")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Gene expression profile of human lung in a relatively early stage of COPD with emphysema
Authors Jeong I, Lim JH, Oh DK, Kim WJ , Oh YM
Received 27 February 2018
Accepted for publication 5 June 2018
Published 28 August 2018 Volume 2018:13 Pages 2643—2655
DOI https://doi.org/10.2147/COPD.S166812
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Ina Jeong,1,* Jae-Hyun Lim,2,* Dong Kyu Oh,3 Woo Jin Kim,4 Yeon-Mok Oh3
1Department of Internal Medicine, National Medical Center, Seoul, Republic of Korea; 2Biomedical Informatics Research Center, Division of Biomedical Informatics, Seoul National University College of Medicine, Seoul, Republic of Korea; 3Department of Pulmonary and Critical Care Medicine, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Republic of Korea; 4Department of Internal Medicine and Environmental Health Center, Kangwon National University Hospital, School of Medicine, Kangwon National University, Chuncheon, Republic of Korea
*These authors contributed equally to this work
Purpose: As only some smokers develop COPD with emphysema, we explored the molecular pathogenesis of early-stage COPD with emphysema using gene expression profiling of human lung tissues.
Patients and methods: First, 110 subjects who had smoked more than ten pack-years were classified into three groups: COPD with emphysema, COPD without emphysema, and healthy smokers. COPD and emphysema were confirmed by post-bronchodilator forced expiratory volume in 1 second/forced vital capacity <0.7 and by chest computed tomography. Lung tissues obtained surgically from the 110 subjects were processed and used for RNA-Seq analysis.
Results: Among the 110 subjects, 29 had COPD with emphysema, 21 had COPD without emphysema, and 60 were healthy smokers; their mean post-bronchodilator forced expiratory volume in 1 second values were 78%, 80%, and 94%, respectively. Using RNA-Seq, we evaluated 16,676 genes expressed in lung tissues. Among them, 1,226 genes in the COPD with emphysema group and 434 genes in the COPD without emphysema group were differentially expressed genes compared to the expression in healthy smokers. In the COPD with emphysema group, ACER2 and LMAN2L were markedly increased and decreased, respectively. In the COPD without emphysema group, the CHRM3 gene, previously reported to be associated with COPD, and HDAC10 were markedly increased and decreased, respectively.
Conclusion: Our study identified differences in gene expression in subjects with COPD according to emphysema status using RNA-Seq transcriptome analysis. These findings may have mechanistic implications in COPD.
Keywords: COPD, emphysema, RNA-Seq, transcriptome, gene
Introduction
COPD is characterized by persistent airflow limitation and is currently the fourth leading cause of death worldwide. COPD is an inflammatory lung disorder with complex pathogenesis and pathological features. Although the mechanisms of COPD are not completely understood, emphysema and small airway disease are the two main characteristics of COPD.1 While emphysema can exist in individuals who do not have airflow obstruction, it is more common among patients who have moderate or severe airflow obstruction and is associated with increased mortality.1–3 The mechanisms involved in emphysema development include protease and anti-protease imbalance, persistent airway inflammation, increased oxidative stress, apoptosis of alveolar cells, and ineffective repair.1,4 However, current therapeutic approaches to COPD mostly focus on targeting chronic bronchiolitis, which appears to be ineffective for emphysema. While cigarette smoking is one of the most well-studied risk factors for COPD, only about 25% of long-term cigarette smokers develop COPD, while the remainder do not.5 The development of COPD is strongly influenced by genetic factors, and genetic variation is also an important determinant of emphysema and airway disease,6 as well as environmental factors.7
Gene expression studies of diseased lungs can provide high-throughput results to better understand the molecular mechanism of COPD. Recent gene expression studies in the lung tissue of emphysema or COPD patients demonstrated and suggested not only several top differentially expressed genes (DEGs) such as known COPD genome-wide association study genes or B-cell-related genes, but also information on pathways related to tissue remodeling and wound repair due to chronic inflammation of COPD.8–10 Next-generation sequencing technology was applied to transcriptomics recently. RNA-Seq technology provides read counts of RNA fragments in each gene.11 Background and cross-hybridization are not issues in RNA-Seq, and the technology can quantify both lowly and highly abundant transcripts.12 The RNA-Seq method has been found to have a high correlation with microarray analyses in gene expression profiles. It was superior at detecting low-abundance transcripts, differentiating biologically critical isoforms, and identifying genetic variants. RNA-Seq was also demonstrated to have a broader dynamic range than microarray, allowing for the detection of more DEGs with higher fold changes.13 In recent studies on lung diseases using RNA-Seq data, the number of COPD subjects was relatively small. In a previous transcriptome study, we reported a method for identifying DEGs between COPD patients and individuals without the disease, suggesting that oxidative phosphorylation, protein degradation, and chromatin modification were dysregulated pathways in the lung tissues of COPD patients.14,15 However, COPD patients diagnosed using decreased lung function parameters can have various phenotypes and are a heterogeneous population. Therefore, in this study, we identified subjects with parenchymal emphysema using computed tomography (CT) imaging and performed gene expression profiling using RNA-Seq of lung tissues. The objective of the current study was to investigate the genetic differences between subjects with emphysema COPD and with non-emphysema COPD.
Patients and methods
Study subjects and specimens
Subjects had been registered at an in-house tissue storage system, the Asan Biobank, between January 2008 and November 2011. The lungs from the subjects were resected due to malignancy. Immediately after the resection, lung tissues were obtained as far away as possible from the cancer tissues and stored in liquid nitrogen tanks in the Asan Biobank.
We selected only subjects who had smoked more than ten pack-years. We defined past smokers as subjects who quit smoking at least 1 year before lung resection surgery. All current smokers stopped smoking at least 1 month before lung resection surgery. On the contrary, we excluded the following subjects: nonsmokers or smokers with less than ten pack-years of smoking, no CT images, or no post-bronchodilator spirometry data. We also excluded subjects in cases where emphysema was uncertain as evaluated by CT (eg, borderline emphysema with a modified Goddard score <1). In addition, we excluded subjects who had been using inhaled or systemic corticosteroids because corticosteroids may affect the expression of genes related to inflammatory and immune reactions.16 The flowchart of the study subjects selection is shown in Figure 1.
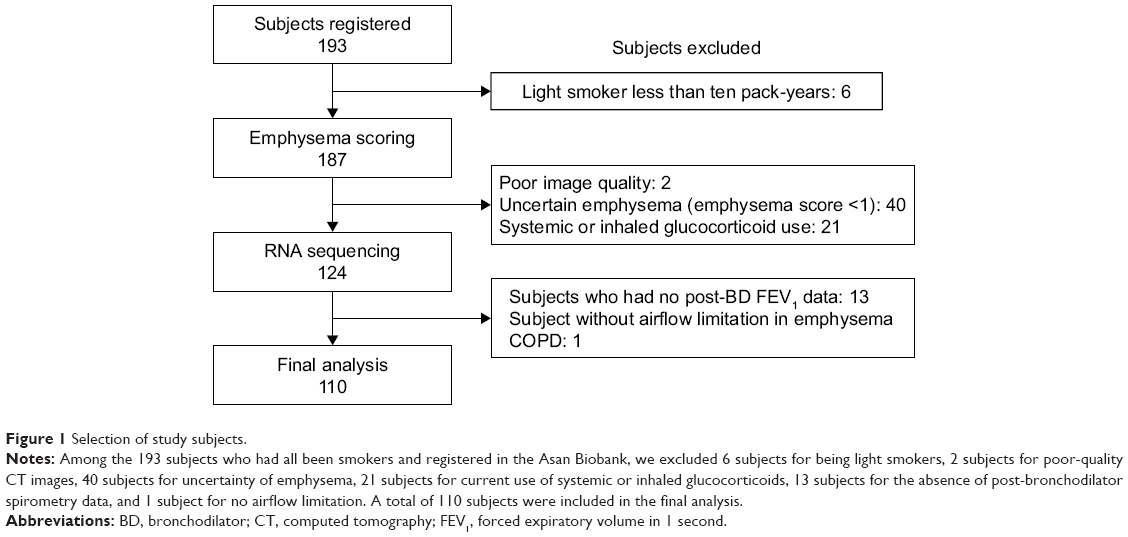 | Figure 1 Selection of study subjects. |
This retrospective study was approved by the institutional review board of Asan Medical Center (protocol no: 2014-0369), and written informed consent was obtained from all subjects.
Classification of groups
We classified the subjects into three groups: COPD with emphysema, COPD without emphysema, and healthy smokers without emphysema or airflow limitation, because these three groups can have DEGs and different involved pathways. We defined the “COPD with emphysema group” as subjects with emphysema and airflow limitation and the “COPD without emphysema group” as subjects without emphysema but with airflow limitation. Airflow limitation was defined as a post-bronchodilator forced expiratory volume in 1 second (FEV1)/forced vital capacity ratio of <0.7 in accordance with the American Thoracic Society/European Respiratory Society criteria.17
Emphysema evaluation
The severity of emphysema was semi-quantitatively assessed by two independent pulmonologists according to the modified Goddard scoring system, a visual scale, in which areas of vascular disruption and low attenuation value were scored for each lung field.18 Six images were evaluated in three slices of the apex, base, and middle of the lungs. Each image was classified as normal (score 0), ≤5% affected (score 0.5), ≤25% affected (score 1), ≤50% affected (score 2), ≤75% affected (score 3), or >75% affected (score 4), giving a minimum score of 0 and maximum of 4 (Figure 2). Moreover, the sum of the six image scores was converted to an average score that was considered the representative value of the severity of emphysema in each person. All images were blindly evaluated twice by two educated readers who are pulmonologists. Because an interclass correlation analysis showed that the emphysema score had good agreement between the two physicians (Cronbach’s alpha value was 0.967), emphysema scoring by one principal physician was finally applied.
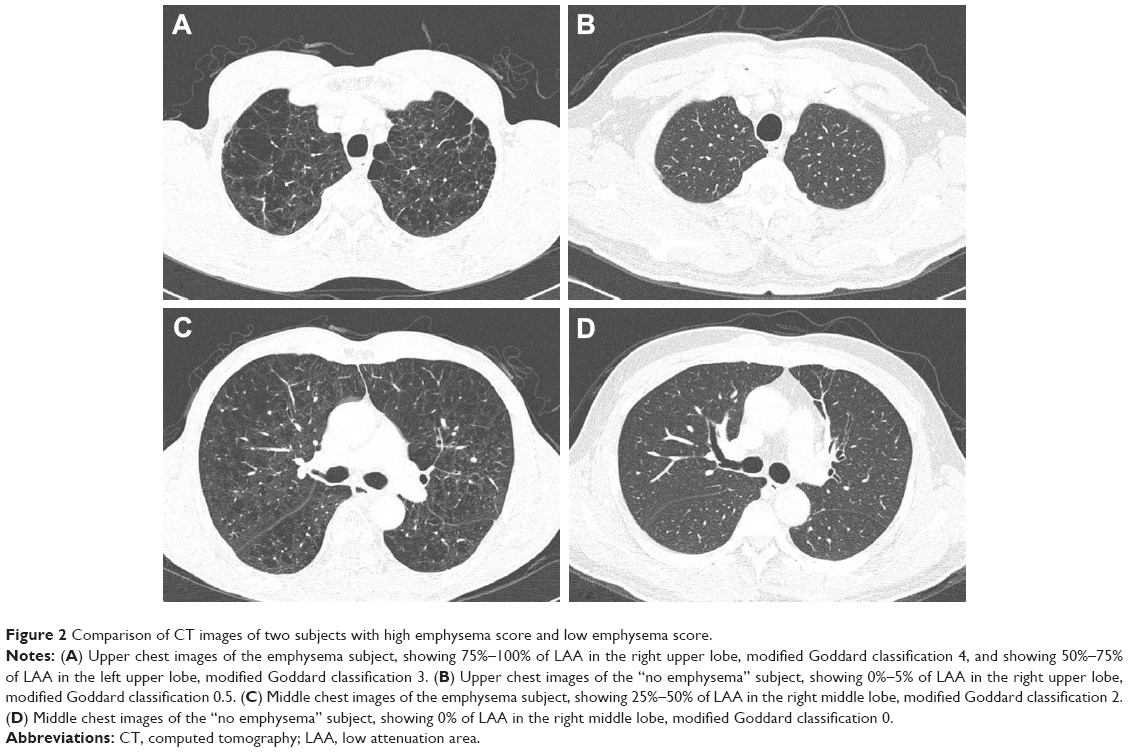 | Figure 2 Comparison of CT images of two subjects with high emphysema score and low emphysema score. |
We categorized the subjects into three groups according to emphysema score. Subjects with a final emphysema score of >1 were defined as having “emphysema”; subjects with an emphysema score ≤0.5 were defined as having “no emphysema”; and the remaining subjects were defined as “uncertainty of emphysema”. We decided to exclude the 40 “uncertainty of emphysema” subjects from the genetic analysis to avoid confusion, particularly with subjects with borderline emphysema.
RNA preparation and sequencing
Total RNA was isolated from apparently normal fresh frozen lung tissue that was remote from the lung cancer. RNA integrity was assessed using an Agilent Bioanalyzer, and RNA purity was assessed using a NanoDrop spectrophotometer. One microgram of total RNA was used to generate cDNA libraries using the TruSeq RNA library kit. The protocol consisted of poly A-selected RNA extraction, RNA fragmentation, reverse transcription using random hexamer primers, and 100 bp paired-end sequencing using the Illumina HiSeq 2000 system. All data have been deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus public repository and can be accessed through the accession number GSE57148.
Quality control and data management
For quality control, read quality was verified using FastQC and read alignment was verified using Picard. DEG analysis was performed using TopHat and Cufflinks software.19 To estimate the expression levels, the RNA-seq reads were mapped to the human genome using TopHat (version 1.4.1)20 and quantified using Cufflinks software 2.0.0.21 Cufflinks software was run with the UCSC hg19 human genome and transcriptome references. The numbers of isoform and gene transcripts were calculated, and the relative abundance of transcripts was measured in fragments per kilobase of exon per million fragments mapped (FPKM). Expression levels were extracted as an FPKM value for each gene of each sample using Cufflinks software. Genes with FPKM values of 0 across all samples were excluded. Filtered data were subjected to upper quantile normalization. Statistical significance was determined using Student’s t-test. The false discovery rate was controlled by adjusting values using the Benjamini–Hochberg algorithm. The analysis steps used are shown in Figure S1.
Pathway analysis
Functional enrichment analysis was performed using gene set enrichment analysis (GSEA; version 2.0.8), which combines information from previously defined gene sets obtained from the Molecular Signature Database (version 6.1). Biological gene functional annotation analysis was performed using DAVID (version 6.7) with a list of DEGs.
Statistical analysis
Clinical statistical analyses were performed using SPSS v18.0 (SPSS Inc., Chicago, IL, USA). The results are expressed as the mean ± SD. One-way analysis of variance (ANOVA) was performed using R software version 3.3.3 to identify DEGs between patients with COPD with emphysema, with COPD without emphysema, and healthy smokers. For genes with adjusted P-values <0.01 in ANOVA, Tukey’s honestly significant difference post hoc test was used for two-group comparisons.
Results
Study subjects
Among the 110 subjects, 29 had COPD with emphysema, 21 had COPD without emphysema, and 60 were healthy smokers. Their mean post-bronchodilator FEV1 values were 78%, 80%, and 94%, respectively (Table 1). Among the three groups of subjects, the group of healthy smokers was significantly younger (67.0 vs 65.7 vs 60.0 years; P<0.001) and smoked less (43.6 vs 49.1 vs 37.0 pack-years; P=0.039). On the contrary, the pre-bronchodilator and post-bronchodilator FEV1 values and the ratio of FEV1 to forced vital capacity were significantly higher in healthy smokers. There were no significant differences in comorbidities or inhaled bronchodilator use among the three groups of subjects (Table 1).
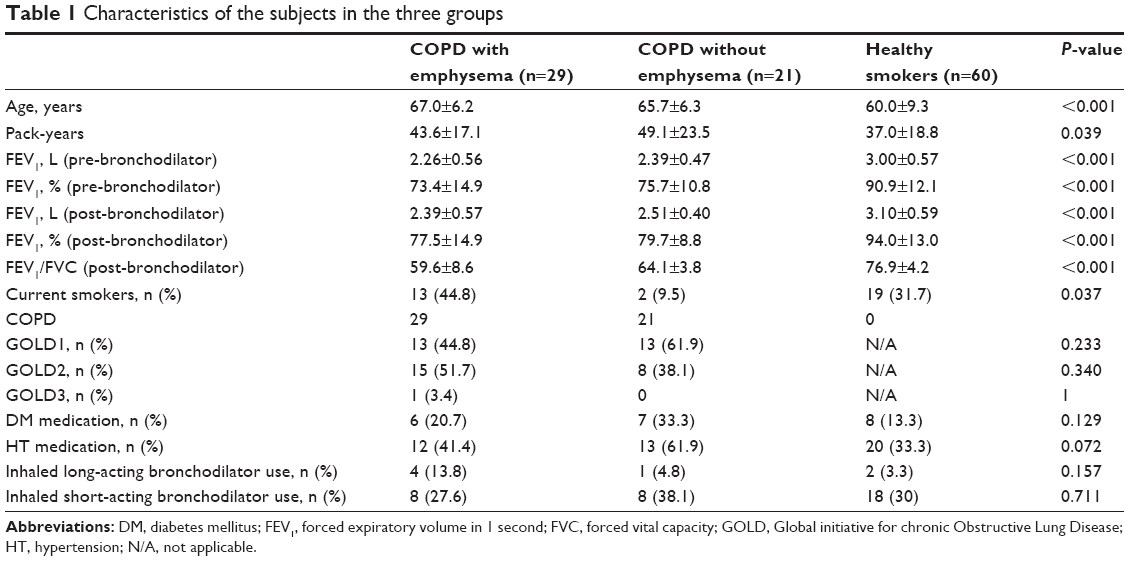 | Table 1 Characteristics of the subjects in the three groups |
Differentially expressed genes
Data about quality control of transcript analysis of RNA-Seq were collected (see Supplementary material). After filtering for genes with zero counts in the whole samples, noncoding genes, and low-variance genes, a total of 16,676 genes were analyzed. Out of these genes, 4,087 genes were differentially expressed among the three groups (Benjamini–Hochberg adjusted). In the COPD with emphysema group, 1,588 genes were upregulated and 1,519 genes were downregulated, compared with those in healthy smokers. In the COPD without emphysema group, 1,257 genes were upregulated and 1,058 genes were downregulated, compared with those in healthy smokers. In both COPD groups, 939 genes were upregulated and 942 genes were downregulated compared with those in healthy smokers, and no genes were reciprocally regulated. Thus, 649 genes were upregulated in the COPD with emphysema group, but not in the COPD without emphysema group, compared with those in healthy smokers, and 577 genes were downregulated in the COPD with emphysema group, but not in the COPD without emphysema group, compared with those in healthy smokers. Moreover, 318 genes were upregulated in the COPD without emphysema group, but not in the COPD with emphysema group, compared with those in healthy smokers, and 116 genes were downregulated in the COPD without emphysema group, but not in the COPD with emphysema group, compared with those in healthy smokers (Table 2; Figure S2). Therefore, a total of 1226 genes in the COPD with emphysema group and 434 genes in the COPD without emphysema group were differentially expressed genes respectively compared to expression in healthy smokers. Among the top DEGs in the COPD with emphysema group, ACER2 and LMAN2L were markedly increased and decreased, respectively. In addition, the CHRM3 gene and HDAC10 were markedly increased and decreased, respectively, in the COPD without emphysema group (Table 3).
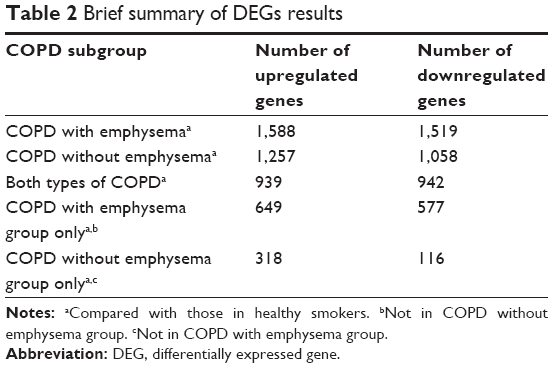 | Table 2 Brief summary of DEGs results |
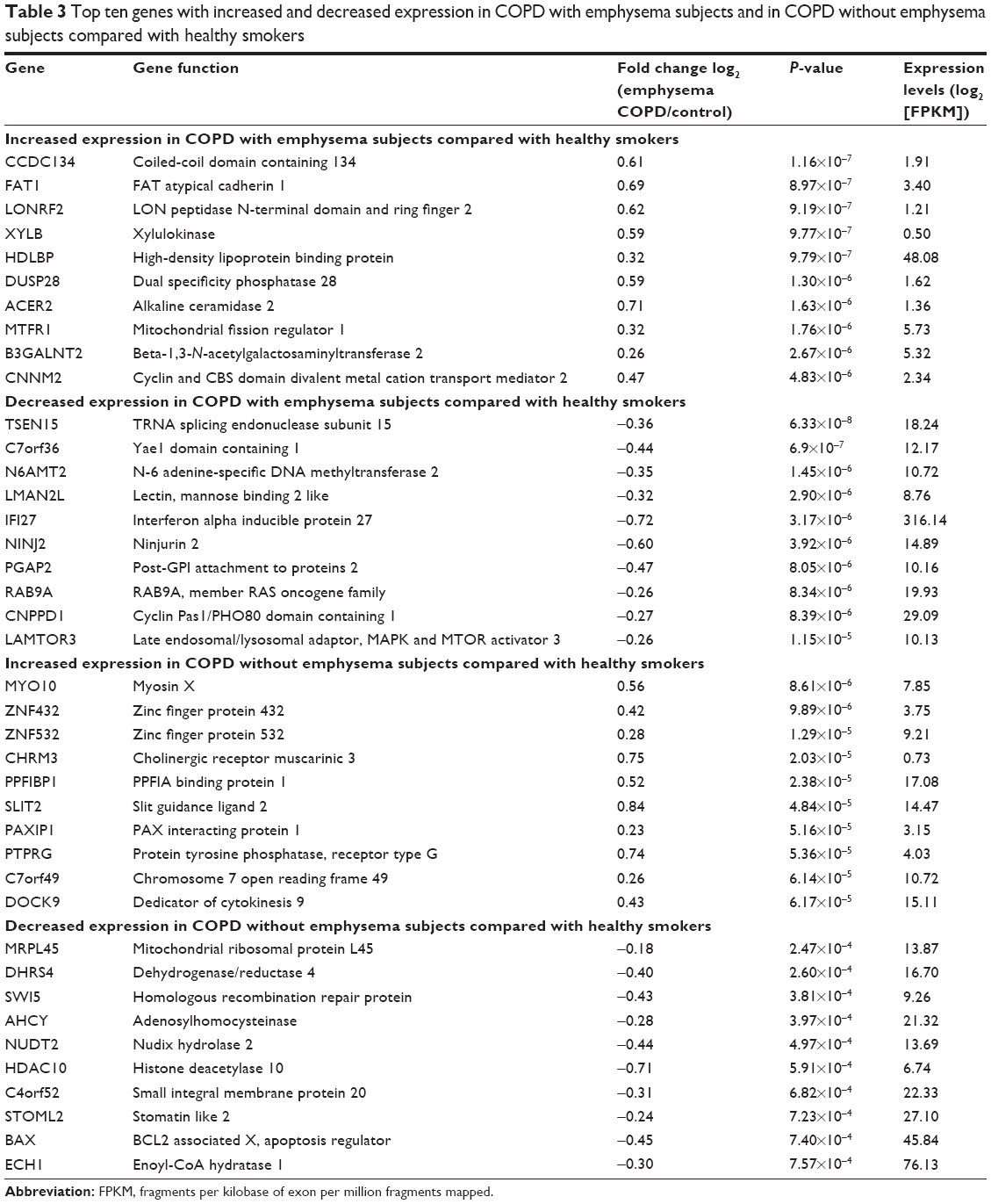 | Table 3 Top ten genes with increased and decreased expression in COPD with emphysema subjects and in COPD without emphysema subjects compared with healthy smokers |
We performed EdgeR analysis to confirm the results, and the results were consistent with 95% or more of the t-test. We also performed two-way ANOVA using the presence and absence of emphysema and current smoking status as independent variables. Moreover, we performed three-way ANOVA including emphysema presence and current smoking interaction term because current smoking status has an important effect on emphysema development. However, for the DEG data, the two-way ANOVA results showed about 90% agreement with the one-way ANOVA results, and the three-way ANOVA results were similar to the two-way ANOVA results.
Pathway analysis
In the pathway analysis using DAVID, mitochondrion genes, response to redox state, and cell redox homeostasis-related pathways decreased in expression in the COPD with emphysema group, compared to those in healthy smokers. Mitochondrion genes, response to oxidative stress, positive regulation of DNA repair, and mismatch repair-related pathways showed decreased expression in the COPD without emphysema group, compared to those in healthy smokers. Lamellipodium, cell–cell adherens junction, cell adhesion, cadherin binding involved in cell–cell adhesion, and focal adhesion-related pathways increased in expression in the COPD without emphysema group, compared to those in healthy smokers (Table 4). Heat maps indicating the genes that were differentially expressed between each group of COPD subjects and healthy smokers are shown in Figures 3 and 4. Also, the additional analysis on the GSEA as well as the pathway analysis showed no significant relation with cancer gene ontology term.
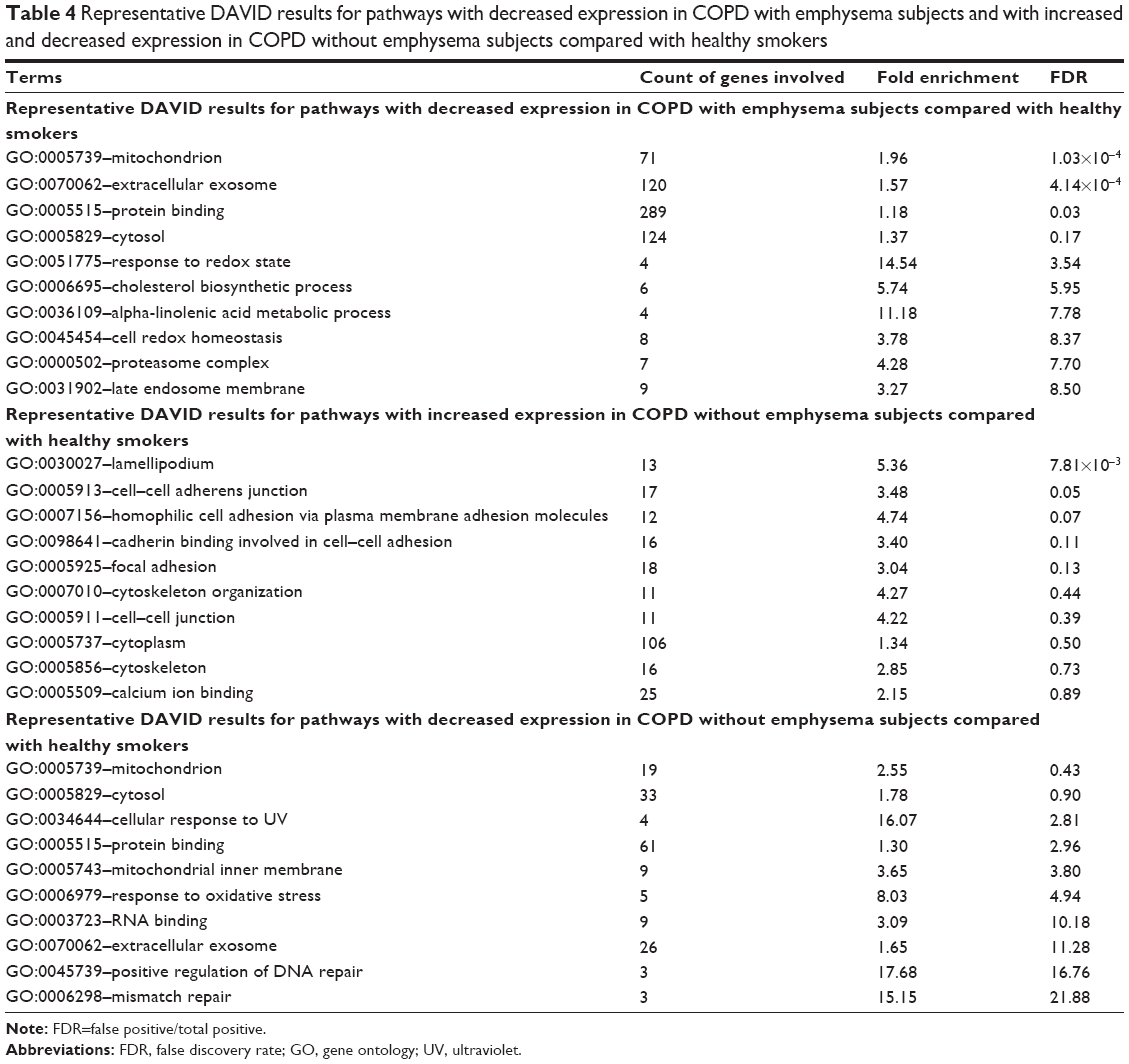 | Table 4 Representative DAVID results for pathways with decreased expression in COPD with emphysema subjects and with increased and decreased expression in COPD without emphysema subjects compared with healthy smokers |
 | Figure 3 Heat map of gene expression in the lung tissues on comparing COPD with emphysema vs healthy smokers. |
 | Figure 4 Heat map of gene expression in the lung tissues on comparing COPD without emphysema vs healthy smokers. |
Discussion
In this study, we identified new genes related to emphysema by using RNA-Seq to analyze the gene expression profiles of human lung tissues from COPD subjects with or without emphysema and comparing them with those from healthy smokers. For proper analysis, we classified the COPD subjects according to emphysema phenotype into two groups, one with emphysema and the other without emphysema, using visual emphysema scoring on CT scans. We found different gene sets in the two groups of COPD with emphysema and without emphysema.
In total, 4,087 genes were identified with a Benjamini–Hochberg adjusted P-value <0.01. We previously validated a subset of RNA-Seq data with quantitative real-time polymerase chain reaction and the results were in good agreement.15 We attempted to identify notable genes related to emphysema or COPD among the 649 upregulated and 577 downregulated genes that were differentially expressed in the COPD with emphysema group. However, the COPD with emphysema group did not exhibit any previously known remarkable DEG. Although emphysema is defined as enlargement of the respiratory acinus, the pattern of enlargement may not be uniform, and different genetic or epigenetic etiologies of emphysema tend to produce different patterns of acinar enlargement. Moreover, some subjects with COPD with emphysema may also overlap with COPD subjects without emphysema, and finally, it is difficult to differentiate exactly between the two groups of COPD, as genes associated with the COPD without emphysema group might also be associated and, therefore, expressed in the COPD with emphysema group. Nonetheless, there are some genes associated with emphysema among the top-rated genes showing increased or decreased expression in the COPD with emphysema group. All new genes related to emphysema have relatively low expression levels, and it could be possible that those of DEGs in the present study have not been found in previous other next-generation sequencing technology experiments other than RNA-seq since the expression level was relatively low.
ACER2, which was one of the genes with a high expression in the COPD with emphysema group, is a critical sphingolipid metabolizing enzyme and has been shown to be markedly upregulated and involved in apoptosis in response to various stimuli such as DNA damage.22,23 Ceramide upregulation was suggested to be a possible crucial mediator of alveolar destruction in an animal emphysema model experiment.23 It is not known whether ACER2 with increased expression was associated with apoptosis of the emphysema in the present study. FAM13A, known as a COPD-related gene, was significantly upregulated in COPD with emphysema group (P-value 0.00096), and this is consistent with many previous genetic studies.9,10
Mannose-binding lectin protein is a known key component of innate immunity that promotes macrophage phagocytosis and clears apoptotic cells. It was shown to decrease in the airway following oxidative stress and be associated with clinical outcomes in COPD in recent studies.24,25 Because LMAN2L also showed decreased expression in the COPD with emphysema group in the present study, it is a possible emphysema-related downregulated gene.
Meanwhile, among the 318 upregulated and 116 downregulated genes that were differentially expressed in the COPD without emphysema group, several genes were remarkable and previously reported to be associated with respiratory disease. The CHRM3 gene showed increased expression in this group of COPD without emphysema subjects compared to that in the healthy smoker group. M3-cholinoreceptors are expressed on macrophages, one of the key cells involved in inflammation, and contribute to their chemotaxis. Although little is known about the role of CHRM3 gene in the development of airway obstruction or COPD, it was recently suggested to be a druggable target candidate gene.26 According to a recent study, the differential expression of the CHRM3 gene might be associated with the clinical phenotypes of COPD with bronchial hyperresponsiveness.27 Significantly lower levels of CHRM3 mRNA were observed in patients with COPD compared with those in asthma patients, and CHRM3 gene expression was significantly elevated in COPD patients with bronchial hyperresponsiveness compared with that in patients without bronchial hyperresponsiveness.27 The SLIT2 gene also showed increased expression in COPD without emphysema subjects compared with that in healthy smokers. SLIT2 is present in murine fetal lungs and adult rat lungs and is expressed by club cells in the adult mouse. However, little is known about the expression of SLIT2 in human lung tissue during inflammation and fibrosis. In another study, SLIT2, which inhibits fibrocyte differentiation, was shown to be produced by fibroblasts and epithelial cells in the lung tissue from patients with relatively normal lung function, whereas less SLIT2 was found in the fibrotic lesions of patients with advanced disease. Whether the upregulated SLIT2 level is augmented by fibroblasts, which modulate the immune system, in a relatively early stage of the disease (COPD without emphysema rather than COPD with emphysema) or there are factors present that upregulate SLIT2 production is unclear. SLIT2 levels seem to be important in the regulation of lung inflammation and fibrosis, and therapeutic use of SLIT2 may be beneficial for regulating fibrosis.28,29
Among the top ten genes with decreased DEGs in the COPD without emphysema group, the expression levels of HDAC10 and STOML2 were noticeable. HDAC2 is known to play a critical role in suppressing inflammatory gene expression in the airways, lung parenchyma, and alveolar macrophages, and HDAC activity was found to be decreased in patients with COPD.29 Histone deacetylation modulates chromatin structure and plays an important role in transcriptional regulation, cell cycle progression, and developmental events. HDAC10 is a novel class IIB histone deacetylase found in both the nucleus and the cytoplasm, and it was reported to function as a transcriptional repressor.30 It was reported that the knockdown of HDAC10 resulted in the accumulation of reactive oxygen species (ROS) and cell death, indicating its importance in the response to oxidative stress in gastric cancer cell lines.31 STOML2 is a gene that plays a key role in mitochondrial function and T-cell activation. Recently, the expression of STOML2 was negatively correlated with that of the IL6R, which has been implicated in asthma pathophysiology, and the increased expression of STOML2 was associated with an increased asthma risk by RNA-seq analysis.32 The exact roles and mechanisms of HDAC10, STOML2, and other genes in COPD, especially in early stages of the disease, need to be clarified in future studies. BAX, an apoptosis regulator, is a marker of upregulated apoptosis in COPD, and it was found to downregulated in the COPD without emphysema group compared with that in healthy smokers in the present study, contrary to a previous report that patients with emphysema showed a significant increase in the Bax index.33 Alterations in the expression of apoptosis-related proteins might be important in determining progression to apoptosis in emphysema pathogenesis.
The major analytic strategy for interpreting hundreds of statistically significant differences in gene expression involves pathway analysis. Previous studies on the COPD transcriptome reported a high degree of overlap in the biological processes affected. In the current study, the most altered pathway in the COPD without emphysema group was lamellipodium organization and cell–cell adherens junctions, possibly providing a clue for the pathogenesis of emphysema. A study found that the genes involved in focal adhesion and lamellipodium organization showed decreased expression with disease severity in emphysema patients.34 Lamellipodium is involved in cell migration, which suggests cytoskeleton reorganization related to epithelial would repair.35 Cell–cell adherens junctions in airway epithelium can function as a barrier in the airway tract. Impairment of the barrier function may increase susceptibility to infection and promote exaggerated and prolonged innate immune responses to pathogens, resulting in chronic inflammation.35 Pathways related to focal adhesion, cytoskeleton or organization, and cell–cell junctions also showed increased expression in subjects with COPD without emphysema. The repair process of airway epithelium after injury in the early stage of COPD might be augmented as a defense mechanism. There was a study about genes decreasing in expression such as transforming growth factor beta pathway, actin organization, and integrin signaling were enriched in tissue repair processes in the lung tissue of emphysematous destruction.9 Also, another recent report about the differential expression of genes associated with tissue repair between small airway obstructive and emphysematous destructive lesions suggested a mechanism for the development of peripheral lesions of COPD.36 The expression of mitochondrial genes was previously shown to be reduced in the lung tissues of COPD subjects using a serial analysis of gene expression,37 which is consistent with the results of the current study for both COPD groups. Pathways related to cytosol, cellular responses to ultraviolet, responses to oxidative stress, positive regulation of DNA repair, and mismatch repair showed decreased expression in the COPD without emphysema group. Mitochondria are multifunctional cellular organelles and play an important role in not only numerous aspects of cell morphology and physiology, but also the innate immune system.38 Oxidant–antioxidant imbalance is recognized as one of the significant factors in COPD pathogenesis. One endogenous source of ROS is mitochondria. Although the leakage of electrons from the electron transport chain and the formation of ROS result from the physiological functioning of mitochondria, there are various intra- and extracellular factors that may increase these amounts and significantly contribute to oxidative–antioxidative imbalance.39 Proteasome complex-related genes were also decreased in expression in the COPD with emphysema group in the present study. The proteasome is not only a multicatalytic enzyme complex that is responsible for the maintenance of cellular proliferation, cell cycling, gene transcription, apoptosis, and antioxidant responses, but also serves as a protein quality control system. Impaired proteasome function can result in the accumulation of altered proteins, resulting in cellular dysfunction and cell death. Recently, decreased proteasomal function was shown to accelerate cigarette smoke-induced pulmonary emphysema in a mouse model.40 Impaired proteasomal expression might be important in the pathogenesis of emphysema.
Pathways with increased expression in the COPD with emphysema group included genes related to adenosine triphosphate binding, helicase activity, nucleoplasm, adenosine triphosphate-dependent RNA helicase activity, membranes, and the endoplasmic reticulum, among others, which could be nonspecifically upregulated in both COPD groups. In addition, we did not find any B-cell-related genes whose expression increased with emphysematous destruction in recently reported studies,8,9 probably because the subjects were in relatively early stages of the disease. The genes with increased expression in the COPD without emphysema group might be associated with, or included among, the genes that increased in the COPD with emphysema group, since small airway obstruction occurred first and emphysema followed. We performed two-way ANOVA and three-way ANOVA to assess the effects of current smoking on gene expression, because it was previously reported that gene expression is associated with smoking in the oxidative stress response, antiapoptosis, and cell death signaling and can be altered by smoking.41 It is possible that genes with increased expression related to current smoking status are more likely to be genes related to emphysema or COPD. However, the overall results after correction for the smoking effect were similar to those from the one-way ANOVA. The total number of patients included in the study was small and the difference by the presence of COPD is evident, so smoking effects may not be reflected. Ultimately, our findings will require replication, ideally in additional large cohorts that include a range of COPD severities.
Limitations
There are several limitations of this study. First, all subjects were men who currently smoked or were former smokers. Healthy smokers also included current or former smokers, and although they did not have emphysema or airflow limitation, it is possible that they had aberrant genetic modifications and expression. According to previous reports regarding sex differences in emphysema and airway disease, male smokers had more emphysema than female smokers, but female smokers did not show increased airway wall thickness compared with men.42,43 However, to date, whether sex-specific genetic risk factors might underlie the differences in COPD susceptibility and severity is largely unknown. According to a recent genetic association study by genome-wide association study, a higher expression of CELSR1, a fetal lung development gene, was observed in female compared with male samples of humans in fetal lung tissue dataset.44 Further studies are needed to explore sex-specific association in the development of emphysema or COPD. Additional gene expression studies that include never smokers and female subjects would improve the RNA-seq analysis of COPD. Second, we used normal-looking tissue adjacent to the lung cancer tissue for RNA-seq analysis. Lung tissue consists of many cell types, including macrophages, epithelial cells, and endothelial cells. Microdissection of lung tissues or single-cell sequencing would be required to determine whether the differential expression is present in all lung cells or only in a specific subset of cells. There might be possible gene expression patterns related to lung cancer. Among DEGs with significant P-values, there are several genes known to be associated with cancer, including lung cancer. PTPRG, which is thought to be a candidate tumor suppressor gene,45 was one of the top ten increased DEG in COPD without emphysema group. IFI27 gene, which is known to be associated with cancer cell apoptosis and lung carcinoma,46 showed decreased expression in COPD with emphysema group (fifth in P-value). However, not only the number of known cancer-related genes is small, but also it is difficult to distinguish whether these genes are associated with emphysema or only cancer. Also, the additional results in the GSEA as well as the pathway analysis showed no relation with cancer gene ontology term.
Finally, it is difficult to determine whether the dysregulated pathways identified in this study are a cause or a consequence of the pathogenesis of emphysema or COPD. Experiments in which the increase/decrease in DEGs is reversed and is shown to slow disease progression are needed to confirm that these pathways are causally involved in the pathogenesis of COPD.
Conclusion
Our study identified differences in gene expression in subjects with COPD according to emphysema status using RNA-Seq transcriptome analysis. These findings may have mechanistic implications in COPD.
Acknowledgments
This work was supported by the Korean Health Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (grant number HC16C2187) and the Global Research Development Center Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (MSIT; NRF-2015K1A4A3046807). The biospecimens and data used in this study were provided by Asan Bio-Resource Center, Korea Biobank Network (2012-9[52]). The authors express their sincere gratitude to Dr Sang-min Lee (Department of Radiology, Asan Medical Center) for his assistance with the radiological analysis.
Author contributions
Study design: YMO and JHK. Clinical data collection and primary analysis: IJ and DKO. Genetic data quality control and analysis: JHL. All authors made substantial contributions to interpretation of data; and took part in drafting the article or revising it critically for important intellectual content.
Disclosure
The authors report no conflicts of interest in this work.
References
Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global strategy for the diagnosis, management and prevention of chronic obstructive pulmonary disease; 2017. Available from: http://goldcopd.org/. Accessed February 10, 2017. | ||
Mcdonough JE, Yuan R, Suzuki M, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med. 2011;365(17):1567–1575. | ||
Haruna A, Muro S, Nakano Y, et al. CT scan findings of emphysema predict mortality in COPD. Chest. 2010;138(3):635–640. | ||
Macnee W, Tuder RM. New paradigms in the pathogenesis of chronic obstructive pulmonary disease I. Proc Am Thorac Soc. 2009;6(6):527–531. | ||
Løkke A, Lange P, Scharling H, Fabricius P, Vestbo J. Developing COPD: a 25 year follow up study of the general population. Thorax. 2006;61(11):935–939. | ||
Cho MH, Castaldi PJ, Hersh CP, et al. A genome-wide association study of emphysema and airway quantitative imaging phenotypes. Am J Respir Crit Care Med. 2015;192(5):559–569. | ||
Kim WJ, Lee CY. Environmental exposures and chronic obstructive pulmonary disease. Mol Cell Toxicol. 2017;13(3):251–255. | ||
Faner R, Cruz T, Casserras T, et al. Network analysis of lung transcriptomics reveals a distinct B-cell signature in emphysema. Am J Respir Crit Care Med. 2016;193(11):1242–1253. | ||
Campbell JD, Mcdonough JE, Zeskind JE, et al. A gene expression signature of emphysema-related lung destruction and its reversal by the tripeptide GHK. Genome Med. 2012;4(8):67. | ||
Morrow JD, Zhou X, Lao T, et al. Functional interactors of three genome-wide association study genes are differentially expressed in severe chronic obstructive pulmonary disease lung tissue. Sci Rep. 2017;7:44232. | ||
Mortazavi A, Williams BA, Mccue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. | ||
Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 2011;12(2):87–98. | ||
Zhao S, Fung-Leung WP, Bittner A, Ngo K, Liu X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS One. 2014;9(1):e78644. | ||
Kim WJ, Lim JH, Hong Y, et al. Altered miRNA expression in lung tissues of patients with chronic obstructive pulmonary disease. Mol Cell Toxicol. 2017;13(2):207–212. | ||
Kim WJ, Lim JH, Lee JS, Lee SD, Kim JH, Oh YM. Comprehensive analysis of transcriptome sequencing data in the lung tissues of COPD subjects. Int J Genomics. 2015;2015:206937. | ||
Adcock IM, Ito K, Barnes PJ. Glucocorticoids: effects on gene transcription. Proc Am Thorac Soc. 2004;1(3):247–254. | ||
Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–338. | ||
Makita H, Nasuhara Y, Nagai K, et al. Characterisation of phenotypes based on severity of emphysema in chronic obstructive pulmonary disease. Thorax. 2007;62(11):932–937. | ||
Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–578. | ||
Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. | ||
Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–515. | ||
Coant N, Sakamoto W, Mao C, Hannun YA. Ceramidases, roles in sphingolipid metabolism and in health and disease. Adv Biol Regul. 2017;63:122–131. | ||
Petrache I, Natarajan V, Zhen L, et al. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med. 2005;11(5):491–498. | ||
Mandal J, Malla B, Steffensen R, et al. Mannose-binding lectin protein and its association to clinical outcomes in COPD: a longitudinal study. Respir Res. 2015;16:150. | ||
Tran HB, Ahern J, Hodge G, et al. Oxidative stress decreases functional airway mannose binding lectin in COPD. PLoS One. 2014;9(6):e98571. | ||
Wain LV, Shrine N, Artigas MS, et al. Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat Genet. 2017;49(3):416–425. | ||
Selivanova PA, Kulikov ES, Kozina OV, et al. Differential expression of the β2-adrenoreceptor and M3-cholinoreceptor genes in bronchial mucosa of patients with asthma and chronic obstructive pulmonary disease. Ann Allergy Asthma Immunol. 2012;108(1):39–43. | ||
Pilling D, Zheng Z, Vakil V, Gomer RH. Fibroblasts secrete Slit2 to inhibit fibrocyte differentiation and fibrosis. Proc Natl Acad Sci U S A. 2014;111(51):18291–18296. | ||
Ito K, Ito M, Elliott WM, et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med. 2005;352(19):1967–1976. | ||
Koeneke E, Witt O, Oehme I. HDAC family members intertwined in the regulation of autophagy: a druggable vulnerability in aggressive tumor entities. Cells. 2015;4(2):135–168. | ||
Lee JH, Jeong EG, Choi MC, et al. Inhibition of histone deacetylase 10 induces thioredoxin-interacting protein and causes accumulation of reactive oxygen species in SNU-620 human gastric cancer cells. Mol Cells. 2010;30(2):107–112. | ||
Revez JA, Matheson MC, Hui J, et al. Identification of STOML2 as a putative novel asthma risk gene associated with IL6R. Allergy. 2016;71(7):1020–1030. | ||
Yokohori N, Aoshiba K, Nagai A; Respiratory Failure Research Group in Japan. Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysema. Chest. 2004;125(2):626–632. | ||
Kantrowitz JJ, Kinose D, Vasilescu DM, et al. Transcriptomic alterations associated with emphysematous lung destruction in centrilobular and panlobular emphysema. Am J Respir Crit Care Med. 2016;193:A4075. | ||
Puchelle E, Zahm JM, Tournier JM, Coraux C. Airway epithelial repair, regeneration, and remodeling after injury in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3(8):726–733. | ||
Gosselink JV, Hayashi S, Elliott WM, et al. Differential expression of tissue repair genes in the pathogenesis of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181(12):1329–1335. | ||
Ning W, Li CJ, Kaminski N, et al. Comprehensive gene expression profiles reveal pathways related to the pathogenesis of chronic obstructive pulmonary disease. Proc Natl Acad Sci U S A. 2004;101(41):14895–14900. | ||
Aravamudan B, Thompson MA, Pabelick CM, Prakash YS. Mitochondria in lung diseases. Expert Rev Respir Med. 2013;7(6):631–646. | ||
Białas AJ, Sitarek P, Miłkowska-Dymanowska J, Piotrowski WJ, Górski P. The role of mitochondria and oxidative/antioxidative imbalance in pathobiology of chronic obstructive pulmonary disease. Oxid Med Cell Longev. 2016;2016:7808576. | ||
Yamada Y, Tomaru U, Ishizu A, et al. Decreased proteasomal function accelerates cigarette smoke-induced pulmonary emphysema in mice. Lab Invest. 2015;95(6):625–634. | ||
Charlesworth JC, Curran JE, Johnson MP, et al. Transcriptomic epidemiology of smoking: the effect of smoking on gene expression in lymphocytes. BMC Med Genomics. 2010;3:29. | ||
Camp PG, Coxson HO, Levy RD, et al. Sex differences in emphysema and airway disease in smokers. Chest. 2009;136(6):1480–1488. | ||
Grydeland TB, Dirksen A, Coxson HO, et al. Quantitative computed tomography: emphysema and airway wall thickness by sex, age and smoking. Eur Respir J. 2009;34(4):858–865. | ||
Hardin M, Cho MH, Sharma S, et al. Sex-based genetic association study identifies CELSR1 as a possible chronic obstructive pulmonary disease risk locus among women. Am J Respir Cell Mol Biol. 2017;56(3):332–341. | ||
Cheung AK, Ip JC, Chu AC, et al. PTPRG suppresses tumor growth and invasion via inhibition of Akt signaling in nasopharyngeal carcinoma. Oncotarget. 2015;6(15):13434–13447. | ||
Cheon H, Borden EC, Stark GR. Interferons and their stimulated genes in the tumor microenvironment. Semin Oncol. 2014;41(2):156–173. |
Supplementary materials
 | Figure S1 Schematic overview of the transcript analysis of the RNA-seq experiment. |
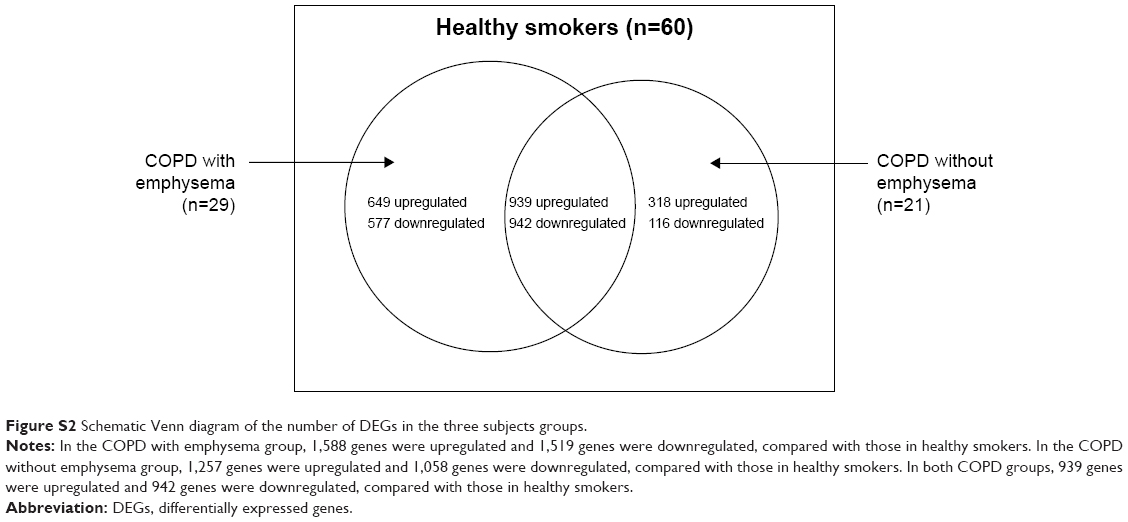 | Figure S2 Schematic Venn diagram of the number of DEGs in the three subjects groups. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.