observed in the LPHNs group and no EGFP signal in the naked-plasmid group. These results indicated that the pCas9/MGMT encapsulated in the LPHNs-cRGD were successfully introduced into the T98G cells.
Genome Editing and Chemosensitivity Testing in vitro
After 48 h of incubation with PBS, LPHNspctrl-cRGD, LPHNspCas9/MGMT, and LPHNspCas9/MGMT-cRGD, Western blotting assays were conducted to determine the MGMT protein expression. As shown in Figure 3A, the LPHNspCas9/MGMT-cRGD group induced significant downregulation of MGMT protein expression, while the LPHNspCas9/MGMT group had a slight decrease, and control groups showed a negligible decrease.
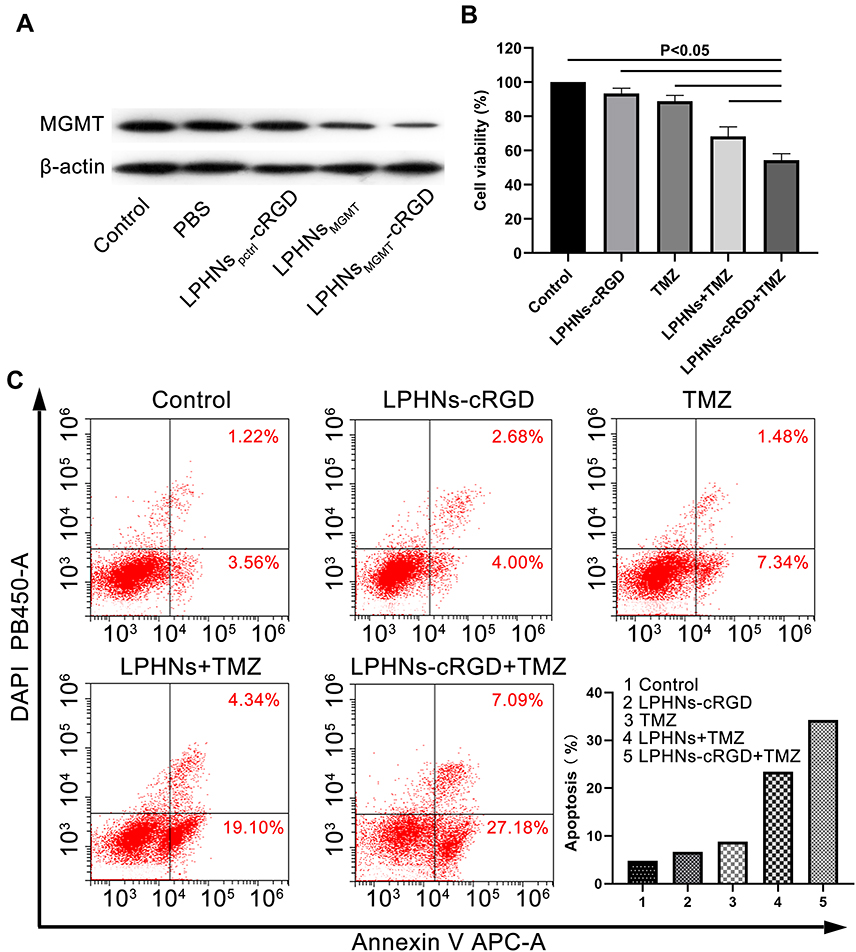 |
Figure 3 Chemosensitivity testing induced by LPHNspCas9/MGMT-cRGD. Abbreviations: LPHNs, lipid–polymer hybrid nanoparticles; pCas9/MGMT, Cas9/MGMT plasmid; cRGD, cyclic arginine-glycine-aspartic acid; MGMT, O6-methylguanine-DNA methyltransferase; TMZ, temozolomide; FCM, flow cytometry. Notes: (A) Western blotting for analyzing the MGMT expression in T98G cells after different treatments. (B) Cell viability of T98G cells after different treatments including control, LPHNspCas9/MGMT-cRGD, TMZ, LPHNspCas9/MGMT + TMZ and LPHNspCas9/MGMT-cRGD + TMZ (n=4). (C) FCM analysis of apoptosis after different treatments. |
Then, we investigated whether the LPHNspCas9/MGMT-cRGD could re-sensitize the T98G cells to TMZ using cell viability and apoptosis assays. As shown in Figure 3B, when only exposed to TMZ or LPHNspCas9/MGMT, the viability of cells did not decrease obviously compared to the negative control. Combination treatment of the LPHNspCas9/MGMT + TMZ or LPHNspCas9/MGMT-cRGD + TMZ resulted in significantly decreased cell viability, especially in the LPHNspCas9/MGMT-cRGD + TMZ group. We also used FCM to quantify the apoptosis percentage of cells with different treatments (Figure 3C). Single-agent treatment caused no significant changes in the apoptosis assay. The percent of apoptotic cells was significantly higher in the combination treatment group in which the LPHNspCas9/MGMT-cRGD + TMZ treatment induced more apoptosis compared with LPHNspCas9/MGMT + TMZ due to the targeting ability of the cRGD (23.44% vs 34.27%).
Local BBB Disruption
Before the BBB-opening experiment, we generated lipid microbubbles (MBs) and combined with LPHNs-cRGD via avidin–biotin interactions.34 Figure 4A shows a microscopy image of the MBs, with a density of 3.95 ± 0.89 × 109/mL, a diameter of 1845.2 ± 366.7 nm, and a zeta potential of -16.2 ± 6.82 mV. The diameter of the MBs-LPHNs-cRGD complexes was 2197.35 ± 448.7 nm, and the zeta potential was -7.49± 3.14 mV (Figure 4C and D). Figure 4B shows a fluorescent microscopy image of the MBs-LPHNs-cRGD complexes illustrating that the LPHNs-cRGD adhered to the MBs’ surface.
 |
Figure 4 BBB-opening experiment in vivo. Abbreviations: MBs, microbubbles; LPHNs, lipid–polymer hybrid nanoparticles; cRGD, cyclic arginine-glycine-aspartic acid; DiO, 3.3’-dioctadecyloxacarbocyanine perchlorate; DiI, 1,1’-dioctadecyl-3,3,3’,3’- tetramethylindocarbocyanine perchlorate; EB, Evan blue; BBB, blood brain barrier; FUS, focused ultrasound; H&E, hematoxylin-eosin. Notes:(A) Bright-field microscopy image of MBs, The scale bar is 10 μm. (B) Fluorescence image of MBs-LPHNs-cRGD complexes. LPHNs-cRGD was stained by DiO (green, left), MBs were stained by DiI (red, middle), the green dots around the red bubble indicated that LPHNs-cRGD and MBs were bound together (merged, right). The scale bar is 1 μm. (C) Particle size and (D) zeta potential of MBs-LPHNs-cRGD complexes. (E) Digital photography of EB leakage (top two rows) and H&E staining of corresponding black rectangle region (bottom two rows) for BBB – opening induced by FUS-assisted MBs-LPHNs-cRGD. White arrow: erythrocyte extravasation. The FUS radiation time and amounts of MBs-LPHNs-cRGD were 3 min and 2 x 106 (line 1); 3 min and 4 x 106 (line 2); 5 min and 4 x1 06 (line 3) and 3 min and 8 x1 06 (line 4), respectively. The scale bar is 2 mm for digital photography and 50 μm for H&E staining, respectively. |
After preparing the MBs-LPHNs-cRGD complexes, we conducted the BBB-opening experiment in vivo. According to our previous studies, we used a focused ultrasound intensity of 1.84 W and investigated the effect of different irradiation times and amounts of MBs-LPHNs-cRGD complexes on the permeability of the BBB via EB dye staining.35 H&E staining was used to observe whether there were any damages to brain tissue (Figure 4E). Upon administration of 2 × 106 MBs-LPHNs-cRGD complexes and 3 min of irradiation, no EB leakage was observed in the brain, indicating a lack of BBB permeability. Increasing the amounts of MBs-LPHNs-cRGD complexes to 4 × 106 resulted in concentrated EB leakage in the irradiated brain region, indicating that the local BBB was opened. The H&E staining showed no erythrocyte extravasation or pathological changes in the irradiated region. At an exposure time of 5 min and 4 × 106 MBs-LPHNs-cRGD complexes, the BBB-opening region extended toward a wider region, with a small amount of erythrocyte extravasation according to the H&E staining. When the radiation conditions were 3 min and 8 × 106 MBs-LPHNs-cRGD, broad BBB-opening was observed in nearly the entire hemisphere, characterized by obvious pathological hemorrhage in the H&E stains.
Targeting Ability, Biodistribution, and Biosafety in vivo
We established a T98G orthotopic xenograft model in NOD-SCID mice. Seven days after cell implantation, tumor formation could be observed on MRI images, with a volume of 3.59 ± 1.22 mm3. Fourteen days later, the tumor volume increased to 18.4 ± 2.05 mm3(Figure 5A).
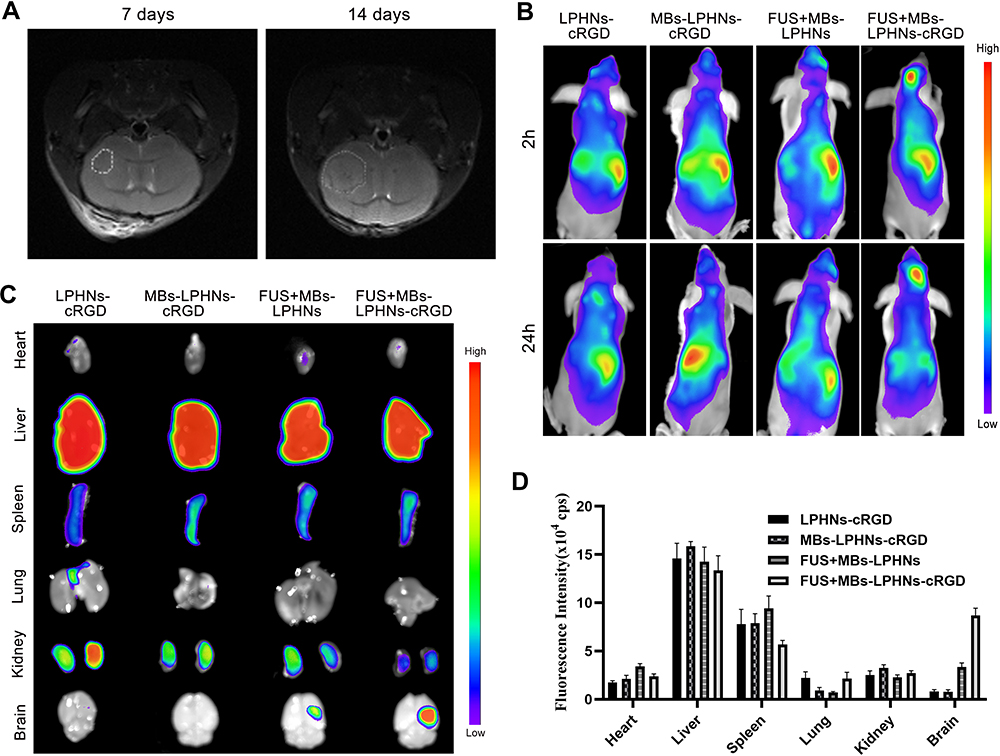 |
Figure 5 Targeting ability and distribution of MBs-LPHNs-cRGD under FUS irradiation in vivo. Abbreviations: MBs, microbubbles; LPHNs, lipid–polymer hybrid nanoparticles; cRGD, cyclic arginine-glycine-aspartic acid; MRI, magnetic resonance imaging; DiR, 1,1’-dioctadecyl-3,3,3’,3’- tetramethylindotricarbocyanine iodide. Notes: (A) MRI images of orthotopic tumor 7 days (left) and 14 days (right) after implantation of T98G cells; (B) in vivo fluorescence images of the distribution of DiR-labeled LPHNs or LPHNs-cRGD in T98G tumor-bearing mice at 2 h and 24 h after different treatments; (C) ex vivo fluorescence images of major organs and brain of mice after 24 h of treatments. (D) Corresponding quantitative analysis of fluorescence intensity in major organs (n=3). |
To further validate the active targeting ability of the cRGD and enhancement effects of the drug delivery induced by FUS in vivo, mice were randomly divided into four groups and given different treatments. Since most studies have shown that FUS-mediated BBB-opening would gradually return to normal within 24 h, we observed the distribution of nanoparticles in the tumor at 2 h and 24 h after FUS exposure.36 As shown in Figure 5B, a significant fluorescence signal was observed in brain after treatment with the FUS-assisted MBs-LPHNs-cRGD complexes, while only a faint signal was found in mice treated with FUS-assisted MBs-LPHNs complexes, and nearly no fluorescence signal appeared in other groups at 2 h and 24 h. For ex vivo fluorescence imaging (Figure 5C and D), parts of the nanoparticles in each group accumulated in the liver and spleen. Between the two groups treated with FUS, the fluorescence intensity in the brain of the MBs-LPHNs-cRGD group was much higher than that of the MBs-LPHNs group. The results proved that FUS in combination with MBs can significantly promote nanoparticle delivery to the brain and LPHNs-cRGD can exert an active targeting effect to further improve nanoparticle delivery in the tumor region in vivo.
After a week of treatment of MBs-LPHNs-cRGD or saline, the H&E staining of major organs (heart, liver, spleen, lungs, and kidneys) showed no significant histological abnormalities between the two groups, indicating the high histocompatibility and biosafety of the nanoparticles in vivo (Figure S8).
Therapeutic Effects of FUS-Assisted MBs-LPHNs-cRGD Complexes in vivo
Seven days after establishing orthotopic xenograft models in NOD-SCID mice, the mice were given different treatments, and the tumor growth was monitored longitudinally using MRI. As shown in Figure 6A, mice treated with FUS-assisted MBs-LPHNspCas9/MGMT-cRGD complexes + TMZ had suppressed tumor growth compared with other groups. According to the tumor volume calculated from the MRI (Figure 6B), the tumor volume of the mice in the FUS-assisted MBs-LPHNspCas9/MGMT-cRGD + TMZ group was smaller than that in other groups in the same treatment period. At the time of death, the final tumor volume in the FUS-assisted MBs-LPHNspCas9/MGMT-cRGD + TMZ group was also smaller than that in other groups. As shown in Figure 6C, the survival of the control group, TMZ group, MBs-LPHNspCas9/MGMT-cRGD + TMZ group, FUS-assisted MBs-LPHNspCas9/MGMT + TMZ group, and FUS-assisted MBs-LPHNspCas9/MGMT-cRGD + TMZ group was 22, 24, 26, 30, and 43 days, respectively. These results indicated that the FUS-assisted MBs-LPHNspCas9/MGMT-cRGD complexes enhanced the therapeutic effect of TMZ in glioblastoma, inhibited the growth of glioblastoma, and prolonged survival in tumor-bearing mice compared with other treatments.
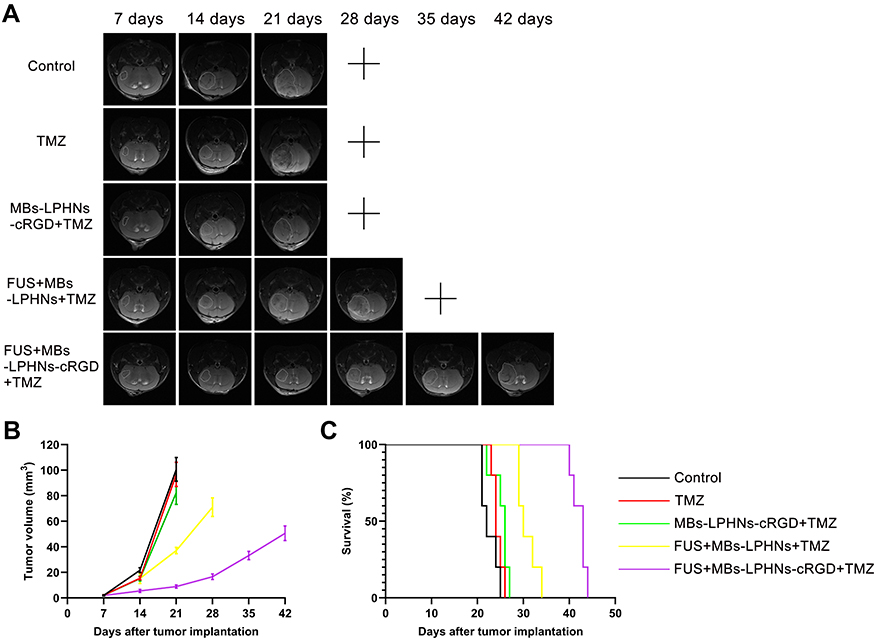 |
Figure 6 Evaluation of antitumor efficacy in vivo. Abbreviation: MRI, magnetic resonance imaging. Notes: (A) MRI images of tumors monitored every 7 days of different treatments. “+” indicates that the mice were sacrificed due to severe neurological symptoms. (B) Tumor volume based on MRI images (n=3, P < 0.05). (C) Kaplan-Meier survival curve of tumor-bearing mice with different treatments (n=5). |
Discussion
The MGMT protein is an important factor in the resistance of tumor cells to alkylating agents (such as TMZ). TMZ induces the O6-methylguanine (O6-MeG) lesion in DNA, resulting in cell cycle arrest and, eventually, apoptosis. MGMT can remove the methyl adducts from DNA and transfer it to the cysteine residue of the MGMT molecule, leading to the repair of DNA damage and irreversible inactivation of MGMT. Therefore, the resistance of tumor cells to TMZ is related to the level of MGMT in the cells.37 Many studies have shown that downregulation of MGMT expression can re-sensitize GBM cells to TMZ.38,39 In previous studies, either signaling pathway interference, or siRNA technology, was used to downregulate the expression of MGMT, but these strategies have obvious disadvantages, such as instability and short working times. We applied CRISPR/Cas9 technology to knock out the expression of MGMT. In the CRISPR/Cas9 system, we used dual-gRNA sequences to obtain efficient gene editing that can recognize two adjacent regions of the target gene and remove large fragments of nucleotides between the two sites.40,41 In order to deliver the Cas9 nucleases and gRNA, we used the “all-in-one” plasmid system containing a Cas9 expression cassette and the dual-gRNA expression cassettes simultaneously instead of Cas9 mRNA or the Cas9 protein/gRNA ribonucleoprotein complexes (Cas9 RNPs). Compared with mRNA- or RNP-based delivery method, plasmid DNA is more stable and cost-effective, and has a more sustained gene expression.14 But due to the large size of the plasmid system, transfection efficiency and editing efficiency may be relatively low, resulting in less satisfactory results, which need to be further optimized in future.
LPHNs nanoparticles have developed into a powerful drug delivery vehicle with many favorable characteristics, such as high structural integrity and stability, controlled drug release, high biocompatibility and bioavailability. In this study, the LPHNs-cRGD were synthesized by nanoprecipitation, an easier and more effective alternative to emulsification-solvent-evaporation (ESE) and two-step methods.42 PLGA, an FDA-approved polymer, was used as the polymer core. DC-chol, a type of cationic lipid, was used to adsorb the pCas9/MGMT by electrostatic action and encapsulate it within the polymer core. PEG chains were conjugated to the lipid (DSPE) to enhance the steric stability and prolong circulation time in vivo.32 To endow the delivery platform with active targeting ability, the cRGD peptide, which targets integrin αvβ3 receptors that are overexpressed in tumor cells, was incorporated with DSPE-PEG.43 The results show that the LPHNs-cRGD have relatively small particle size (179 nm) and typical core-shell structure. The preparation process does not affect the plasmid structure, and LPHNs-cRGD can protect the plasmid against enzyme degradation. But as shown in Figure 1D, the nanoparticles have a relatively wide size distribution. Now some strategies have been used to improve the size homogeneity of the LPHNs, such as optimizing formulation parameters, microfluidic nanoprecipitation process and so on21.
The FUS in combination with MBs plays an important role in drug delivery in the CNS. In this study, LPHNs-cRGD were bound with MBs to obtain effective nanoparticle delivery. Under FUS irradiation, focused ultrasound energy interacts with MBs-LPHNs-cRGD complexes and triggers MBs’ vibrations (expansions/contractions, stable cavitation), forming shear force, which can induce temporal disassembling of BBB as well as separation of LPHNs-cRGD from MBs. When accumulated enough energy, the MBs will rupture (inertial cavitation) to further promote BBB-opening, and at the same time all LPHNs-cRGD are released from the MBs, enter the brain and target tumor cells via cRGD-αvβ3 reaction. Since a host of factors can influence the outcome of FUS-MBs on BBB-opening, including the duration, burst length, pulse frequency of FUS, and the composition, size, concentration, dose of MBs and so on, it is recommended that those factors should be carefully adjusted according to the specific situational requirements to obtain safe and effective BBB-opening, especially when bound other nanoparticles to MBs, which might significantly change the surface structure of the MBs.44
Tumors in the treatment group were not completely eliminated during the course of treatment. This was possibly because the CRISPR/Cas9 system cannot effectively edit genes in all GBM cells due to the relatively low transfection efficiency and editing efficiency. It is also plausible that the presence of glioma stem cells (GSCs) and genotype heterogeneity could protect part of the GBM population from treatment.45,46 These residual tumor cells can continue to proliferate, leading to tumor recurrence. Lastly, TMZ-resistance in glioblastoma is a complicated process whose mechanism has not been fully elucidated. MGMT is one of the many important factors, and other mechanisms such as the base excision repair (BER) and the DNA mismatch repair (MMR) system also play important roles in the TMZ resistance of glioblastoma.3,47,48 In the future study, the CRISPR/Cas9 system can be optimized to achieve an enhanced gene editing efficiency. Additionally, several gRNAs that target key proteins in the TMZ-resistance pathway can be co-encapsulated in LPHNs-cRGD to further restore the sensitivity of glioblastoma to TMZ. We will also explore the potential off-target effects of CRISPR/Cas9, which is one of the main concerns in the clinical application of this system. Recently, many efforts have been made to improve the specificity and reduce the off-target effects of the CRISPR/Cas9 system, such as dCas9-FokI,49 SpCas9-HF1,50 and truncated guide RNAs.51 With these modified strategies, we hope that the FUS-assisted MBs-LPHNs-cRGD delivery system can significantly improve the specificity and reduce the off-target effects of CRISPR/Cas9 in glioblastoma treatment.
Conclusions
In summary, we developed a multi-functional (MBs-LPHNs-cRGD complexes) CRISPR/Cas9 delivery system and demonstrated the inhibition effect on the tumor growth in an orthotopic model of glioblastoma with FUS-assisted BBB-opening. The LPHNs-cRGD could efficiently deliver CRISPR/Cas9 plasmids into GBM cells and downregulate the expression of MGMT, resulting in an increased sensitivity of the cells to TMZ. Subsequently, the FUS-MBs parameters were evaluated for effective BBB-opening to ensure biosafety. Finally, the FUS-assisted MBs-LPHNspCas9/MGMT-cRGD + TMZ treatment efficiently inhibited tumor growth and prolonged survival in tumor-bearing mice. Importantly, the biotoxicity study also demonstrated the delivery platform had high biocompatibility and biosafety, which indicates its potential use in further clinical applications. These results also highlight the synergistic targeting ability of the physically site-specific characteristics of FUS and the biologically active targeting ability of cRGD peptide, which can significantly improve nanoparticle delivery into the CNS. This study provides a promising strategy for drug-resistant glioblastoma treatment via a safe, effective and targeted CRISPR/Cas9 delivery system, and further illustrates the therapeutic applications of FUS-assisted BBB-opening in CNS diseases.
Acknowledgments
We thank the Chongqing Key Laboratory of Ultrasound Molecular Imaging of Chongqing Medical University for their assistance on this project. We also thank Dr. Yi Liu for helpful discussions on this project. This work was supported by the National Natural Science Foundation of China (Grant Nos.81771961).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised Phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi:10.1016/S1470-2045(09)70025-7
2. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi:10.1056/NEJMoa043330
3. Lee SY. Temozolomide resistance in glioblastoma multiforme. Genes Diseases. 2016;3:198–210. doi:10.1016/j.gendis.2016.04.007
4. Sarkaria JN, Kitange GJ, James CD, et al. Mechanisms of chemoresistance to alkylating agents in malignant glioma. Clinical Cancer Res. 2008;14:2900–2908. doi:10.1158/1078-0432.CCR-07-1719
5. Villano JL, Seery TE, Bressler LR. Temozolomide in malignant gliomas: current use and future targets. Cancer Chemother Pharmacol. 2009;64:647–655. doi:10.1007/s00280-009-1050-5
6. Wang K, Chen D, Qian Z, Cui D, Gao L, Lou M. Hedgehog/Gli1 signaling pathway regulates MGMT expression and chemoresistance to temozolomide in human glioblastoma. Cancer Cell Int. 2017;17:117. doi:10.1186/s12935-017-0491-x
7. Papachristodoulou A, Signorell RD, Werner B, et al. Chemotherapy sensitization of glioblastoma by focused ultrasound-mediated delivery of therapeutic liposomes. J Controlled Release. 2019;295:130–139. doi:10.1016/j.jconrel.2018.12.009
8. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi:10.1126/science.1225829
9. Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi:10.1126/science.1231143
10. Mout R, Ray M, Lee YW, Scaletti F, Rotello VM. In vivo delivery of CRISPR/Cas9 for therapeutic gene editing: progress and challenges. Bioconjug Chem. 2017;28:880–884. doi:10.1021/acs.bioconjchem.7b00057
11. Sanchez-Rivera FJ, Jacks T. Applications of the CRISPR-Cas9 system in cancer biology. Nat Rev Cancer. 2015;15:387–395. doi:10.1038/nrc3950
12. Chiou SH, Winters IP, Wang J, et al. Pancreatic cancer modeling using retrograde viral vector delivery and in vivo CRISPR/Cas9-mediated somatic genome editing. Genes Dev. 2015;29:1576–1585. doi:10.1101/gad.264861.115
13. Chen X, Gonçalves MA. Engineered viruses as genome editing devices. Molecular Therapy. 2016;24:447–457. doi:10.1038/mt.2015.164
14. Wang HX, Li M, Lee CM, et al. CRISPR/Cas9-based genome editing for disease modeling and therapy: challenges and opportunities for nonviral delivery. Chem Rev. 2017;117:9874–9906. doi:10.1021/acs.chemrev.6b00799
15. Madni A, Sarfraz M, Rehman M, et al. Liposomal drug delivery: a versatile platform for challenging clinical applications. J Pharm Pharmaceutical Sci. 2014;17:401–426. doi:10.18433/J3CP55
16. Pridgen EM, Alexis F, Farokhzad OC. Polymeric nanoparticle technologies for oral drug delivery. Clin Gastroenterol Hepatol. 2014;12:1605–1610. doi:10.1016/j.cgh.2014.06.018
17. Gaspar R, Duncan R. Polymeric carriers: preclinical safety and the regulatory implications for design and development of polymer therapeutics. Adv Drug Deliv Rev. 2009;61:1220–1231. doi:10.1016/j.addr.2009.06.003
18. Duncan R. Polymer therapeutics as nanomedicines: new perspectives. Curr Opin Biotechnol. 2011;22:492–501. doi:10.1016/j.copbio.2011.05.507
19. Zhang L, Chan JM, Gu FX, et al. Self-assembled lipid–polymer hybrid nanoparticles: a robust drug delivery platform. ACS Nano. 2008;2:1696–1702.
20. Salvador-Morales C, Zhang L, Langer R, Farokhzad OC. Immunocompatibility properties of lipid-polymer hybrid nanoparticles with heterogeneous surface functional groups. Biomaterials. 2009;30:2231–2240. doi:10.1016/j.biomaterials.2009.01.005
21. Mukherjee A, Waters AK, Kalyan P, Achrol AS, Kesari S, Yenugonda VM. Lipid-polymer hybrid nanoparticles as a next-generation drug delivery platform: state of the art, emerging technologies, and perspectives. Int J Nanomedicine. 2019;14:1937–1952. doi:10.2147/IJN.S198353
22. Gao F, Zhang J, Fu C, et al. iRGD-modified lipid-polymer hybrid nanoparticles loaded with isoliquiritigenin to enhance anti-breast cancer effect and tumor-targeting ability. Int J Nanomedicine. 2017;12:4147–4162. doi:10.2147/IJN.S134148
23. Zheng M, Yue C, Ma Y, et al. Single-step assembly of DOX/ICG loaded lipid–polymer nanoparticles for highly effective chemo-photothermal combination therapy. ACS Nano. 2013;7:2056–2067. doi:10.1021/nn400334y
24. Yang XZ, Dou S, Sun TM, Mao CQ, Wang HX, Wang J. Systemic delivery of siRNA with cationic lipid assisted PEG-PLA nanoparticles for cancer therapy. J Controlled Release. 2011;156:203–211. doi:10.1016/j.jconrel.2011.07.035
25. Mieszawska AJ, Gianella A, Cormode DP, et al. Engineering of lipid-coated PLGA nanoparticles with a tunable payload of diagnostically active nanocrystals for medical imaging. Chemical Communications. 2012;48:5835–5837. doi:10.1039/c2cc32149a
26. Zhan C, Lu W. The blood-brain/tumor barriers: challenges and chances for malignant gliomas targeted drug delivery. Curr Pharm Biotechnol. 2012;13:2380–2387. doi:10.2174/138920112803341798
27. Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2:3–14. doi:10.1602/neurorx.2.1.3
28. Etame AB, Diaz RJ, Smith CA, Mainprize TG, Hynynen K, Rutka JT. Focused ultrasound disruption of the blood-brain barrier: a new frontier for therapeutic delivery in molecular neurooncology. Neurosurg Focus. 2012;32:E3.
29. Tsai HC, Tsai CH, Chen WS, Inserra C, Wei KC, Liu HL. Safety evaluation of frequent application of microbubble-enhanced focused ultrasound blood-brain-barrier opening. Sci Rep. 2018;8:17720. doi:10.1038/s41598-018-35677-w
30. Liu HL, Fan CH, Ting CY, Yeh CK. Combining microbubbles and ultrasound for drug delivery to brain tumors: current progress and overview. Theranostics. 2014;4:432–444. doi:10.7150/thno.8074
31. Pramual S, Lirdprapamongkol K, Svasti J, et al. Polymer-lipid-PEG hybrid nanoparticles as photosensitizer carrier for photodynamic therapy. J Photochem Photobiol B. 2017;173:12–22. doi:10.1016/j.jphotobiol.2017.05.028
32. Dave V, Tak K, Sohgaura A, Gupta A, Sadhu V, Reddy KR. Lipid-polymer hybrid nanoparticles: synthesis strategies and biomedical applications. J Microbiol Methods. 2019;160:130–142. doi:10.1016/j.mimet.2019.03.017
33. Szabo E, Schneider H, Seystahl K, et al. Autocrine VEGFR1 and VEGFR2 signaling promotes survival in human glioblastoma models in vitro and in vivo. Neuro-Oncology. 2016;18:1242–1252. doi:10.1093/neuonc/now043
34. Yan F, Li L, Deng Z, et al. Paclitaxel-liposome-microbubble complexes as ultrasound-triggered therapeutic drug delivery carriers. J Controlled Release. 2013;166:246–255. doi:10.1016/j.jconrel.2012.12.025
35. Zhao G, Huang Q, Wang F, et al. Targeted shRNA-loaded liposome complex combined with focused ultrasound for blood brain barrier disruption and suppressing glioma growth. Cancer Lett. 2018;418:147–158. doi:10.1016/j.canlet.2018.01.035
36. O’Reilly MA, Hough O, Hynynen K. Blood-brain barrier closure time after controlled ultrasound-induced opening is independent of opening volume. J Ultrasound Med. 2017;36:475–483. doi:10.7863/ultra.16.02005
37. Kaina B, Margison GP, Christmann M. Targeting O6-methylguanine-DNA methyltransferase with specific inhibitors as a strategy in cancer therapy. Cellular Molecular Life Sci. 2010;67:3663–3681.
38. Zhu Z, Du S, Ding F, Guo S, Ying G, Yan Z. Ursolic acid attenuates temozolomide resistance in glioblastoma cells by downregulating O (6)-methylguanine-DNA methyltransferase (MGMT) expression. Am J Transl Res. 2016;8:3299–3308.
39. Huang H, Lin H, Zhang X, Li J. Resveratrol reverses temozolomide resistance by downregulation of MGMT in T98G glioblastoma cells by the NF-κB-dependent pathway. Oncol Rep. 2012;27:2050–2056.
40. Zhao Y, Zhang C, Liu W, et al. An alternative strategy for targeted gene replacement in plants using a dual-sgRNA/Cas9 design. Sci Rep. 2016;6:23890. doi:10.1038/srep23890
41. Hara S, Kato T, Goto Y, et al. Microinjection-based generation of mutant mice with a double mutation and a 0.5 Mb deletion in their genome by the CRISPR/Cas9 system. J Reprod Dev. 2016;62:531–536. doi:10.1262/jrd.2016-058
42. Hadinoto K, Sundaresan A, Cheow WS. Lipid-polymer hybrid nanoparticles as a new generation therapeutic delivery platform: a review. European j Pharm Biopharmaceutics. 2013;85:427–443. doi:10.1016/j.ejpb.2013.07.002
43. Bello L, Francolini M, Marthyn P, et al. Alpha (v) beta3 and alpha (v) beta5 integrin expression in glioma periphery. Neurosurgery. 2001;49:
44. Chen KT, Wei KC, Liu HL. Theranostic strategy of focused ultrasound induced blood-brain barrier opening for CNS disease treatment. Front Pharmacol. 2019;10:86. doi:10.3389/fphar.2019.00086
45. Patel AP, Tirosh I, Trombetta JJ, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi:10.1126/science.1254257
46. Auffinger B, Spencer D, Pytel P, Ahmed AU, Lesniak MS. The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert Rev Neurother. 2015;15:741–752. doi:10.1586/14737175.2015.1051968
47. Zhang J, Stevens MF, Bradshaw TD. Temozolomide: mechanisms of action, repair and resistance. Curr Mol Pharmacol. 2012;5:102–114. doi:10.2174/1874467211205010102
48. Kaina B, Christmann M. DNA repair in resistance to alkylating anticancer drugs. Int J Clin Pharmacol Ther. 2002;40:354–367. doi:10.5414/CPP40354
49. Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol. 2014;32:577–582. doi:10.1038/nbt.2909
50. Kleinstiver BP, Pattanayak V, Prew MS, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi:10.1038/nature16526
51. Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279–284. doi:10.1038/nbt.2808
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.