Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 14
Gene Expression Profiling of Apoptotic Proteins in Circulating Peripheral Blood Mononuclear Cells in Type II Diabetes Mellitus and Modulation by Metformin
Authors Al Dubayee M, Alshahrani A, Aljada D, Zahra M ![]() , Alotaibi A
, Alotaibi A ![]() , Ababtain I
, Ababtain I ![]() , Alnaim M, Alahmari A
, Alnaim M, Alahmari A ![]() , Aljarallah A
, Aljarallah A ![]() , Elahi MA, Fakhoury HMA
, Elahi MA, Fakhoury HMA ![]()
Received 17 January 2021
Accepted for publication 23 February 2021
Published 15 March 2021 Volume 2021:14 Pages 1129—1139
DOI https://doi.org/10.2147/DMSO.S300048
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Mohammed Al Dubayee,1– 3 Awad Alshahrani,1– 3 Dana Aljada,4 Mahmoud Zahra,5 Ahmed Alotaibi,1 Ibrahim Ababtain,1 Malik Alnaim,1 Ali Alahmari,1 Abdullah Aljarallah,1 Muhammad Affan Elahi,5 Hana MA Fakhoury5
1College of Medicine, King Saud Bin Abdulaziz University for Health Sciences (KSAU-HS), Riyadh, Saudi Arabia; 2King Abdullah International Medical Research Center (KAIMRC), Riyadh, Saudi Arabia; 3Department of Medicine, Ministry of National Guard Health Affairs (MNG-HA), Riyadh, Saudi Arabia; 4College of Liberal Arts and Sciences, Hofstra University, Hempstead, NY, USA; 5Department of Biochemistry and Molecular Medicine, College of Medicine, Alfaisal University, Riyadh, Saudi Arabia
Correspondence: Mohammed Al Dubayee
College of Medicine, King Saud Bin Abdulaziz University for Health Sciences (KSAU-HS), P.O. Box 22490, Riyadh, Saudi Arabia
Tel +966 11 801 1111 ext: 53551
Email [email protected]
Introduction: Insulin resistance in obesity and type 2 diabetes mellitus (T2DM) is associated with cardiovascular complications such as atherosclerosis. On the other hand, the reduction of apoptosis in macrophages has been linked with accelerated atherosclerosis. Apoptosis is controlled by a different family of proteins including Bcl-2 and caspases.
Methods: To examine apoptosis in insulin resistance, we assessed the mRNA expression by qRT-PCR of several Bcl-2 family members, as well as caspase-3, − 7, − 8, and − 9 in peripheral blood mononuclear cells (PBMCs) isolated from lean, obese, diabetic, and diabetic on metformin individuals.
Results: PBMCs of diabetic individuals exhibited reduced expression of caspase-7 and increased expression of Bcl-10, Bad, Bax, Bid, and caspase-3. T2DM on metformin group had significantly higher Bad, Bax, and caspase-7 expression.
Discussion: The moderate up-regulation of pro-apoptotic Bcl-10, Bax, Bad, Bid, and the effector caspase-3 coupled with inhibition of caspase-7 in circulating PBMCs of T2DM could be the result of increased inflammation in T2DM. Metformin treatment significantly inhibited the expression of Bcl-10, Bid, and caspase-3 and upregulated Bad/Bax/caspase-7 pathway suggesting the activation of Bad/Bax/caspase-7 apoptotic pathway. Further studies are warranted to elicit the underlying apoptotic pathways of PBMCs in T2DM and following metformin treatment.
Keywords: apoptosis, T2DM, peripheral blood mononuclear cells, PBMC, caspase, BCl-10, BCl-2
Introduction
T2DM is a multifactorial metabolic and inflammatory disorder characterized by hyperglycemia.1 The incidence of T2DM has increased four-fold globally making it the ninth major cause of mortality.2 Atherosclerosis has been seen to prevail in diseases such as T2DM, as a result of increased inflammatory, and oxidative stress.3–5 Atherosclerosis is a complex process involving a focal accumulation of lipid, leucocytes, smooth muscle cells, and extracellular matrix in the intima of large arteries. Adherence of circulating monocytes and lymphocytes to the arterial endothelial lining is one of the earliest detectable events in both human and experimental atherosclerosis.6 These adherent cells subsequently transmigrate, accumulate in the intima, transform into lipid-engorged “foam cells”, and secrete cytokines and growth factors for the initiation and progression of atherosclerotic plaques.7,8 Apoptosis in atherosclerotic lesions has been studied in the last few years. Reduced Apoptosis in macrophages has been linked with accelerated atherosclerosis in mice.9
The apoptotic intrinsic pathway is regulated by a balance of pro-and anti-apoptotic Bcl-2 family members.10 The Bcl-2 protein family comprises inhibitor of apoptosis (Bcl-2, Bcl-w, Bcl-xL, Mcl-1, and Bcl-2 related protein A1), BH3-only proteins that bind and regulate the anti-apoptotic Bcl-2 proteins (Bad, Bik/Nbk, Bid, Bim, Hrk/DP5, Blk, Bmf), and activators of apoptosis (Bak, Bax, Bcl-rambo, Bcl-xS, and Bok/Mtd). Bcl-2 proteins tend to regulate the mitochondrial pathway, glucose metabolism, ß-cell homeostasis, and apoptosis.11 Cytochrome C release is directly promoted by the pro-apoptotic members (Bax and Bak) leading to the activation of initiator caspases such as caspase-9.12,13 APAF-1 is a multidomain adapter protein containing an N-terminal caspase recruitment domain (CARD) that interacts with Bcl-2 family members. The binding of cytochrome C to APAF-1 and procaspase 9 generates the formation of a complex (the apoptosome) which then leads to the initiation of the caspase cascade, leading to irreversible cell death.14
Apoptosis can affect all major cell types in atherosclerotic lesions (e.g. endothelial and vascular smooth muscle cells, as well as inflammatory cells, such as T-lymphocytes and macrophages). Apoptosis is mediated through extrinsic (death-receptor induced) and intrinsic (B-cell lymphoma 2 [Bcl-2] regulated or mitochondrial) pathways.15 Caspases are evolutionarily conserved cysteine-aspartyl-specific proteases involved in apoptosis and cytokine activation.16 Caspase-8 and −9 are upstream caspases whereas caspase-3 and −7 are downstream effector caspases of both pathways.17 The extrinsic pathway is mediated by cell death-receptors like Fas and the pro-inflammatory mediator tumor necrosis factor-alpha (TNF-α).15 NF-κB activation pathways include the classical pathway triggered in response to microbial infections or exposure to proinflammatory cytokines that activate the IκB kinase complex (IKK) and the alternative pathway triggered by members of the TNF cytokine family. The classical pathway is responsible for the inhibition of programmed cell death under most conditions, while the alternative pathway is important for the survival of premature B cells and the development of secondary lymphoid organs.18 The prosurvival NF-κB transcription factor has been reported to be induced in human PBMCs of obese and diabetic individuals, a phenomenon that could lead to inhibition of PBMCs apoptosis and thus increased atherosclerosis.19–21 The proapoptotic Bcl-10 functions through CARD. On the other hand, Bcl-10 is involved in the formation of complexes that antagonize apoptosis and contribute to cell survival after DNA damage.22 NF-κB and Bcl-10 have special significance since they can determine if the cell commits to or aborts apoptosis. Bcl-10 activates NF-κB via NF-κB-inducing kinase (NIK) and IKK.23
In this study, the expression of several apoptotic proteins in PBMCs of obese and diabetic individuals was studied to examine the effect of insulin resistance on apoptosis. We hypothesize that insulin resistance and hyperglycemia in T2DM are associated with decreased apoptotic proteins in PBMCs and thus increased atherogenicity. Additionally, we investigated the effect of metformin, a prominent biguanide class of anti-diabetic drug, with documented protective cardiovascular diseases (CVD) effect, on the expression of apoptotic genes in PBMCs of diabetic individuals.
Materials and Methods
Study Participants
Thirty lean (group I), 30 obese with normal glucose and on no medications (group II), 30 obese newly diagnosed with T2DM (group III), and 30 obese T2DM on metformin (group IV) adult volunteers, were recruited for the study. The duration of metformin treatment for group IV ranged between 9 months and 15 years. Most metformin regimen were 1000–2000 mg. However, 5 patients had dosages of 100–500 mg daily, and 3 had 3000–4500 mg daily. Only 5 subjects from group IV were on insulin therapy. The study was conducted in accordance with the Helsinki Declaration and was approved by the IRB at King Abdullah International Medical Research Center (Protocol NO. SP19/474/R). All study participants signed written consent forms.
Isolation of PBMC
Blood samples were collected in Na EDTA as an anticoagulant as described previously.24 Briefly, 3.5 mL of anticoagulated blood sample was layered carefully over 3.5 mL of PMN medium (Robbins Scientific Corp, Sunnyvale, CA). Samples were centrifuged and by the end of the centrifugation, two bands separated at the top of the RBC. The top band consisted of PBMC while the bottom consisted of PMN. The bands were harvested, repeatedly washed with Hank’s balanced salt solution (HBSS). This method provides a yield greater than 95% pure PBMC suspension. Qiagen RNALater solution (Qiagen Inc., CA, USA) was added to the pellet, and samples were frozen at −80°C.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) Analysis
Total RNA isolation was carried using an Ambion isolation kit according to the manufacturer’s instructions (Ambion). One µg of isolated total RNA was reverse transcribed into cDNA using Advantage RT-for-PCR kit following the manufacturer standard (Clontech, CA). qRT-PCR was performed using Applied Biosystems 7900HT analyzer (Applied Biosystems, USA) in which 2 µL cDNA, 10 µL Sybergreen Master mix [150 mM Tris, pH 9.2, 40 mM (NH4)2SO4, 5 mM MgCl2, 0.02% Tween-20, 0.4 mM dNTPs, 1.25 Units Taq Polymerase, 1 X Sybergreen] along with 0.5 µL of 20 µM gene-specific primers (Table 1). Theoretical optimal conditions of primers including melting temperature, annealing temperature, CG content, cross homology, and secondary structure were considered when designing primers of interest. The specificity and size of the PCR products were tested by adding a melt curve at the end of the amplification, followed by band analysis on a 2% agarose gel. The reaction protocol consisted of one activation cycle of 50°C for 2 min followed by 95°C for 15 s. Subsequently, 40 cycles of denaturation at 95°C for 15 s, and 60°C annealing/extension for 2 min were performed. All values were normalized to housekeeping genes RPL13, ubiquitin C, and cyclophilin A expressions. Although normalization to RPL13 and Ubiquitin C showed similar trends, all values were normalized to cyclophilin A. The 2−ΔΔCT method was used for relative quantification for qRT-PCR experiments.25
 |
Table 1 Primer Sequences for All Primers Used in RT-qPCR |
Statistical Analysis
SigmaStat software ver. 3.0 (Jandel Scientific, San Rafael, CA) was utilized for statistical analysis. One Way ANOVA comparing the four independent groups followed by (Holm-Sidak method) for pairwise comparisons were applied to analyze the calculated fold change of mRNA expression from qRT-PCR experiments. Upon failure of normality distribution, One Way ANOVA on Ranks was employed and trailed by Dunn’s test for pairwise comparisons. P-value < 0.05 was considered significant for all statistical analyses. Results were presented as mean ± standard error of the mean (S.E.M.).
Results
Demographic Data
Despite significant differences in the mean age among the four groups (Table 2), there was no correlation between mRNA levels of the measured apoptotic markers and age. Moreover, a preliminary analysis was conducted after excluding 5 individuals in group IV who were on insulin therapy. However, the gene expression levels measured from those individuals did not affect the results, therefore they were included in the final analysis.
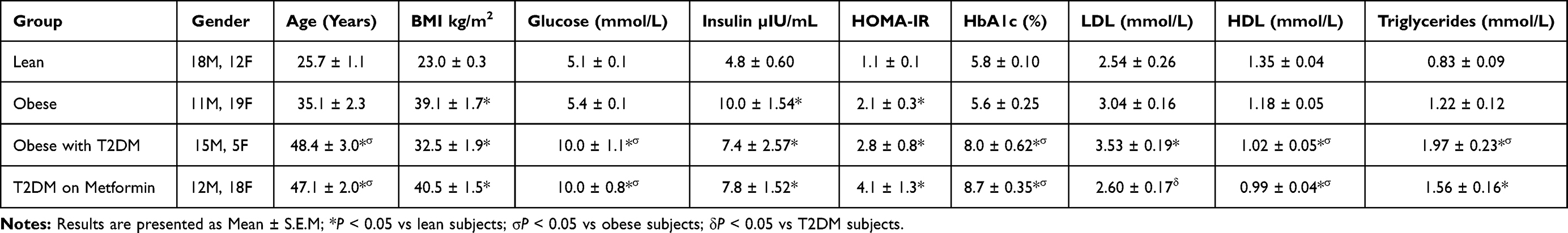 |
Table 2 Demographic Data of the Study Subjects |
mRNA Expression Analysis of Bcl-2 Family Members
No significant difference in the expression level of Bcl-2 was observed among the four studied groups (Figure 1). Similarly, there was no significant difference in Bak expression (Figure 2A). Conversely, we found significant up-regulation of Bax and Bcl-10 genes in the diabetic groups III and IV (Figure 2B and C) in comparison to the non-diabetic groups I and II. Indeed, Bax mRNA transcripts were absent in most non-diabetic PBMCs (CT < 40) but were highly detected in the diabetic groups (P<0.001; Figure 2B). On the other hand, Bcl-10 mRNA expression was significantly higher in T2DM in comparison to T2DM on metformin group (P<0.01; Figure 2C). Furthermore, the expression of Bad gene was elevated in diabetics, with a more pronounced increase in T2DM on metformin (P<0.001; Figure 3A). Conversely, Bid expression was significantly increased in T2DM but was reverted to normal following metformin (group IV) treatment (P<0.01; Figure 3B). In sharp contrast, Bim expression was only inhibited in the T2DM group (P<0.05; Figure 3C).
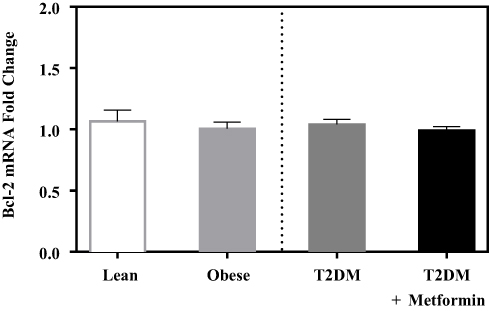 |
Figure 1 mRNA expression of Bcl-2 in PBMC of lean, obese, T2DM, and T2DM on metformin. Results are presented as mean ± S.E.M. |
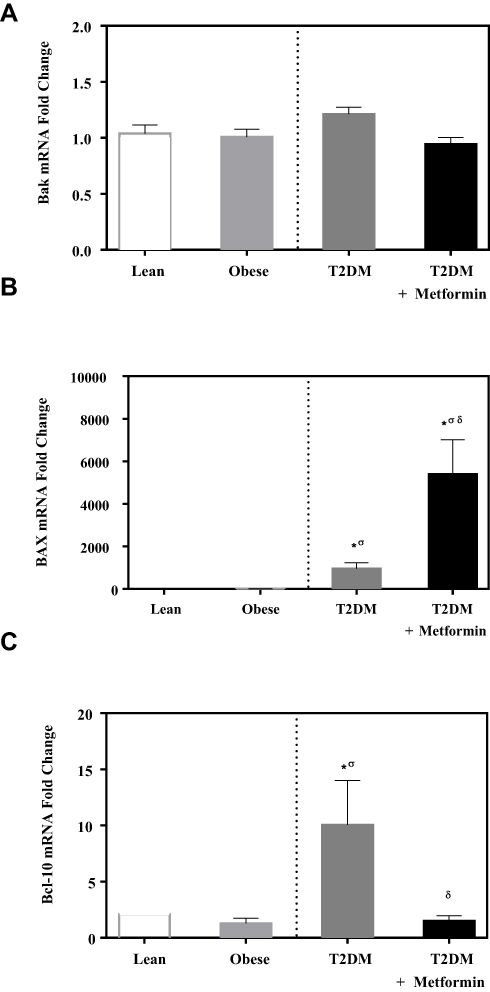 |
Figure 2 mRNA expression of three members of the Bcl-2 family that promote apoptosis in PBMC of lean, obese, T2DM, and T2DM on metformin: (A) Bak; (B) Bax; and (C) Bcl-10. Results are presented as mean ± S.E.M. *P < 0.05 vs lean subjects; σP < 0.05 vs obese subjects; δP < 0.05 vs T2DM subjects. |
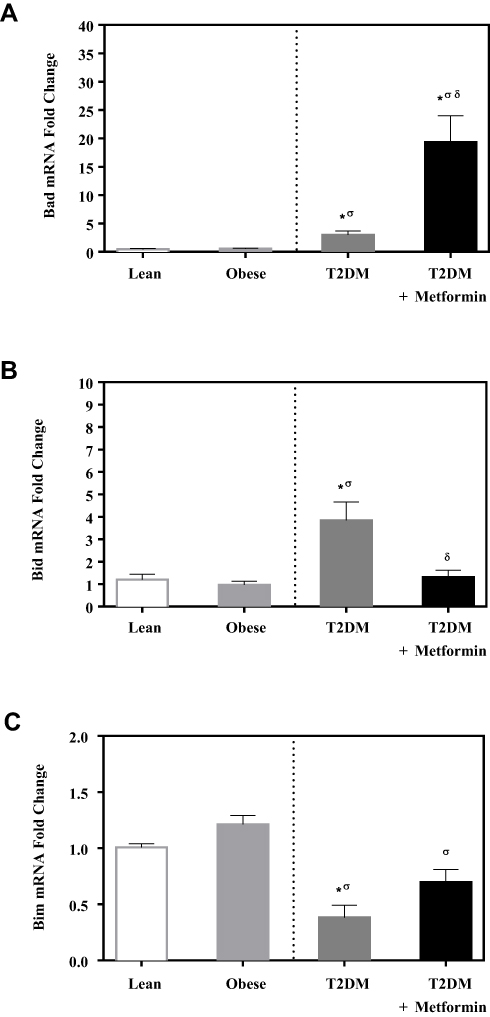 |
Figure 3 mRNA expression of three members of apoptotic BH3 domain-only proteins that bind and regulate the anti-apoptotic Bcl-2 proteins in PBMC of lean, obese, T2DM, and T2DM on metformin: (A) Bad; (B) Bid; and (C) Bim. Results are presented as mean ± S.E.M. *P < 0.05 vs lean subjects; σP < 0.05 vs obese subjects; δP < 0.05 vs T2DM subjects. |
Expression Analysis of Caspases
Caspase-3 expression level was not significantly different between lean and obese groups (Figure 4A). However, significant up-regulation was observed in the T2DM group (P<0.05; Figure 4A). Metformin treatment reduced caspase-3 expression in T2DM on the metformin group.
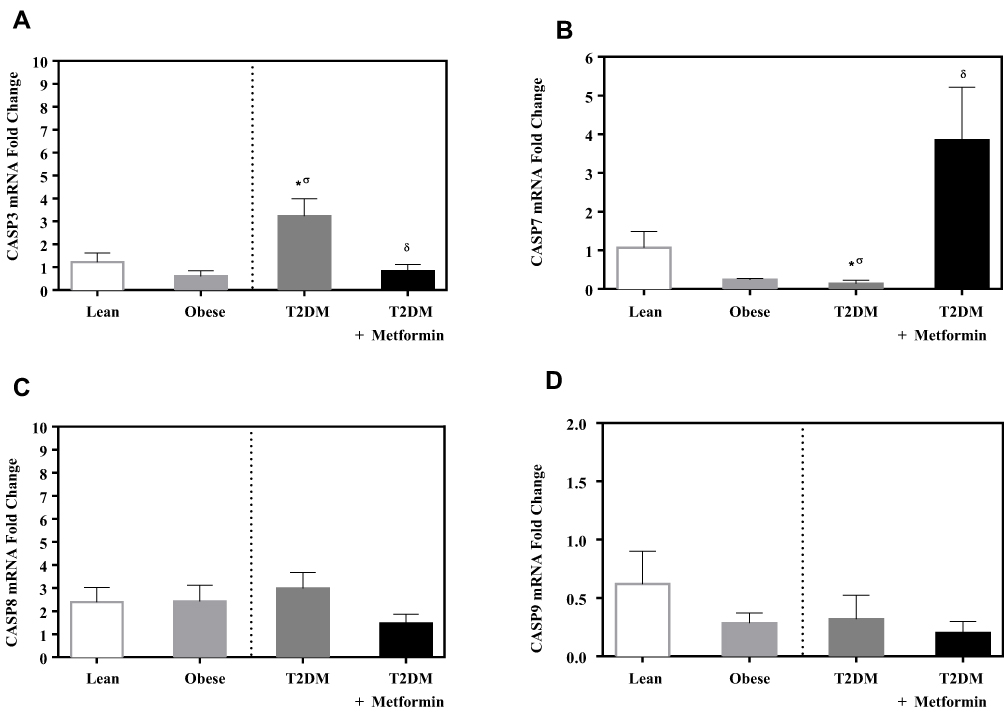 |
Figure 4 mRNA expression of three caspases in PBMC of lean, obese, T2DM and T2DM on metformin: (A) CASP3; (B) CASP7; (C) CASP8; and (D) CASP9. Results are presented as mean ± S.E.M. *P < 0.05 vs lean subjects; σP < 0.05 vs obese subjects; δP < 0.05 vs T2DM subjects. |
On the other hand, the expression level of caspase-7 was significantly inhibited in T2DM compared to the non-diabetic groups. Interestingly, metformin was associated with a significant increase in caspase-7 expression in PBMCs of T2DM on metformin (P<0.05; Figure 4B).
In contrast, the expression levels of caspase-8 and −9 did not differ significantly between the 4 groups (Figure 4C and D). Similarly, the expression levels of APAF-1 did not differ significantly between the studied groups (Figure 5).
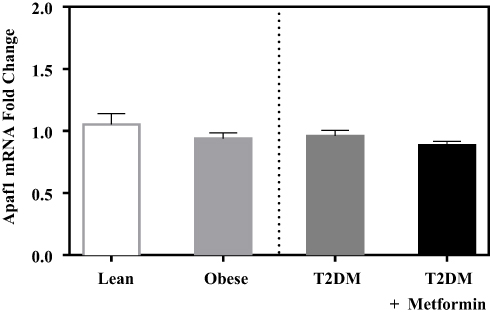 |
Figure 5 Apoptotic protease activating factor 1 (Apaf1) mRNA expression in PBMC of lean, obese, T2DM, and T2DM on metformin. Results are presented as mean ± S.E.M. |
Discussion
Given the complexity of atherosclerotic plaque, apoptosis in leukocytes, endothelial, and smooth muscle cells may play different roles in atherogenesis.26,27 Two different pathways for apoptosis have been identified, the mitochondrial pathway where multidomain Bcl-2 family members play pivotal roles and the death receptor pathway.28 Activation of Bax is essential to mitochondrial dysfunction and cell death.29,30 Bax is predominantly a cytosolic monomer, and stimulation of Bax results in conformational changes and translocation to the outer mitochondrial membrane or endoplasmic reticulum where it oligomerizes.31,32 Activation of Bax and other related apoptotic proteins have been reported in human atherosclerotic plaques, resulting from uptake of free cholesterol by macrophages or after treatment of macrophages with cytotoxic oxysterols components of LDL.33–37 Activation of Bax in atherosclerotic plaques increases the susceptibility of macrophages and smooth muscle cells to undergo apoptosis. Bax is assumed to exert at least part of its apoptosis-inducing function by facilitating mitochondrial permeability transition pore opening, a process that is supposedly antagonized by Bcl-2 and Bcl-xL.38 Bax activation is induced through cleavage and mitochondrial translocation of Bid which represents an integration of two apoptotic pathways on mitochondria. Truncated Bid (tBid) interacts with pro-apoptotic (Bax, Bak) or anti-apoptotic members (Bcl-2, Bcl-xL) of Bcl-2 family proteins and stimulates cytochrome C release from mitochondria.39 However, a non-apoptotic role of Bid in inflammation and innate immunity has been reported.40 Similarly, the pro-apoptotic protein Bad has been reported to play a role in atherosclerosis.41,42 Bad competes for the binding to Bcl-2, Bcl-xL, and Bcl-W, thereby affecting the level of heterodimerization of these proteins with Bax.43 Bad can be either pro-apoptotic or pro-survival depending on the phosphorylation state of three specific serine residues.44 Expression analysis comparison among the four different groups studied (lean, obese, T2DM, and T2DM on metformin) showed the association of T2DM with increased expression levels of Bad, Bax, Bid, and caspase-3 and lower caspase-7 expression in PBMCs of T2DM patients. These results are consistent with the previously reported higher levels of caspase-3 cleavage in PBMC from T2DM individuals.45 Bad/Bax/Bid/caspase-3 cascade upregulation in T2DM may result from increased inflammation present in T2DM and could indicate a proinflammatory role, possibly linked to Bcl-10/NF-κB pathway. Higher expression of Bax in regulatory T cells has been reported to increase vascular inflammation and Bid plays a role in inflammation and innate immunity.40,46 On the other hand, the anti-inflammatory metformin reverted Bid, Bcl-10, and caspase-3 expressions to normal while it was associated with upregulation of Bax, Bad and caspase-7 expressions. These data are consistent with the hypothesis that macrophage apoptosis suppresses the development of atherosclerosis as macrophages death may reduce growth factors and inflammatory cytokines production indicating that macrophage apoptosis provides a critical self-defense mechanism in suppressing atherosclerosis.47 Metformin anti-inflammatory and anti-atherosclerotic effect could be a result of reduced proinflammatory Bad/Bax/Bid/caspase-3 cascade activation, especially through Bcl-10/NF-κB and by increased apoptosis of these inflammatory vascular cells, including PBMCs, by upregulation of the pro-apoptotic Bad/Bax/caspase-7 pathway.
The pro-survival NF-κB transcription factor is activated in PBMCs of obese and T2DM and the expression of the pro-apoptotic Bcl-10 is upregulated in PBMCs of T2DM.19–21 Bcl-10 activates NF-κB via NIK and IKK.23 Bcl-10 interacts with MALT1 proteins and CARMA1, as part of a signaling complex (CBM signalosome) that mediates antigen receptor-dependent NF-κB activation.48 CARMA3-containing CBM signalosome can operate in cells outside the hematopoietic system and mediate NF-κB activation machinery in endothelial and vascular smooth muscle cells, thereby contributing to pro-inflammatory signaling within the vessel wall. Bcl-10−/− mice are reported to be protected from developing both Ang II-dependent atherosclerotic lesions and Ang II-dependent abdominal aortic aneurysms.49 Bcl-10−/- mouse showed also better glucose control in glucose tolerance test as compared to wild type surprisingly with less insulin production.50 Thus, Bcl-10 induces vascular inflammation possibly by mediating NF-κB activation machinery. Several reports demonstrated the cardioprotective effects of metformin.51,52 Metformin inhibits NF-κB pathway in vascular wall cells and experimental atherogenesis of rabbits.53,54 In this study, metformin treatment reduced expression levels of pro-apoptotic gene Bcl-10 which is also consistent with its inhibitory effect of the NF-κB pathway reported previously.21,53,54
Bim has been reported to increase in atherosclerosis.41 Bim serves a pivotal role in regulating endothelial cell apoptosis induced by oxidative stress which is an early event in the development of atherosclerosis.55 However, leukocyte Bim deficiency in ldlr −/− mice results in increased activated T-cell content in circulation, lymphoid organs, and atherosclerotic lesions.56 Urokinase-type plasminogen activator (uPA) has been reported to stimulate the differentiation of monocytes into macrophages and attenuates ox-LDL-induced macrophage apoptotic death via Bim downregulation.57 These data are consistent with our data, as Bim expression levels in PBMCs were inhibited in T2DM while metformin reverted Bim expression levels to normal.
The observed increase in expression of pro-apoptotic genes in T2DM patients was associated with the up-regulation of effector caspase-3 and inhibition of caspase-7 in T2DM patients only. Once caspase-3 is activated, it will cleave key structural proteins, cell cycle proteins, and DNase proteins, such as poly(ADP-ribose) polymerase, and DNA-dependent kinases.58,59 Moreover, high expression of caspase-3 has been detected in the absence of apoptosis, indicating that caspase-3 may participate in processes other than apoptosis.60 Caspase-3 may be involved in inflammatory processes by cleaving cytokines like IL-16 and IL-18 and the upregulation of caspase-3 in T2DM could be a result of the increased inflammation present in T2DM.61,62 Deletion of caspase-3 promotes plaque growth and plaque necrosis in ApoE−/− mice.63 In this study, caspase-3 was the only upregulated caspase among the ones measured (caspase −3, −7, −8, and −9) in T2DM while caspase-7 was downregulated. Previous reports demonstrated that caspase-3, −7, and −9 have distinct roles during intrinsic apoptosis and that caspase-3 and −7 functions are not limited to apoptosis, with their activation linked to proliferation or differentiation.64,65 Metformin reduced the expression levels of caspase-3 seen in T2DM, possibly due to its anti-inflammatory effect, while induced caspase-7 expression levels, possibly due to its pro-apoptotic properties. The increased caspase-7 mediated apoptosis could be a defensive mechanism to reduce inflammation present in T2DM.
Although metformin reduces cardiovascular complications in patients with diabetes,66 this protective mechanism remains poorly understood. Metformin is known to inhibit macrophage apoptosis induced by ox-LDL and reduces mitochondrial fragmentation which are critical integrators of apoptosis.67,68 In addition, metformin inhibits cardiac remodeling such as inhibiting cardiomyocytes apoptosis and cardiac fibrosis.69 Moreover, endoplasmic reticulum (ER) stress that is prolonged or severe can lead to apoptosis. Metformin differentially activates ER stress signaling pathways without inducing apoptosis.70 On the other hand, metformin exerted pro-apoptotic properties in several preclinical studies. It reduced cell proliferation, caused cell cycle arrest, reduced incidence and growth of experimental tumors, and induced apoptosis.71,72 Since reduced apoptosis in macrophages has been linked with accelerated atherosclerosis and metformin has anti-atherosclerotic properties, we could infer that metformin induces apoptosis, possibly through Bax/Bad/caspase-7 mediated pathway.9 Bax/Bad/caspase-7 mediated apoptosis could be a defensive mechanism that targets activated leucocytes to hamper inflammation. This would require further investigation.
PBMCs represent a mixture of cells (monocytes, B cells, and T cells). This heterogeneity of cells utilized in this study represents a significant shortcoming in this study and purified monocytes could be a better approach to examine the expression of apoptotic proteins in monocytes, B cells, and T cells. However, purification of monocytes could activate monocytes resulting in changes in the expression of apoptotic proteins.
In conclusion, the moderate up-regulation of pro-apoptotic Bcl-10, Bax, Bad, Bid, and the effector caspase-3 coupled with inhibition of caspase-7 in circulating PBMCs of T2DM could be the result of increased inflammation in T2DM. Metformin treatment significantly inhibited the expression of Bcl-10, Bid, and caspase-3 and upregulated Bad/Bax/caspase-7 pathway (Figure 6). Future studies are warranted to understand the underlying signaling network associated with diabetes-apoptotic pathways in PBMCs.
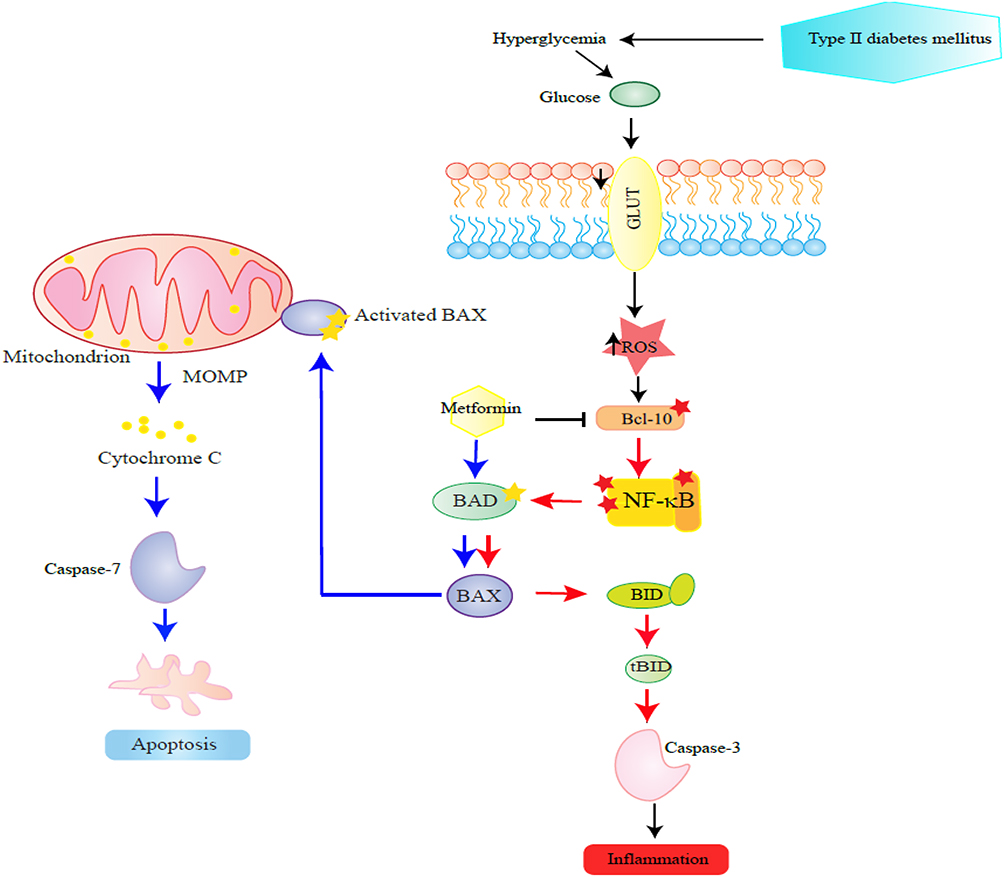 |
Figure 6 This figure schematizes steps in the proposed effect of metformin on apoptotic proteins in PBMCs examined in this study. The steps are depicted in a sequence proceeding from right to left. Hyperglycaemia presents in diabetes induces NF-κB pathway by reactive oxygen species (ROS). The low to moderate up-regulation of inflammatory Bcl-10, Bax, Bad, Bid, and the effector caspase-3 coupled with inhibition of caspase-7 in circulating PBMCs of T2DM could be a result of activated NF-κB pathway in T2DM. |
Funding
This work was supported by King Abdullah International Medical Research Center (KAIMRC), Riyadh, Kingdom of Saudi Arabia (KAIMRC, Grant # SP10/474/R) and conducted at College of Medicine, King Saud bin Abdulaziz University for Health Sciences, Riyadh, Kingdom of Saudi Arabia.
Disclosure
The authors report no conflicts of interest for this work and declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
References
1. Anuradha R, Saraswati M, Kumar KG, Rani SH. Apoptosis of beta cells in diabetes mellitus. DNA Cell Biol. 2014;33(11):743–748. doi:10.1089/dna.2014.2352
2. Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol. 2018;14(2):88–98. doi:10.1038/nrendo.2017.151
3. Beckman JA, Creager MA, Libby P. Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. JAMA. 2002;287(19):2570–2581. doi:10.1001/jama.287.19.2570
4. Kanter JE, Averill MM, Leboeuf RC, Bornfeldt KE. Diabetes-accelerated atherosclerosis and inflammation. Circ Res. 2008;103(8):e116–e117. doi:10.1161/CIRCRESAHA.108.182642
5. Santini SA, Marra G, Giardina B, et al. Defective plasma antioxidant defenses and enhanced susceptibility to lipid peroxidation in uncomplicated IDDM. Diabetes. 1997;46(11):1853–1858. doi:10.2337/diab.46.11.1853
6. DiCorleto PE. Cellular mechanisms of atherogenesis. Am J Hypertens. 1993;6(11 Pt 2):314S–318S. doi:10.1093/ajh/6.11.314S
7. Faruqi RM, DiCorleto PE. Mechanisms of monocyte recruitment and accumulation. Br Heart J. 1993;69(1 Suppl):S19–S29. doi:10.1136/hrt.69.1_Suppl.S19
8. Raines EW, Ross R. Biology of atherosclerotic plaque formation: possible role of growth factors in lesion development and the potential impact of soy. J Nutr. 1995;125(3 Suppl):624S–630S. doi:10.1093/jn/125.3_Suppl.624S
9. Liu J, Thewke DP, Su YR, Linton MF, Fazio S, Sinensky MS. Reduced macrophage apoptosis is associated with accelerated atherosclerosis in low-density lipoprotein receptor-null mice. Arterioscler Thromb Vasc Biol. 2005;25(1):174–179. doi:10.1161/01.ATV.0000148548.47755.22
10. Tomita T. Apoptosis in pancreatic β-islet cells in type 2 diabetes. Bosn J Basic Med Sci. 2016;16(3):162. doi:10.17305/bjbms.2016.919
11. Gurzov EN, Eizirik DL. Bcl-2 proteins in diabetes: mitochondrial pathways of β-cell death and dysfunction. Trends Cell Biol. 2011;21(7):424–431. doi:10.1016/j.tcb.2011.03.001
12. Thomas HE, Biden TJ. Bad news for β-cell apoptosis. Diabetes. 2009;58(8):1725–1727. doi:10.2337/db09-0732
13. Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol. 2007;8(5):405–413. doi:10.1038/nrm2153
14. Kavurma MM, Bhindi R, Lowe HC, Chesterman C, Khachigian LM. Vessel wall apoptosis and atherosclerotic plaque instability. J Thromb Haemost. 2005;3(3):465–472. doi:10.1111/j.1538-7836.2005.01120.x
15. Thomas HE, McKenzie MD, Angstetra E, Campbell PD, Kay TW. Beta cell apoptosis in diabetes. Apoptosis. 2009;14(12):1389. doi:10.1007/s10495-009-0339-5
16. Creagh EM, Conroy H, Martin SJ. Caspase‐activation pathways in apoptosis and immunity. Immunol Rev. 2003;193(1):10–21. doi:10.1034/j.1600-065X.2003.00048.x
17. Liadis N, Murakami K, Eweida M, et al. Caspase-3-dependent β-cell apoptosis in the initiation of autoimmune diabetes mellitus. Mol Cell Biol. 2005;25(9):3620–3629. doi:10.1128/MCB.25.9.3620-3629.2005
18. Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death–a new approach to cancer therapy. J Clin Invest. 2005;115(10):2625–2632. doi:10.1172/JCI26322
19. Ghanim H, Aljada A, Hofmeyer D, Syed T, Mohanty P, Dandona P. Circulating mononuclear cells in the obese are in a proinflammatory state. Circulation. 2004;110(12):1564–1571. doi:10.1161/01.CIR.0000142055.53122.FA
20. Aljada A, Garg R, Ghanim H, et al. Nuclear factor-kappaB suppressive and inhibitor-kappaB stimulatory effects of troglitazone in obese patients with type 2 diabetes: evidence of an antiinflammatory action? J Clin Endocrinol Metab. 2001;86(7):3250–3256. doi:10.1210/jcem.86.7.7564
21. Patel S, Santani D. Role of NF-kappa B in the pathogenesis of diabetes and its associated complications. Pharmacol Rep. 2009;61(4):595–603. doi:10.1016/s1734-1140(09)70111-2
22. Luo Y, Wu J, Zou J, et al. BCL10 in cell survival after DNA damage. Clin Chim Acta. 2019;495:301–308. doi:10.1016/j.cca.2019.04.077
23. Bhattacharyya S, Borthakur A, Tyagi S, et al. B-cell CLL/lymphoma 10 (BCL10) is required for NF-kappaB production by both canonical and noncanonical pathways and for NF-kappaB-inducing kinase (NIK) phosphorylation. J Biol Chem. 2010;285(1):522–530. doi:10.1074/jbc.M109.050815
24. Dandona P, Aljada A, Mohanty P, et al. Insulin inhibits intranuclear nuclear factor kappaB and stimulates IkappaB in mononuclear cells in obese subjects: evidence for an anti-inflammatory effect? J Clin Endocrinol Metab. 2001;86(7):3257–3265. doi:10.1210/jcem.86.7.7623
25. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25(4):402–408. doi:10.1006/meth.2001.1262
26. Lusis AJ. Atherosclerosis. Nature. 2000;407(6801):233–241. doi:10.1038/35025203
27. Kockx MM, Herman AG. Apoptosis in atherosclerosis: beneficial or detrimental? Cardiovasc Res. 2000;45(3):736–746. doi:10.1016/s0008-6363(99)00235-7
28. Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13(15):1899–1911. doi:10.1101/gad.13.15.1899
29. Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292(5517):727–730. doi:10.1126/science.1059108
30. Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of bax and bak. Genes Dev. 2001;15(12):1481–1486. doi:10.1101/gad.897601
31. Hsu YT, Wolter KG, Youle RJ. Cytosol-to-membrane redistribution of bax and bcl-X(L) during apoptosis. Proc Natl Acad Sci U S A. 1997;94(8):3668–3672. doi:10.1073/pnas.94.8.3668
32. Antonsson B, Montessuit S, Sanchez B, Martinou JC. Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J Biol Chem. 2001;276(15):11615–11623. doi:10.1074/jbc.M010810200
33. Kockx MM, De Meyer GR, Muhring J, Jacob W, Bult H, Herman AG. Apoptosis and related proteins in different stages of human atherosclerotic plaques. Circulation. 1998;97(23):2307–2315. doi:10.1161/01.cir.97.23.2307
34. Saxena A, McMeekin JD, Thomson DJ. Expression of bcl-x, bcl-2, bax, and bak in endarterectomy and atherectomy specimens. J Pathol. 2002;196(3):335–342. doi:10.1002/path.1040
35. Martinet W, Schrijvers DM, De Meyer GR, et al. Gene expression profiling of apoptosis-related genes in human atherosclerosis: upregulation of death-associated protein kinase. Arterioscler Thromb Vasc Biol. 2002;22(12):2023–2029. doi:10.1161/01.atv.0000041843.44312.12
36. Yao PM, Tabas I. Free cholesterol loading of macrophages is associated with widespread mitochondrial dysfunction and activation of the mitochondrial apoptosis pathway. J Biol Chem. 2001;276(45):42468–42476. doi:10.1074/jbc.M101419200
37. Panini SR, Sinensky MS. Mechanisms of oxysterol-induced apoptosis. Curr Opin Lipidol. 2001;12(5):529–533. doi:10.1097/00041433-200110000-00008
38. Zamzami N, Brenner C, Marzo I, Susin SA, Kroemer G. Subcellular and submitochondrial mode of action of Bcl-2-like oncoproteins. Oncogene. 1998;16(17):2265–2282. doi:10.1038/sj.onc.1201989
39. Kutuk O, Basaga H. Bcl-2 protein family: implications in vascular apoptosis and atherosclerosis. Apoptosis. 2006;11(10):1661–1675. doi:10.1007/s10495-006-9402-7
40. Yeretssian G, Correa RG, Doiron K, et al. Non-apoptotic role of BID in inflammation and innate immunity. Nature. 2011;474(7349):96–99. doi:10.1038/nature09982
41. Rusinol AE, Thewke D, Liu J, Freeman N, Panini SR, Sinensky MS. AKT/protein kinase B regulation of BCL family members during oxysterol-induced apoptosis. J Biol Chem. 2004;279(2):1392–1399. doi:10.1074/jbc.M308619200
42. Napoli C, Quehenberger O, De Nigris F, Abete P, Glass CK, Palinski W. Mildly oxidized low density lipoprotein activates multiple apoptotic signaling pathways in human coronary cells. FASEB J. 2000;14(13):1996–2007. doi:10.1096/fj.99-0986com
43. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. doi:10.1080/01926230701320337
44. Bui NL, Pandey V, Zhu T, Ma L, Basappa LPE. Bad phosphorylation as a target of inhibition in oncology. Cancer Lett. 2018;415:177–186. doi:10.1016/j.canlet.2017.11.017
45. Bhattacharya D, Dutta M, Mukhopadhyay M, Bhattacharyya M, Chowdhury S, Karmakar P. The protective role of metformin in autophagic status in peripheral blood mononuclear cells of type 2 diabetic patients. Cell Biol Int. 2020;44(8):1628–1639. doi:10.1002/cbin.11355
46. Xiong Z, Song J, Yan Y, et al. Higher expression of bax in regulatory T cells increases vascular inflammation. Front Biosci. 2008;13:7143–7155. doi:10.2741/3217
47. Rayner KJ. Cell death in the vessel wall: the good, the bad, the ugly. Arterioscler Thromb Vasc Biol. 2017;37(7):e75–e81. doi:10.1161/ATVBAHA.117.309229
48. Rawlings DJ, Sommer K, Moreno-Garcia ME. The CARMA1 signalosome links the signalling machinery of adaptive and innate immunity in lymphocytes. Nat Rev Immunol. 2006;6(11):799–812. doi:10.1038/nri1944
49. McAllister-Lucas LM, Jin X, Gu S, et al. The CARMA3-Bcl10-MALT1 signalosome promotes angiotensin II-dependent vascular inflammation and atherogenesis. J Biol Chem. 2010;285(34):25880–25884. doi:10.1074/jbc.C110.109421
50. Van Beek M, Oravecz-Wilson KI, Delekta PC, et al. Bcl10 links saturated fat overnutrition with hepatocellular NF-κB activation and insulin resistance. Cell Rep. 2012;1(5):444–452.
51. Maruthur NM, Tseng E, Hutfless S, et al. Diabetes medications as monotherapy or metformin-based combination therapy for type 2 diabetes: a systematic review and meta-analysis. Ann Intern Med. 2016;164(11):740–751. doi:10.7326/M15-2650
52. UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet. 1998;352(9131):854–865. doi:10.1016/S0140-6736(98)07037-8
53. Isoda K, Young JL, Zirlik A, et al. Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Arterioscler Thromb Vasc Biol. 2006;26(3):611–617. doi:10.1161/01.ATV.0000201938.78044.75
54. Li SN, Wang X, Zeng QT, et al. Metformin inhibits nuclear factor kappaB activation and decreases serum high-sensitivity C-reactive protein level in experimental atherogenesis of rabbits. Heart Vessels. 2009;24(6):446–453. doi:10.1007/s00380-008-1137-7
55. Shi X, Chen X. Effect of microRNA-370 on coronary atherosclerosis and its underlying mechanism. Exp Ther Med. 2019;17(1):115–122. doi:10.3892/etm.2018.6961
56. Temmerman L, Westra MM, Bot I, et al. Leukocyte bim deficiency does not impact atherogenesis in ldlr (-/-) mice, despite a pronounced induction of autoimmune inflammation. Sci Rep. 2017;7(1):3086. doi:10.1038/s41598-017-02771-4
57. Paland N, Aharoni S, Fuhrman B. Urokinase-type plasminogen activator (uPA) modulates monocyte-to-macrophage differentiation and prevents Ox-LDL-induced macrophage apoptosis. Atherosclerosis. 2013;231(1):29–38. doi:10.1016/j.atherosclerosis.2013.08.016
58. Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391(6662):43–50. doi:10.1038/34112
59. Liu X, Zou H, Slaughter C, Wang X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell. 1997;89(2):175–184. doi:10.1016/s0092-8674(00)80197-x
60. Miossec C, Dutilleul V, Fassy F, Diu-Hercend A. Evidence for CPP32 activation in the absence of apoptosis during T lymphocyte stimulation. J Biol Chem. 1997;272(21):13459–13462. doi:10.1074/jbc.272.21.13459
61. Zhang Y, Center DM, Wu DM, et al. Processing and activation of pro-interleukin-16 by caspase-3. J Biol Chem. 1998;273(2):1144–1149. doi:10.1074/jbc.273.2.1144
62. Akita K, Ohtsuki T, Nukada Y, et al. Involvement of caspase-1 and caspase-3 in the production and processing of mature human interleukin 18 in monocytic THP.1 cells. J Biol Chem. 1997;272(42):26595–26603. doi:10.1074/jbc.272.42.26595
63. Grootaert MO, Schrijvers DM, Hermans M, Van Hoof VO, De Meyer GR, Martinet W. Caspase-3 deletion promotes necrosis in atherosclerotic plaques of ApoE knockout mice. Oxid Med Cell Longev. 2016;2016:3087469. doi:10.1155/2016/3087469
64. Brentnall M, Rodriguez-Menocal L, De Guevara RL, Cepero E, Boise LH. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013;14(1):32. doi:10.1186/1471-2121-14-32
65. Svandova E, Lesot H, Vanden Berghe T, et al. Non-apoptotic functions of caspase-7 during osteogenesis. Cell Death Dis. 2014;5(8):e1366. doi:10.1038/cddis.2014.330
66. Hong J, Zhang Y, Lai S, et al. Effects of metformin versus glipizide on cardiovascular outcomes in patients with type 2 diabetes and coronary artery disease. Diabetes Care. 2013;36(5):1304–1311. doi:10.2337/dc12-0719
67. Huangfu N, Wang Y, Cheng J, Xu Z, Wang S. Metformin protects against oxidized low density lipoprotein-induced macrophage apoptosis and inhibits lipid uptake. Exp Ther Med. 2018;15(3):2485–2491. doi:10.3892/etm.2018.5704
68. Wang Q, Zhang M, Torres G, et al. Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of Drp1-mediated mitochondrial fission. Diabetes. 2017;66(1):193–205. doi:10.2337/db16-0915
69. Sasaki H, Asanuma H, Fujita M, et al. Metformin prevents progression of heart failure in dogs: role of AMP-activated protein kinase. Circulation. 2009;119(19):2568–2577. doi:10.1161/CIRCULATIONAHA.108.798561
70. Quentin T, Steinmetz M, Poppe A, Thoms S. Metformin differentially activates ER stress signaling pathways without inducing apoptosis. Dis Model Mech. 2012;5(2):259–269. doi:10.1242/dmm.008110
71. Alimova IN, Liu B, Fan Z, et al. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle. 2009;8(6):909–915. doi:10.4161/cc.8.6.7933
72. Ben Sahra I, Laurent K, Loubat A, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27(25):3576–3586. doi:10.1038/sj.onc.1211024
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.