Back to Journals » Journal of Hepatocellular Carcinoma » Volume 8
Gene Expression Profile and Prognostic Value of m6A RNA Methylation Regulators in Hepatocellular Carcinoma
Authors Chang X, Lv YF, He J ![]() , Cao Y, Li CQ, Guo QN
, Cao Y, Li CQ, Guo QN ![]()
Received 17 December 2020
Accepted for publication 23 February 2021
Published 12 March 2021 Volume 2021:8 Pages 85—101
DOI https://doi.org/10.2147/JHC.S296438
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Ahmed Kaseb
Xian Chang,1,* Yang-Fan Lv,2,* Jing He,3,* Ya Cao,2 Chang-Qing Li,1 Qiao-Nan Guo2
1Department of Orthopedics, Xinqiao Hospital, Third Military Medical University (Army Medical University), Chongqing, 400037, People’s Republic of China; 2Department of Pathology, Xinqiao Hospital, Third Military Medical University (Army Medical University), Chongqing, 400037, People’s Republic of China; 3Department of Pediatric Surgery, Guangzhou Institute of Pediatrics, Guangdong Provincial Key Laboratory of Research in Structural Birth Defect Disease, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangzhou, 510623, Guangdong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qiao-Nan Guo; Chang-Qing Li Email [email protected]; [email protected]
Background: N6-methyladenosine (m6A) RNA methylation is the most prevalent modification of mammalian RNA, and it is associated with tumorigenesis and cancer progression. Its regulation is mediated via m6A-related regulators, including “erasers,” “readers,” and “writers”. The present study evaluated the expression profile, risk signature and prognostic value of 13 m6A regulators in hepatocellular carcinoma (HCC) using different datasets, including The Cancer Genome Atlas (TCGA), Gene Expression Omnibus (GEO) and clinical samples.
Methods: We used 374 HCC samples derived from the TCGA database, 569 HCC samples from 2 GEO datasets, and clinical tumour and nontumour tissues derived from 60 patients with HCC who underwent surgery in Xinqiao Hospital Chongqing to assess the gene expression profiles and prognostic values of m6A-related regulators in HCC.
Results: Eight of 13 core m6A-related regulators were overexpressed in all databases, including TCGA, GSE, clinical tumour and nontumour tissues of HCC. Two clusters (Cluster 1 and Cluster 2) were identified via consensus clustering. Cluster 2 was associated with poorer prognosis, higher tumour grade, higher AFP levels, and worse outcome compared to Cluster 1, which indicates that these m6A-related regulators are highly correlated with HCC malignancy. We performed survival analyses using the Log rank tests and a Cox regression model. Gene enrichment analysis was used to detect the related KEGG and GO pathways. We derived a prognostic risk signature using five selected m6A-related regulators.
Conclusion: Our work suggested that m6A-related regulators might be key participants in the tumour progression of HCC and potential biomarkers with prognostic value.
Keywords: RNA modification, N6-methyladenosine, m6A, hepatocellular carcinoma, prognosis, RNA methylation regulators
Introduction
Hepatocellular carcinoma (HCC) is the third most common cause of cancer-related death, and its incidence is increasing globally.1–3 Similar to other malignant tumours, the tumorigenesis of HCC is a multistep process involving genetic, epigenetic, and transcriptomic alterations. The current 5-year survival rate is approximately 18% partially because 50% of a late-stage diagnosis, tumour metastasis, postoperative recurrence, and chemotherapeutic drug resistance.4 Despite the low survival rate, the detailed mechanisms underlying HCC progression are not known, and the identification of effective molecular targets in carcinogenesis and metastasis to improve the outcomes for patients with HCC remains a challenging issue.
Epigenetics, which involves alterations of DNA or histones, has been widely studied in tumour progression. Epigenetic changes are potentially reversible and independent of DNA sequences and may lead to the development of several therapeutic modalities. RNA modification was first described in 1974, but it was not regarded as another meaningful epigenetic layer until recently.5,6 RNA modifications include pseudouridine, N6-methyladenosine (m6A), N1-methyladenosine, 5-methylcytosine, and 2′-O-methylation. M6A is the most common and abundant modified base in eukaryotic cell mRNA. M6A is present in >300 noncoding RNAs and 7600 mRNAs, and it is widespread throughout the transcriptome.7 It is located in 3′-UTRs around long internal exons and stop codons and results in alterations of RNA stability, intracellular distribution, splicing, and translation. The primary m6A-related regulators consist of 13 genes: “writers” [methyltransferases, including methyltransferase-like 3 (METTL3), zinc finger CCCH domain-containing protein 13 (ZC3H13), methyltransferase-like 14 (METTL14), RNA-binding protein 15 (RBM15), vir-like m6A methyltransferase associated (VIRMA, also known as KIAA1429), and Wilms’ tumour 1 associating protein (WTAP)]; “erasers” [demethylases, including RNA demethylase ALKBH5 (ALKBH5) and alpha-ketoglutarate dependent dioxygenase (FTO)]; and “readers” [binding proteins, including YTH domain containing 1 (YTHDC1), YTH domain family protein 1 (YTHDF1), YTH domain containing 2 (YTHDC2), YTH domain family protein 2 (YTHDF2), and heterogeneous nuclear ribonucleoproteins C1/C2 (HNRNPC)]. Writers up-regulate m6A levels, which form a methyltransferase enzyme complex. Erasers act as demethylases and downregulate the level of m6A. Therefore, m6A levels are dynamic in nature and levels are likely regulated and determined via the interplay between “writers” and “erasers.” The “readers” act as effectors to decode the m6A RNA methylation information.5,8 These findings considerably increased our understanding of the mechanisms underlying m6A modifications.
M6A dysregulation is vital in many key biological processes, including cell proliferation, cell self-renewal capacity, stress responses, and cell death.9 The important role of m6A methylation in tumorigenesis recently emerged. Alterations in m6A-related genes promote the progression of gliomas, breast cancer, haematologic malignancies, and clear-cell renal cell carcinoma.10–13 METTL14 is the most common m6A regulatory gene in aberrant HCC m6A modification, which suppresses tumour metastasis via modulation of microRNA-126 in a manner that is dependent on DGCR8.14 Zhou et al demonstrated the role of 12 m6A-related genes in HCC from 4 datasets and found that two genes, METTL3 and YTHDF1, were associated with OS in HCC.15 Recent studies examined the role of m6A regulatory genes in HCC prognosis.16–19 The present study systematically analysed the clinical and RNA-seq pertaining to HCC from The Cancer Genome Atlas (TCGA) database and evaluated the alterations of 13 m6A regulatory genes in HCC and their association with clinicopathological features. All of the results were validated in the Gene Expression Omnibus (GEO) database and clinical samples. We report that m6A-related regulators are vital in tumorigenesis in HCC, and five m6A regulators were chosen to analyse the prognosis of HCC.
Materials and Methods
Data Resources
RNA sequencing and corresponding clinical data for 374 patients with HCC and 50 healthy (control) individuals were retrieved from TCGA, which is open to the public under related guidelines. The datasets GSE62232 and GSE14520 were downloaded from the GEO database. GSE62232 contained 91 samples, including 81 HCC tumour and 10 adjacent nontumour tissues. GSE14520 contained 488 clinical samples, including 241 tumour and adjacent tissues. The other 247 were tumour samples of HCC. R language was used to normalize the expression data.14 The clinicopathological characteristics of patients from the TCGA and GEO databases are listed in Table 1.
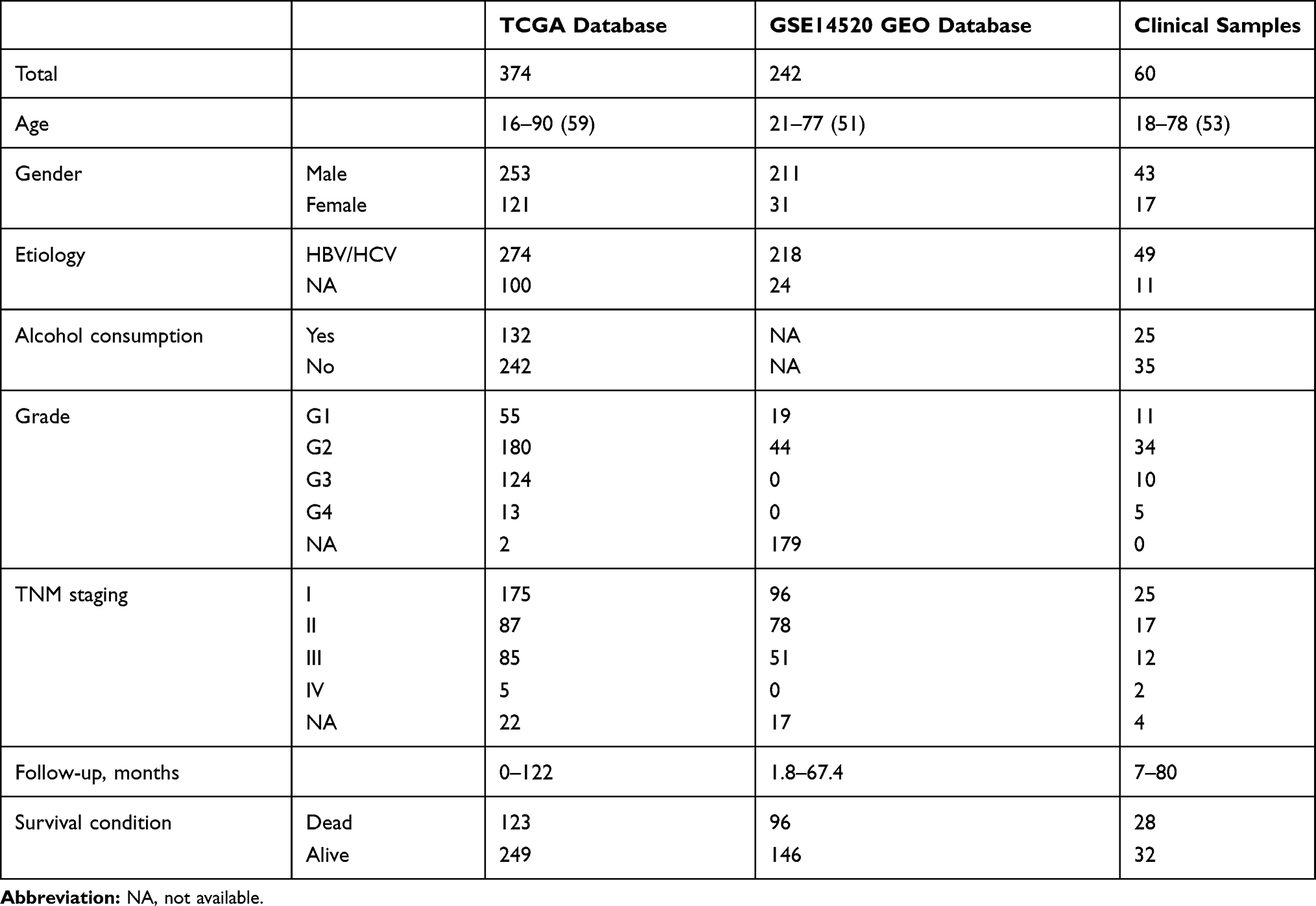 |
Table 1 Clinicopathological Features of Patients Included in This Study |
Patients and Tissue Specimens
Sixty pairs of fresh tissue samples comprised of HCC and adjacent noncancerous tissues were obtained from patients who underwent surgery and pathological biopsy in Xinqiao Hospital in Chongqing, China between 2011 and 2018. Samples were stored in liquid nitrogen. All enrolled patients were Asian.
qRT-PCR Analysis
Total RNA was extracted from frozen tissues using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Quantitative real-time reverse transcription PCR (qRT-PCR) was performed exactly as previously described.20 Briefly, mRNA reverse transcription and qRT-PCR were performed using a SYBR Green Kit (Invitrogen) with an ABI PRISM 7500 system (Applied Biosystems, Foster City, CA, USA). The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used as an internal control, and the 2-∆∆Ct method was used for relative quantification. The primers used are shown in Supplementary Table 1. The experiment was repeated 3 times with 4 replicates. Student’s t-test was used for comparisons between these two groups, and P < 0.05 was considered statistically significant.
Immunohistochemistry
Immunohistochemistry was performed as described previously.21 The primary antibodies used were METTL3 (GeneTex, USA, 1:150), RBM15 (Abcam Inc., USA, 1:50), YTHDC1 (Abcam Inc., USA, 1:000), YTHDF1 (Abcam Inc., USA, 1:100), and YTHDF2 (Abcam Inc., USA, 1:50).
Bioinformatics Analyses
The association between the expression of 13 m6A-related regulators and tumour stage was evaluated using Gene Expression Profiling Interactive Analysis (GEPIA), which is an online website for tumour- and nontumour related gene expression data, using profiling from TCGA. To systematically and effectively investigate the roles and functions of m6A-related regulators, we clustered liver cancer into different subgroups using consensus clustering. We used principal component analysis in the R package v3.4.1 to analyse the gene expression patterns in different HCC clusters. Interactions among the 13 m6A-related regulators were detected using the STRING database, and GSEA was used to determine functions that potentially correlated with HCC clusters.
Univariate Cox regression analysis was used to evaluate the potential prognostic value of m6A-related regulators in HCC. Nine genes were related to overall survival (OS), and these genes were chosen for functional analyses and the establishment of a risk score using LASSO Cox regression analysis.22,23 Five m6A-related regulators and the coefficients obtained based on the minimum criteria were introduced to determine the risk score. The following formula was used to determine the risk score:
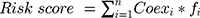
Using GO and KEGG pathway enrichment analyses, functional annotation was performed on the differentially expressed genes in each group.
Statistical Analyses
Patients were divided into two subgroups via consensus clustering of different m6A-related regulators. High- and low-risk categories were identified using a cut-off value from the median risk score, which was derived from the indicated risk signature. Multivariate and univariate Cox regression analyses were used to assess the prognostic value of the risk signature and the indicated clinicopathological parameters. Kaplan–Meier survival curves were used to compare the OS of different subgroups of HCC patients. Statistical analyses were performed using R v3.5.1 and SPSS 20.0 (version 20.0, SPSS Inc., Chicago, IL, USA). P < 0.05 was considered statistically significant.
Results
The Correlation Between m6A-Related Regulators and HCC Malignancy
Gene expression profiles were downloaded from TCGA [data for 374 HCC patients and 50 healthy (control) individuals] and GEO databases (GSE62232, data for 81 patients with HCC and 10 nontumour tissues), and the expression of m6A-related regulators was analysed. As shown in Figure 1A and B, 11 of the 13 m6A-related regulators in the TCGA database were significantly upregulated in HCC: FTO (P<0.001), YTHDC1 (P<0.001), YTHDC2 (P<0.001), ALKBH5 (P<0.001), KIAA1429 (P<0.001), METTL3 (P<0.001), HNRNPC (P<0.001), YTHDF2 (P<0.001), RBM15 (P<0.001), WTAP (P<0.001), and YTHDF1 (P<0.001). Figure 1C shows that 10 m6A-related regulators in the GSE62232 dataset were significantly upregulated in HCC: WTAP (P<0.01), YTHDC2 (P<0.001), RBM15 (P<0.001), KIAA1429 (P<0.001), METTL3 (P<0.001), METTL14 (P<0.001), YTHDF1 (P<0.001), YTHDC1 (P<0.001), HNRNPC (P<0.001), and FTO (P<0.05). Therefore, 8 genes, METTL3, WTAP, RBM15, KIAA1429, YTHDF1, YTHDC1, YTHDC2 and HNRNPC, were upregulated in both databases. We selected 60 patients with hepatocellular carcinoma that was diagnosed by pathology for subsequent experimental studies. The qRT-PCR results showed that 10 of the 13 m6A-related regulators were significantly upregulated in HCC clinical samples (Figure 1D): YTHDC1 (P<0.05), YTHDC2 (P<0.05), ALKBH5 (P<0.01), KIAA1429 (P<0.01), METTL3 (P<0.001), HNRNPC (P<0.01), YTHDF2 (P<0.01), RBM15 (P<0.01), WTAP (P<0.01), and YTHDF1 (P<0.05, Figure 1D). With tumour progression from stage I to III, the expression of five m6A-related regulators (two “writers” and three “readers”) gradually increased: METTL3 (P<0.05), RBM15 (P<0.01), YTHDC1 (P<0.05), YTHDF1 (P<0.001), and YTHDF2 (P<0.05, Figure 2A). The expression of the other eight genes was not associated with tumour staging (Supplementary Figure 1). Although the expression of these genes in stage IV tumours notably decreased, the overall trend was statistically significant. The immunohistochemical staining results of the 5 m6A-related regulators in different stages of HCC are shown in Figure 2B. METTL3, YTHDF1, YTHDC1, YTHDF2 and RBM15 were all positively expressed in HCC specimens. METTL3, YTHDC1 and RBM15 were primarily expressed in the nucleus, and YTHDF1 and YTHDF2 were primarily expressed in the cytoplasm. The expression intensity and positive rate of these regulators increased with tumour stage progression from stage I to III. To examine the prognostic value of m6A-related regulators in HCC, we analysed the effects of the 13 genes on the OS of patients with HCC. The results indicated that HCC samples with higher expression levels of KIAA1429 (P<0.01), YTHDF1 (P<0.01), METTL3 (P<0.001), WTAP (P<0.01), or YTHDF2 (P<0.01, three “writers” and two “readers”) showed poorer OS (Figure 3A), but no significant differences were observed for the remaining eight m6A-related regulators (Supplementary Figure 2). We further validated the association of these five genes with patient OS in the 60 clinical HCC samples. Except for YTHDF2 (P=0.05), patients with higher expression levels of KIAA1429 (P<0.05), METTL3 (P<0.05), WTAP (P<0.05), or YTHDF1 (P<0.05) showed poorer OS (Figure 3B).
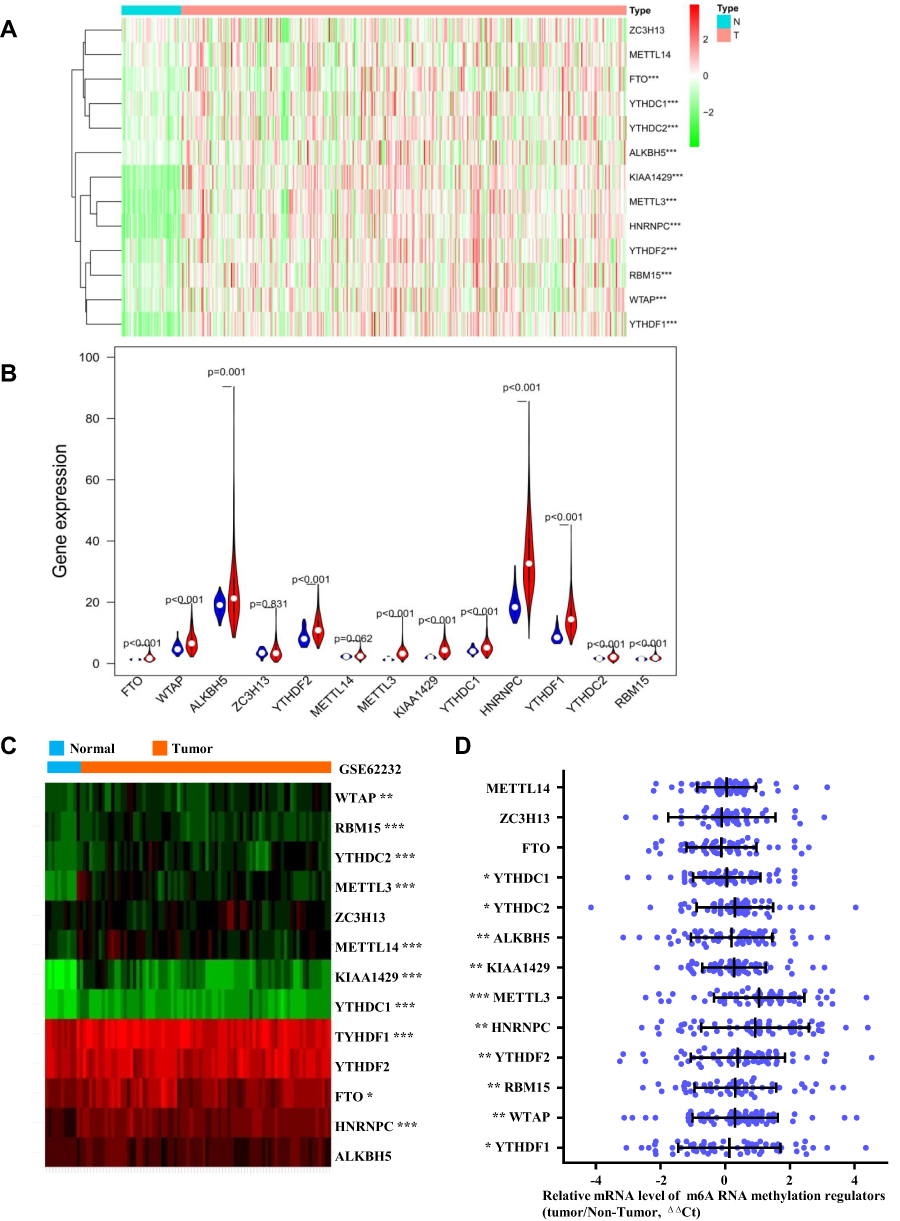 |
Figure 1 Expression of m6A RNA methylation regulators in HCC samples vs in normal tissues in different database. (A and B) Expression levels of 13 m6A-related genes in TCGA database. (C) Expression heatmap plotting of m6A-related genes in GSE62232 dataset. (D) Expression levels of m6A-related genes in 60 paired HCC tissues and adjacent nontumor tissues (Xinqiao Hospital clinical samples) was examined by qRT-PCR, in which GAPDH was used as internal control. Relative gene expression was determined using the comparative delta-delta CT method. Abbreviation: ΔΔCt, delta-delta CT. Note: *P < 0.05, **P < 0.01, ***P < 0.001. |
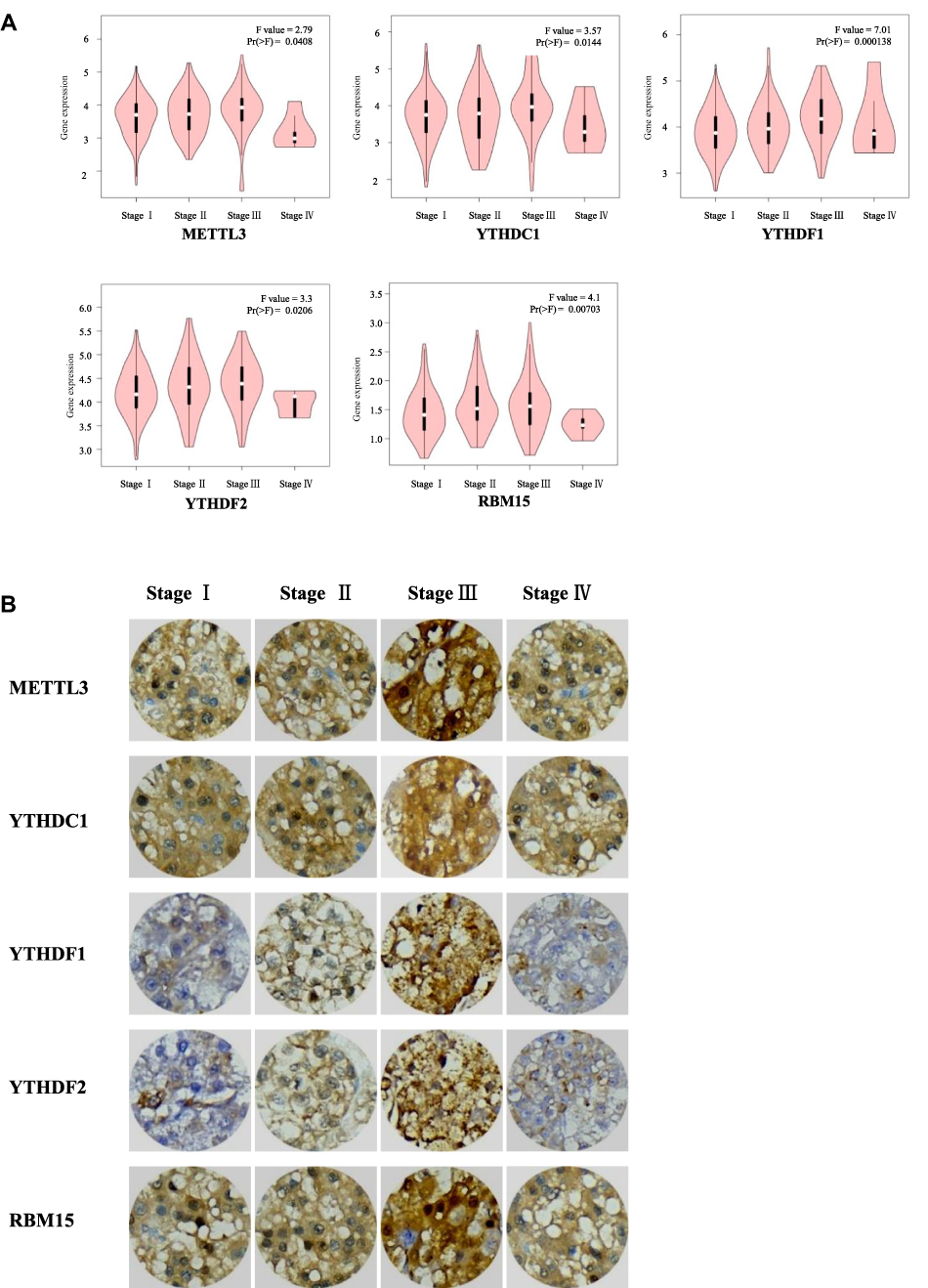 |
Figure 2 Expression levels of m6A RNA methylation regulators in HCC at different tumor stages. (A) The association between the expression of five m6A RNA methylation regulators and tumor stage was evaluated by GEPIA. (B) Representative immunohistochemistry images of the five m6A RNA methylation regulators at different tumor stages. |
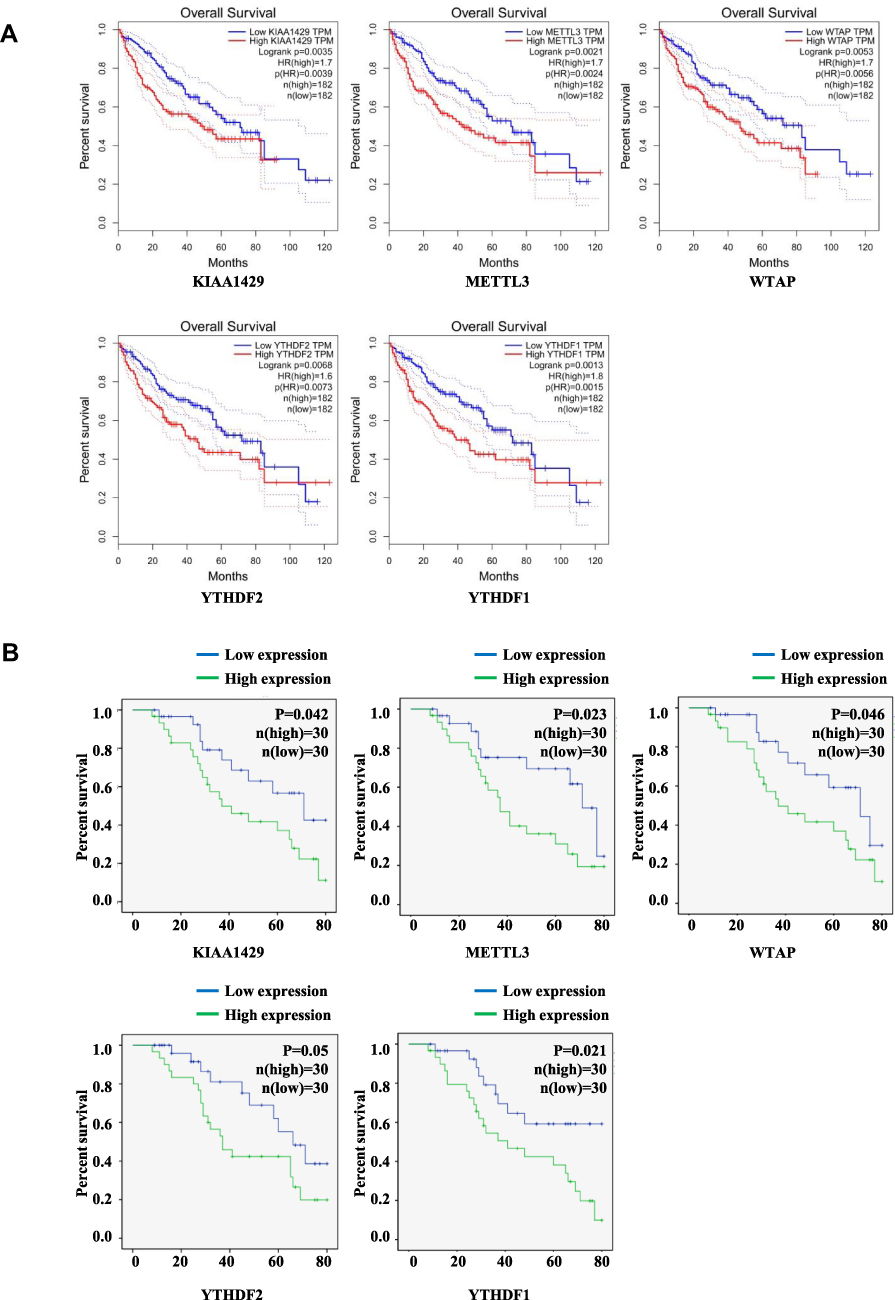 |
Figure 3 Kaplan–Meier OS curve for patients with HCC with high or low expression levels of m6A RNA methylation regulators. (A) Results from GEPIA. (B) Results from analysis of clinical 60 HCC samples. |
HCC Subgroups Defined by Consensus Cluster of m6A-Related Regulators
The clinical features of HCC associated with m6A-related regulators may provide a new perspective for the molecular classification of liver cancer. Therefore, we performed further studies using consensus clustering. We used the following principles in the process of choosing the proper k value: 1) the cluster number was determined by the K value with the lowest proportion of ambiguous clustering (PAC) because this value indicated a low rate of discordant assignments across different clusters; 2) we ensured the lowest intra-group differences and greatest inter-group differences; and 3) we ensured that no group had a particularly small sample size. Consequently, k=2 was the comprehensive choice based on Figure 4A–C and Supplementary Figure 3. Principal component analysis was used to compare the transcriptional profile between Clusters 1 and 2, and a clear distinction was noted (Figure 4D). We systematically compared the clinicopathological characteristics of Clusters 1 and 2 (Figure 4E). The clinicopathological features of patients from cluster 1 and 2 are listed in Table 2. Cluster 1 was significantly associated with lower α-fetoprotein (AFP) levels (P<0.001), male sex (P<0.01), low tumour grade (P<0.05), and better outcomes (P<0.05). Cluster 2 primarily correlated with higher AFP levels, female sex, higher tumour grade, and worse outcomes. We observed significantly better OS in Cluster 1 than Cluster 2 (Figure 4F, P < 0.001). These results showed that the clusters classified using consensus clustering were closely related to HCC malignancy. We validated our results of consensus clustering in GSE62232, and the clinicopathological characteristics of Clusters 1 and 2 were systematically compared (Supplementary Figure 4). Consistent with the TCGA findings, the GSE62232 results also indicated that Cluster 1 was notably associated with low tumour grade (P<0.05).
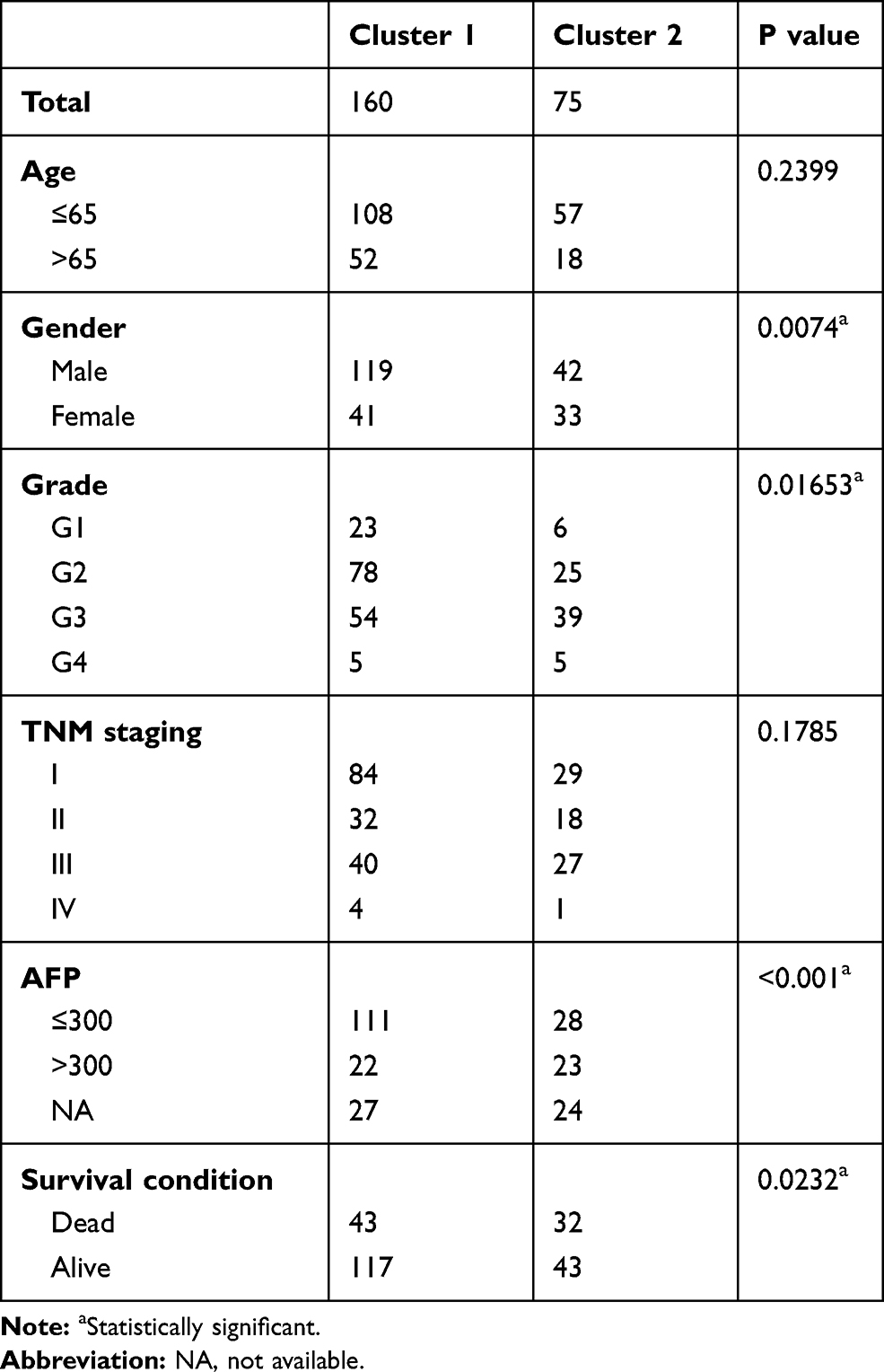 |
Table 2 Clinicopathological Features of Cluster 1 and 2 |
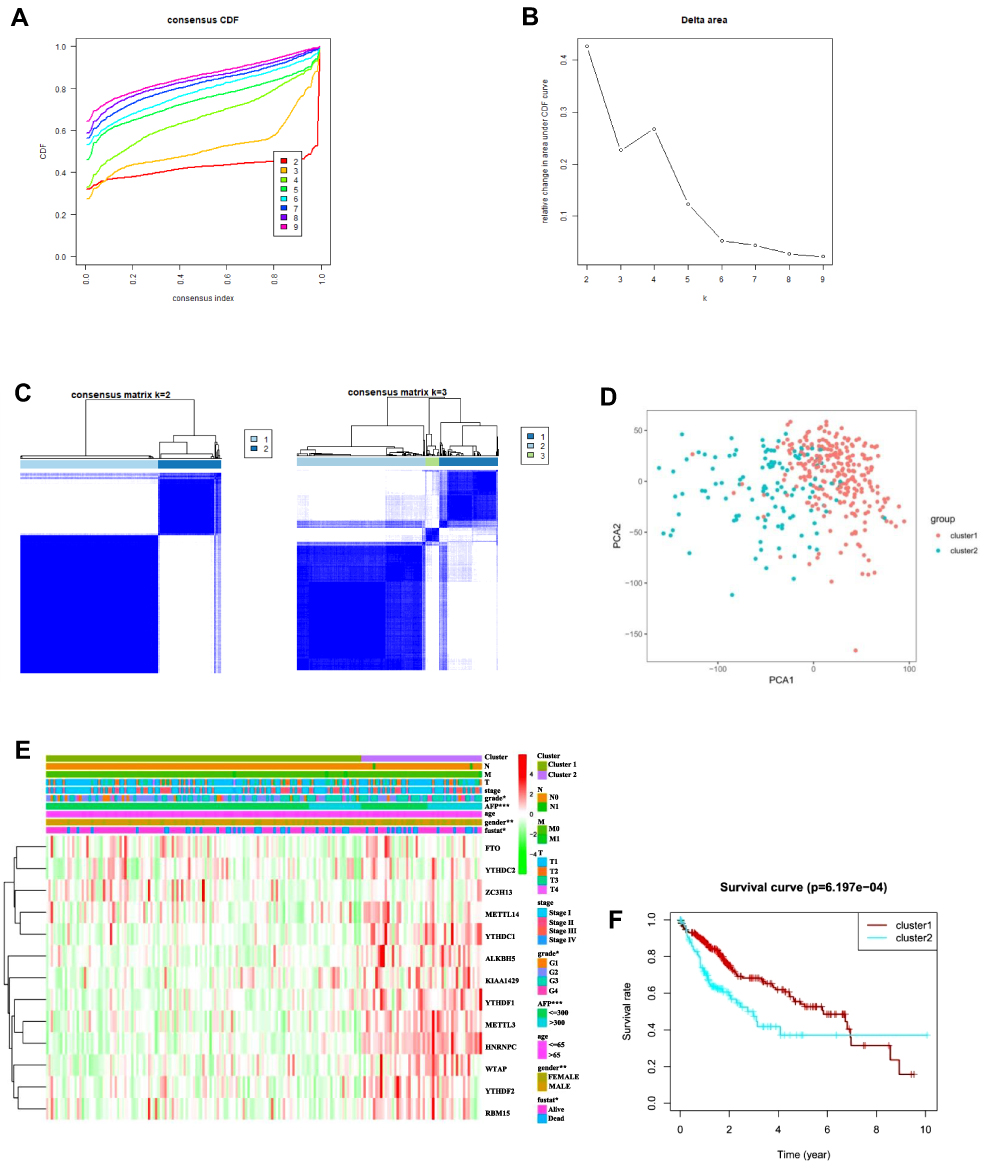 |
Figure 4 Differential clinicopathological features and OS of patients with HCC in the Cluster 1 and 2 subgroups. (A) Consensus clustering CDF for k = 2 to 9. (B) Relative change in the area under CDF curve for k = 2 to 9. (C) Consensus clustering matrix for k = 2 and k = 3. (D) Principal component analysis of the total RNA expression profile in the TCGA dataset. (E) Heatmap and clinicopathological features of the two clusters defined by the consensus expression of m6A RNA methylation regulators. (F) Kaplan–Meier OS curves for patients with HCC in Cluster 1 and 2. Abbreviation: CDF, cumulative distribution function. |
Prognostic Value of m6A-Related Regulators in HCC and Risk Signature Construction
Univariate Cox regression analysis was performed to detect the potential prognostic role of m6A-related regulators in HCC. Analysis of data from the TCGA database showed that nine of the 13 m6A-related regulators were closely correlated with OS (Figure 5A). Among the nine m6A-related regulators, FTO (P=0.245), WTAP (P<0.01), YTHDF2 (P<0.001), METTL3 (P<0.001), KIAA1429 (P<0.001), YTHDC1 (P<0.05), HNRNPC (P<0.01), YTHDF1 (P<0.001), and RBM15 (P<0.05) were risk factors with hazard ratios > 1, but only ZC3H13 (P<0.05) was a protective factor with a hazard ratio < 1. To assess the unpredictable role of m6A-related regulators in the prognosis of HCC, LASSO Cox regression analysis was used, and the coefficients obtained were introduced to determine the risk score (Figure 5B). Based on the minimum criteria, five genes, ZC3H13, KIAA1429, YTHDF2, YTHDF1, and METTL3, were used to establish the risk signature, and HCC patients derived from the TCGA dataset were classified into high- and low-risk categories. The high- and low-risk categories exhibited significantly different OS (Figure 5C, P<0.001)). The receiver operating characteristic curve showed that the risk signature was somewhat reliable in predicting the 5-year survival rates of HCC patients (area under the curve = 0.614; Figure 5D).
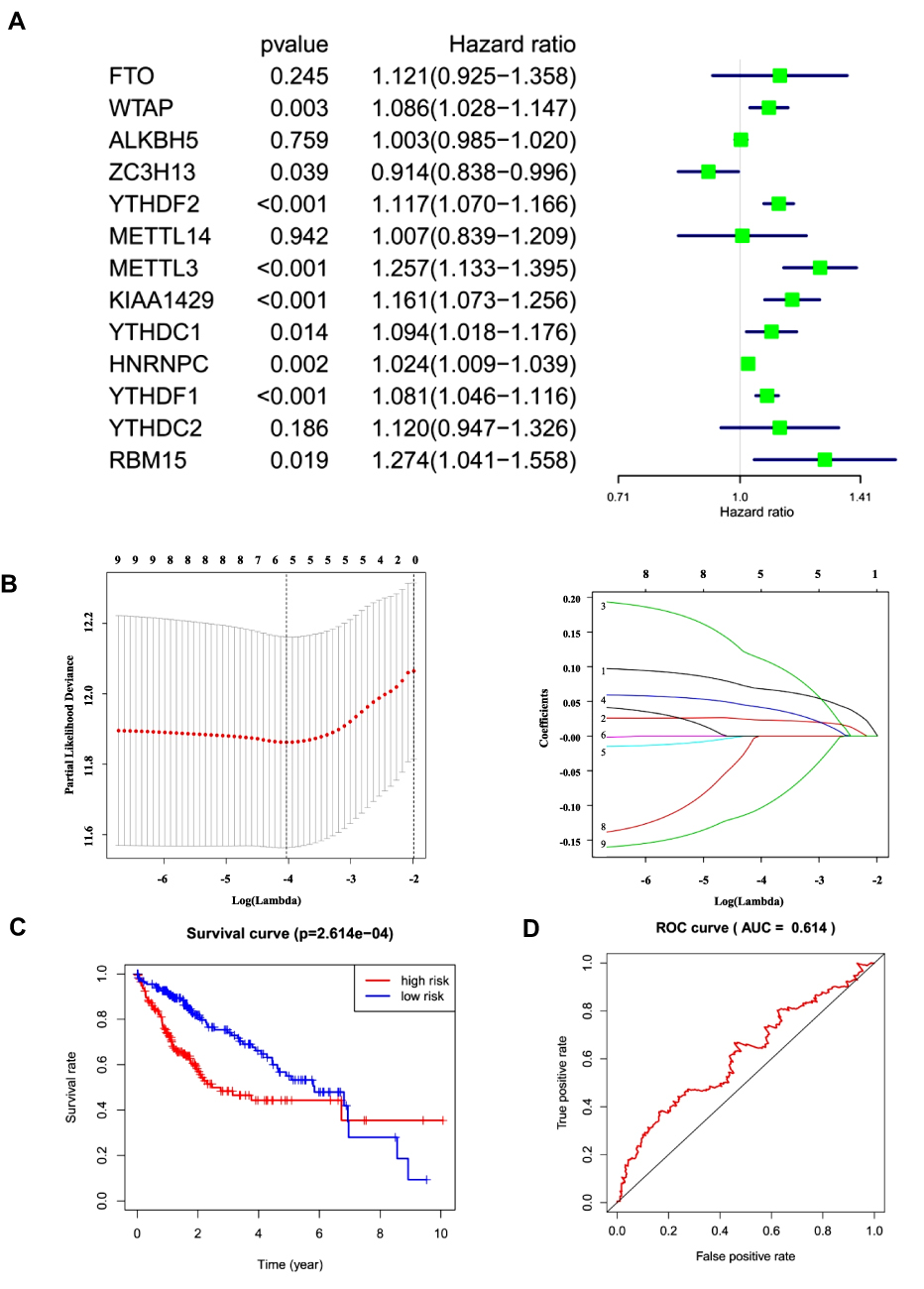 |
Figure 5 Risk signature with five selected m6A RNA methylation regulators. (A and B) Process of building the signature with five m6A RNA methylation regulators. HR and 95% CI calculated by univariate Cox regression and coefficients calculated by multivariate Cox regression using LASSO. (C) OS curves for patients in the TCGA dataset assigned to high- and low-risk groups based on the risk score. (D) Receiver operating characteristic curves showing the predictive efficiency of the risk signature. Abbreviations: HR, hazard ratio; CI, confidence interval. |
The heat map in Figure 6A shows the expression of these five m6A-related regulator genes in the high-risk and low-risk subgroups. The high- and low-risk categories differed significantly in T (P<0.05) of the TNM system, stage (P<0.01), grade (P<0.05) and AFP (P<0.001) levels of HCC. We used Cox regression analyses (univariate and multivariate analyses) to detect whether the risk model we established could indicate prognosis in HCC independently. Univariate analyses revealed that tumour stage (P<0.0001), T (P<0.001) of the TNM system, and risk score (P<0.001) were significantly associated with OS. When all these factors were included in the multivariate analysis, the risk signature (P<0.001) continued to be significantly associated with OS (Figure 6B and C, Tables 3 and 4). Analysis of Figure 6A–C was also performed in GSE14520 and the clinical samples to validate the results of the risk signature. The risk score in GSE14520 significantly correlated with tumour stage (P<0.05), sex (P<0.05) and the ultimate survival state (P<0.01, Supplementary Figure 5A). Univariate and multivariate analyses indicated that risk score (P<0.01) and stage (P<0.001) were strongly associated with OS (Supplementary Figure 5B and C, Supplementary Tables 2 and 3). To further validate the risk score derived from these five genes in another database, a GEO dataset with prognostic data, GSE14520, was used. Patients were classified into low- and high-risk subgroups based on the expression of risk-related genes. Kaplan–Meier results indicated that patients in the low-risk subgroup had notably increased survival (P<0.01, Supplementary Figure 5D). The risk signature in the clinical samples was significantly associated with tumour stage (P<0.001) and patient survival state (P<0.05, Supplementary Figure 6A). The results of univariate analyses and multivariate analysis were consistent with the GSE14520 dataset (Supplementary Figure 6B and C, Supplementary Tables 4 and 5). The clinical HCC samples collected in Xinqiao Hospital were also classified into high- and low-risk categories. A significant difference in OS was noted between the high- and low-risk categories (P<0.01, Supplementary Figure 6D). These findings validate that the risk signature based on m6A-related regulators independently predicted the clinical outcome in HCC patients.
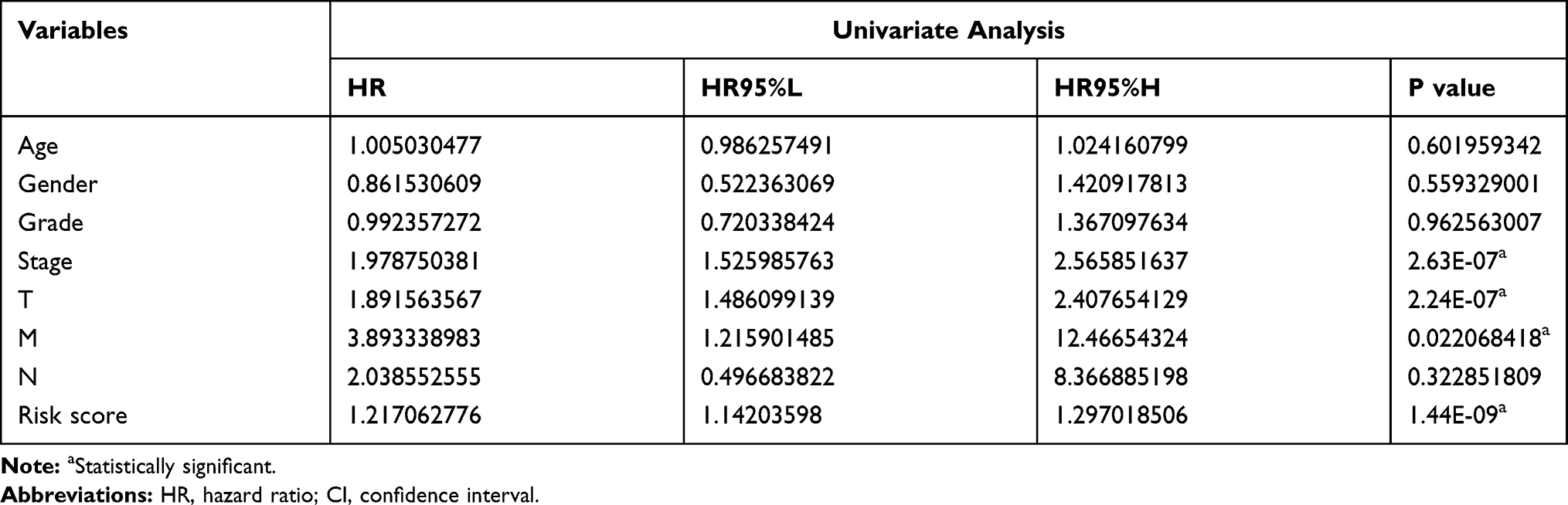 |
Table 3 Univariate Analysis for Survival Analysis |
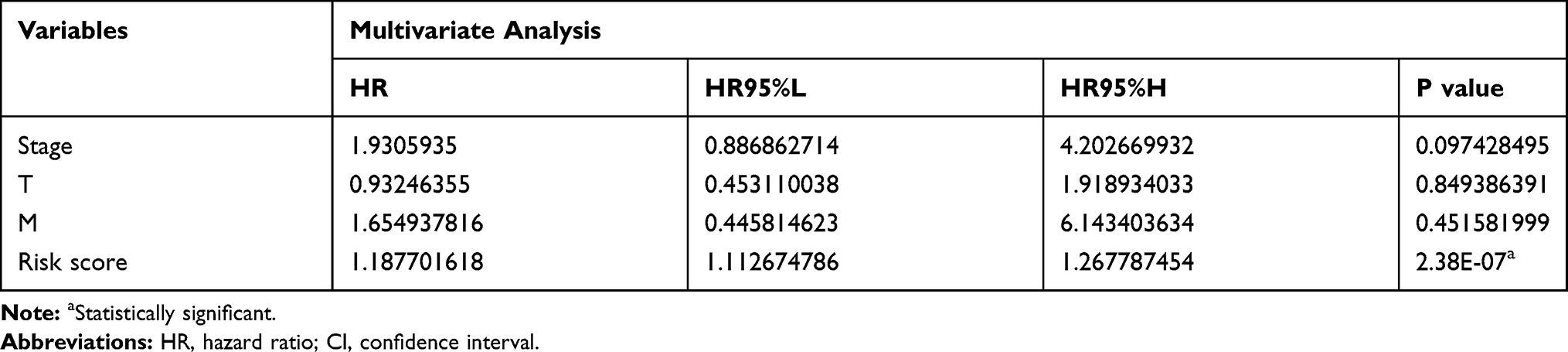 |
Table 4 Multivariate Analysis for Survival Analysis |
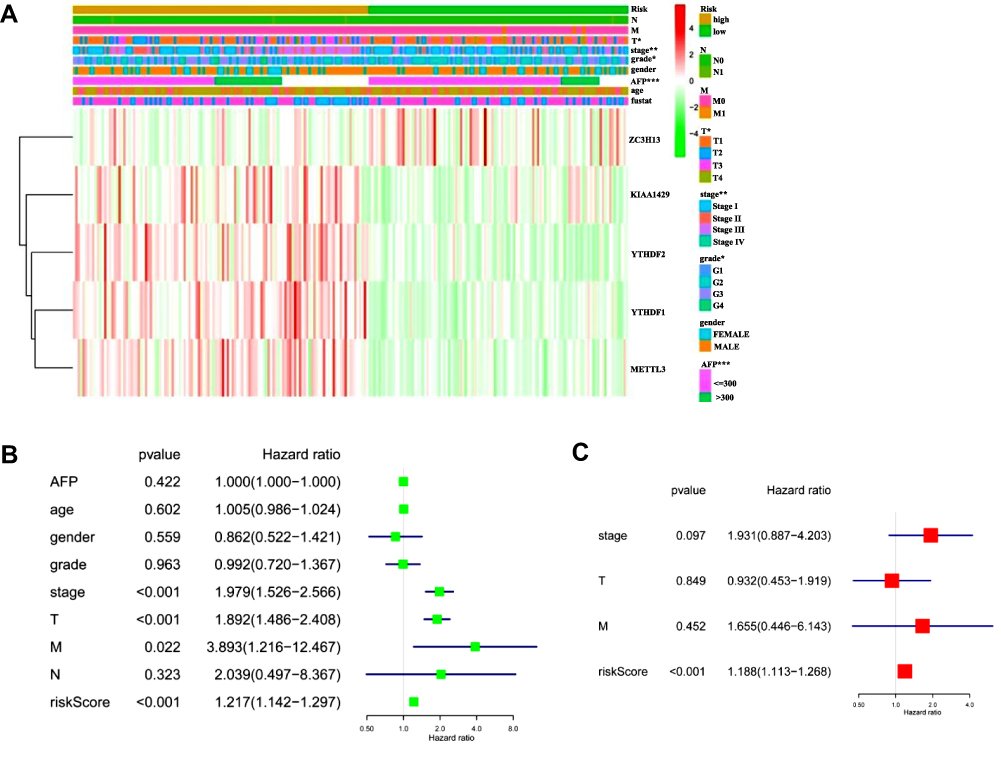 |
Figure 6 Prognostic value of m6A RNA methylation regulators in HCC from TCGA database. (A) The heatmap shows the expression of the five m6A RNA methylation regulators in low- and high-risk groups. The distribution of clinicopathological features was compared between these two groups. (B and C) Univariate and multivariate Cox regression analyses of the association between clinicopathological factors (including the risk score) and OS of patients with HCC. Notes: *P < 0.05, **P < 0.01, ***P < 0.001. |
We screened the enriched KEGG and GO pathways to identify correlation with the five genes we used to establish the risk signature (Figure 7). The expression of the five genes was closely related to diverse biological processes, such as endothelial-to-haematopoietic transition, gamete generation, the developmental process, negative regulation of the Notch signalling pathway, multicellular organism development, and cellular response to DNA damage stimulus.
 |
Figure 7 Gene set enrichment analysis for five m6A RNA methylation regulators used in risk signature building. (A) GO terms of biological processes and (B) KEGG pathway analysis. |
Discussion
RNA m6A modification, similar to DNA histone modifications, was first described in the 1970s, but it was not regarded as another meaningful epigenetic layer until recently. The spread and development of the epitranscriptome in the last decade facilitated scientific research on RNA epigenetic modification.7,24–27
The dynamic regulation of m6A modification is achieved via the functional collaboration between m6A “writers”, “readers” and “erasers”. M6A modification is critical for various cellular processes, including RNA splicing, cell proliferation, embryonic development, stem cell renewal, neural development, protein translation, cell death, and stress responses. However, the genes participating in m6A dysregulation are distinct in tumours, partially due to tissue specificity and tumour heterogeneity. The m6A-Seq and m6A/MeRIP-seq methods are used for the direct detection of m6A, but both of these techniques are somewhat complicated. Therefore, some studies used an indirect approach to examine alterations in m6A regulatory genes and evaluate the relevance between m6A status and human diseases.5,13,28 Recent papers also examined the role of m6A-regulated regulators in HCC prognosis.16–19 However, the present study systematically analysed the prognostic value and expression profile of m6A regulators in HCC from the TCGA database and validated the results in the GEO database and clinical surgical samples.
The present study used data downloaded from TCGA, GEO datasets and clinical surgical samples collected in Xinqiao Hospital Chongqing to comprehensively analyse the expression levels of 13 m6A-related regulators in HCC. Eight m6A were significantly upregulated in HCC in all datasets. Analyses of the online databases showed that the expression of five m6A-related regulators (METTL3, YTHDF1, YTHDC1, YTHDF2 and RBM15) gradually increased with tumour progression from stage I to III. This result was verified by immunohistochemical staining of paraffin sections of HCC tissue. Notably, the expression of these five m6A-related regulators decreased in stage IV in the TCGA database and immunohistochemical staining results. We posit the following hypotheses to explain these results. First, the number of HCC samples in stage IV was too small: there were 5 stage IV patients in the 374 samples from TCGA; and only one of the 60 clinical HCC tissue samples was in stage IV, which may have introduced bias in the statistical results. It is difficult for researchers to obtain clinical HCC samples of stage IV because the patients’ conditions are not suitable for surgical treatment. Second, patients in stage IV may have totally disordered immune functions and epigenetic regulations that are completely different from, or even opposite to, the early and middle stages of the disease. M6A RNA methylation may also contribute to tumour immunity.29 Third, necrosis of the tumour is often observed in stage IV tissues. Epigenetic regulation may also be different between the necrotic tissue, the surrounding tissues, and tissue that is far removed from the necrotic tissue. The aetiology of HCC includes HBV, HCV, and alcohol consumption. Data from the TCGA database allowed us to perform an analysis of the expression of m6A methylation regulators in HCC with different aetiologies (data not shown). YTHDF1 expression was notably increased in patients with hepatitis (HBV/HCV) compared to patients without hepatitis. This finding may be related to liver immunology because YTHDF1 controls antigen presentation by dendritic cells (DCs). YTHDF1 in DCs suppresses the cross-presentation of tumour antigens to CD8+ T cells by inhibiting the translation of lysosomal cathepsins.29 For patients with alcohol consumption, 1 m6A methylation regulator (FTO) was expressed differently, but the remaining 12 regulators showed no significant difference. The expression of FTO in patients with alcohol consumption was significantly lower in HCC than patients without alcohol consumption. FTO increases the expression of lipid metabolism genes (FASN, SCD1, MGAT1) but decreases the expression of lipid transport genes (MTTP, APOB, LIPC), which results in lipid accumulation.29 Alcoholism alters hepatic metabolism and leads to lipid accumulation and hepatitis. The underlying reason for the consistency requires further studies using more adequate and accurate clinical data. Therefore, we will continue to collect clinical HCC samples, and their epitranscriptomic information should be collected carefully and thoroughly to fully evaluate the underlying mechanism and dynamic alterations in RNA modifications during liver disease progression. Survival analyses revealed that patients with HCC with high expression levels of KIAA1429, METTL3, WTAP, YTHDF1, or YTHDF2 (three “writers” and two “readers”) showed poorer OS. These results suggest that m6A RNA methylation regulators are notably related to the malignancy and prognosis of HCC, which was further confirmed in subsequent qRT-PCR assays in HCC clinical surgical specimens. Consensus clustering classified two HCC clusters. Both of these subgroups distinctly influenced tumour prognosis and clinicopathological characteristics. Cluster 1 was significantly correlated with lower AFP levels, male sex, lower tumour grade, and better outcomes. Consensus clustering was based on systematic analysis of the expression of 13 m6A-related genes in HCC and further demonstrated that the analysis of m6A-related regulators is valuable for clinical and scientific research. The molecular classification of liver cancer received increasing attention from researchers worldwide, and this research may provide a new perspective for the molecular classification of liver cancer.
Whether m6A-related regulators are valuable prognostic biomarkers with clinical significance is a meaningful research hotspot for future studies.30 Our HCC prognostic signature derived from the five m6A-related regulators had clinical significance in the prognostic value of HCC stage and grade, AFP levels and T of the TNM system.
The primary m6A-related regulators consist of 13 genes, including “writers,” “erasers,” and “readers.” Among the “writers,” METTL3 is highly conserved in eukaryotes and acts as a cancer suppressor gene in glioblastoma and colorectal cancer, but it is an oncogene in acute myelocytic leukaemia, gastric cancer, and HCC. METTL3 and METTL14 are key components of the m6A methyltransferase complex,31,32 in which they cooperate to form a heterocomplex that substantially enhances methylation efficiency.31,33,34 These molecules were markedly overexpressed in HCC, and their expression positively correlated to each other (Supplementary Figure 7). However, a recent study demonstrated that METTL14 acted as a tumour suppressor in HCC and suppressed tumour metastasis by interacting with DGCR8 and positively regulating miR-126 in an m6A-dependent manner.14 Therefore, further in-depth studies are warranted to comprehensively examine the complicated role of these two m6A writers in HCC.
METTL3 knockout remarkably suppressed HCC carcinogenesis in a manner dependent on YTHDF2, which suggests the efficient cooperation of m6A-related regulatory genes.35 Proteins that contain the YTH domain have a conserved m6A-binding domain and are direct “readers” of m6A. Different “readers” have different functions.32 For example, YTHDC1 and YTHDC2 are located in the nucleus, and YTHDF1 and YTHDF2 are located in the cytoplasm.34,35 YTHDC1 binds to m6A and recruits alternative splicing factors to affect target mRNA splicing.36 YTHDF2 destabilizes alternative m6A-containing RNA via direct recruitment of the CCR4-NOT deadenylase complex.37 YTHDF2 also acts as an oncogene in pancreatic cancer by orchestrating the epithelial-to-mesenchymal transition and tumour cell proliferation.38 Notably, we found that all five readers were overexpressed in HCC compared to normal tissues, which indicates a common activation in HCC. YTHDF1 and YTHDF2 had significant prognostic value for clinical outcomes (eg, OS) in patients with HCC. Further investigations are needed to completely and systematically address their role in HCC.
ALKBH5 and FTO are “erasers” that act as oncogenes in several types of cancers, including glioma, breast cancer and acute myelocytic leukaemia.11,39,40 We noted that ALKBH5 and FTO were overexpressed in HCC. However, these molecules were not independent valuable prognostic factors for HCC. Correlation analysis showed a close association between these two “erasers” and other m6A-related regulators, particularly FTO (Supplementary Figure 7), which suggests that these factors work in collaboration with other factors to regulate m6A RNA methylation. These findings collectively suggest that the dysregulation of m6A-related regulators correlates with dysregulated RNAs in cancer. The same m6A-related regulator may have entirely different features and functions in diverse types of cancer due to tumour heterogeneity.
The results also suggested a correlation between m6A-related regulators and the pathophysiological function and related signal routing of HCC progression. M6A-related regulators are involved in many important biological functions, including self-renewal and pluripotent regulation of stem cells, such as the directional differentiation of haematopoietic stem cells,12,41 the various processes of RNA processing and metabolism,42 DNA damage response,43 biorhythm, and early embryonic development in mice. Our findings demonstrated that m6A-related regulators were closely correlated with biological processes, such as endothelial-to-haematopoietic transition, gamete generation, regulation of haematopoietic stem cell differentiation, developmental process, negative regulation of the Notch signalling pathway, multicellular organism development, and regulation of primary metabolic process.
In conclusion, the present study comprehensively demonstrated the expression profile and prognostic value of 13 primary m6A-related regulators in HCC. Their expression was closely related to HCC malignancy, and some regulators also had potential prognostic value. These findings are meaningful for the development of novel therapeutic strategies directed toward the expression profile of m6A-related regulators in HCC. Agents targeting m6A RNA methylation are one potential approach for tumour therapy.
Data Sharing Statement
This manuscript contains previously unpublished data. The name of the repository and accession number(s) are not available.
Ethics Statement
Written informed consent for the experimental studies was obtained from the patients. The Institutional Ethics Committee of Xinqiao Hospital, Military Medical University approved all experiments (2018-sci 069-01), and this study complied with the Declaration of Helsinki.
Author Contributions
All authors made a significant contribution to the work, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by grants from the National Natural Science Foundation Project of China (81672653, 81602591, and 81972113).
Disclosure
The authors disclose no potential conflicts of interest.
References
1. Carr BI, D’Alessandro R, Refolo MG, et al. Effects of low concentrations of regorafenib and sorafenib on human HCC cell AFP, migration, invasion, and growth in vitro. J Cell Physiol. 2013;228(6):1344–1350. doi:10.1002/jcp.24291
2. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–2576. doi:10.1053/j.gastro.2007.04.061
3. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. doi:10.1002/ijc.29210
4. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi:10.3322/caac.20107
5. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–206. doi:10.1038/nature11112
6. Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat Rev Genet. 2014;15(5):293–306. doi:10.1038/nrg3724
7. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3ʹ UTRs and near stop codons. Cell. 2012;149(7):1635–1646. doi:10.1016/j.cell.2012.05.003
8. Ke S, Alemu EA, Mertens C, et al. A majority of m6A residues are in the last exons, allowing the potential for 3ʹ UTR regulation. Genes Dev. 2015;29(19):2037–2053. doi:10.1101/gad.269415.115
9. Niu Y, Zhao X, Wu YS, Li MM, Wang XJ, Yang YG. N6-methyl-adenosine (m6A) in RNA: an old modification with a novel epigenetic function. Genomics Proteomics Bioinformatics. 2013;11(1):8–17. doi:10.1016/j.gpb.2012.12.002
10. Kwok CT, Marshall AD, Rasko JE, Wong JJ. Genetic alterations of m(6)A regulators predict poorer survival in acute myeloid leukemia. J Hematol Oncol. 2017;10(1):39. doi:10.1186/s13045-017-0410-6
11. Pan Y, Ma P, Liu Y, Li W, Shu Y. Multiple functions of m(6)A RNA methylation in cancer. J Hematol Oncol. 2018;11(1):48. doi:10.1186/s13045-018-0590-8
12. Wang S, Sun C, Li J, et al. Roles of RNA methylation by means of N(6)-methyladenosine (m(6)A) in human cancers. Cancer Lett. 2017;408:112–120. doi:10.1016/j.canlet.2017.08.030
13. Zhou J, Wang J, Hong B, et al. Gene signatures and prognostic values of m6A regulators in clear cell renal cell carcinoma - a retrospective study using TCGA database. Aging (Albany NY). 2019;11(6):1633–1647. doi:10.18632/aging.101856
14. Ma JZ, Yang F, Zhou CC, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary microRNA processing. Hepatology. 2017;65(2):529–543. doi:10.1002/hep.28885.
15. Zhou Y, Yin Z, Hou B, et al. Expression profiles and prognostic significance of RNA N6-methyladenosine-related genes in patients with hepatocellular carcinoma: evidence from independent datasets. Cancer Manag Res. 2019;11:3921–3931. doi:10.2147/CMAR.S191565
16. Li G, Zhang Y, Du X, et al. N6-methyladenosine (m6A) RNA methylation regulators are associated with clinical prognosis in hepatocellular carcinoma. Transl Cancer Res. 2020;9(1):323–334. doi:10.21037/tcr.2019.12.84
17. Wu X, Zhang X, Tao L, et al. Prognostic value of an m6A RNA methylation regulator-based signature in patients with hepatocellular carcinoma. Biomed Res Int. 2020;2020(8):1–11. doi:10.1155/2020/2053902
18. Li W, Q F C, Huang T, et al. Profiles of m6A RNA methylation regulators for the prognosis of hepatocellular carcinoma. Oncol Lett. 2020;19(4). doi:10.3892/ol.2020.11435
19. Zhao Z, Yang L, Fang S, et al. The effect of m6A methylation regulatory factors on the malignant progression and clinical prognosis of hepatocellular carcinoma. Front Oncol. 2020;10:1435. doi:10.3389/fonc.2020.01435
20. Lv YF, Yan GN, Meng G, Zhang X, Guo QN. Enhancer of zeste homolog 2 silencing inhibits tumor growth and lung metastasis in osteosarcoma. Sci Rep. 2015;5(1):12999. doi:10.1038/srep12999
21. Lv YF, Dai H, Yan GN, Meng G, Zhang X, Guo QN. Downregulation of tumor suppressing STF cDNA 3 promotes epithelial-mesenchymal transition and tumor metastasis of osteosarcoma by the Wnt/GSK-3beta/beta-catenin/snail signaling pathway. Cancer Lett. 2016;373(2):164–173. doi:10.1016/j.canlet.2016.01.046
22. Hu X, Martinez-Ledesma E, Zheng S, et al. Multigene signature for predicting prognosis of patients with 1p19q co-deletion diffuse glioma. Neuro Oncol. 2017;19(6):786–795. doi:10.1093/neuonc/now285
23. Zhou Z, Huang R, Chai R, et al. Identification of an energy metabolism-related signature associated with clinical prognosis in diffuse glioma. Aging (Albany NY). 2018;10(11):3185–3209. doi:10.18632/aging.101625
24. Bhutani N, Burns DM, Blau HM. DNA demethylation dynamics. Cell. 2011;146(6):866–872. doi:10.1016/j.cell.2011.08.042
25. He C. Grand challenge commentary: RNA epigenetics? Nat Chem Biol. 2010;6(12):863–865. doi:10.1038/nchembio.482
26. Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–45. doi:10.1038/47412
27. Wei CM, Gershowitz A, Moss B. Methylated nucleotides block 5ʹ terminus of HeLa cell messenger RNA. Cell. 1975;4(4):379–386. doi:10.1016/0092-8674(75)90158-0
28. Chai RC, Wu F, Wang QX, et al. m(6)A RNA methylation regulators contribute to malignant progression and have clinical prognostic impact in gliomas. Aging (Albany NY). 2019;11(4):1204–1225. doi:10.3389/fonc.2019.01038
29. Zhao Z, Meng J, Su R, et al. Epitranscriptomics in liver disease: basic concepts and therapeutic potential. J Hepatol. 2020;73(3):3. doi:10.1016/j.jhep.2020.04.009
30. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi:10.3322/caac.21442
31. Liu J, Yue Y, Han D, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10(2):93–95. doi:10.1038/nchembio.1432
32. Wang X, Zhao BS, Roundtree IA, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161(6):1388–1399. doi:10.1016/j.cell.2015.05.014
33. Wang P, Doxtader KA, Nam Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell. 2016;63(2):306–317. doi:10.1016/j.molcel.2016.05.041
34. Wang X, Feng J, Xue Y, et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature. 2016;534(7608):575–578. doi:10.1038/nature18298
35. Zhu T, Roundtree IA, Wang P, et al. Crystal structure of the YTH domain of YTHDF2 reveals mechanism for recognition of N6-methyladenosine. Cell Res. 2014;24(12):1493–1496. doi:10.1038/cr.2014.152
36. Xiao W, Adhikari S, Dahal U, et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61(4):507–519. doi:10.1016/j.molcel.2016.01.012
37. Du H, Zhao Y, He J, et al. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun. 2016;7(1):12626. doi:10.1038/ncomms12626
38. Chen J, Sun Y, Xu X, et al. YTH domain family 2 orchestrates epithelial-mesenchymal transition/proliferation dichotomy in pancreatic cancer cells. Cell Cycle. 2017;16(23):2259–2271. doi:10.1080/15384101.2017.1380125
39. Wei J, Liu F, Lu Z, et al. Differential m(6)A, m(6)Am, and m(1)A demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol Cell. 2018;71(6):973–985. doi:10.1016/j.molcel.2018.08.011
40. Zhang S, Zhao BS, Zhou A, et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31(4):591–606. doi:10.1016/j.ccell.2017.02.013
41. Cui Q, Shi H, Ye P, et al. m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18(11):2622–2634. doi:10.1016/j.celrep.2017.02.059
42. Dai D, Wang H, Zhu L, Jin H, Wang X. N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 2018;9(2):124. doi:10.1038/s41419-017-0129-x
43. Xiang Y, Laurent B, Hsu CH, et al. m6A RNA methylation regulates the UV-induced DNA damage response. Nature. 2017;543(7646):573. doi:10.1038/nature24007
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.