Back to Journals » Drug Design, Development and Therapy » Volume 11
Gastroretentive behavior of orally administered radiolabeled tamarind seed formulations in rabbits validated by gamma scintigraphy
Authors Razavi M, Karimian H, Yeong CH, Fadaeinasab M, Khaing SL, Chung LY ![]() , Mohamad Haron DE, Noordin MI
, Mohamad Haron DE, Noordin MI
Received 21 June 2016
Accepted for publication 22 August 2016
Published 19 December 2016 Volume 2017:11 Pages 1—15
DOI https://doi.org/10.2147/DDDT.S115466
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Manfred Ogris
Mahboubeh Razavi,1 Hamed Karimian,1 Chai Hong Yeong,2 Mehran Fadaeinasab,3 Si Lay Khaing,4 Lip Yong Chung,1 Didi Erwandi B Mohamad Haron,5 Mohamed Ibrahim Noordin1,6
1Department of Pharmacy, 2University Malaya Research Imaging Centre and Department of Biomedical Imaging, Faculty of Medicine, 3Center for Natural Product Research and Drug Discovery (CENAR), 4Department of Obstetrics & Gynaecology, 5Shimadzu-UMMC Centre for Xenobiotics Studies, Pharmacology Department, Faculty of Medicine, University of Malaya, 6Malaysian Institute of Pharmaceuticals and Nutraceuticals (IPharm), National Institutes of Biotechnology Malaysia, Ministry of Science, Technology and Innovation, Penang, Malayasia
Abstract: This study aimed to formulate floating gastroretentive tablets containing metformin hydrochloric acid (HCl), using various grades of hydrogel such as tamarind powders and xanthan to overcome short gastric residence time of the conventional dosage forms. Different concentrations of the hydrogels were tested to determine the formulation that could provide a sustained release of 12 h. Eleven formulations with different ratios of tamarind seed powder/tamarind kernel powder (TKP):xanthan were prepared. The physical parameters were observed, and in vitro drug-release studies of the prepared formulations were carried out. Optimal formulation was assessed for physicochemical properties, thermal stability, and chemical interaction followed by in vivo gamma scintigraphy study. MKP,3 formulation with a TKP:xanthan ratio of 3:2 was found to have 99.87% release over 12 h. Furthermore, in vivo gamma scintigraphy study was carried out for the optimized formulation in healthy New Zealand White rabbits, and the pharmacokinetic parameters of developed formulations were obtained. 153Sm2O3 was used to trace the profile of release in the gastrointestinal tract of the rabbits, and the drug release was analyzed. The time (Tmax) at which the maximum concentration of metformin HCl in the blood (Cmax) was observed, and it was extended four times for the gastroretentive formulation in comparison with the formulation without polymers. Cmax and the half-life were found to be within an acceptable range. It is therefore concluded that MKP,3 is the optimal formulation for sustained release of metformin HCl over a period of 12 h as a result of its floating properties in the gastric region.
Keywords: drug delivery systems, floating system, gastric retention, sustained release natural polymers, evaluation in vitro and in vivo
Introduction
Polymers have various applications in pharmaceutical sciences, often being used as gel-forming agents, binders, diluents, thickeners, and protective colloids in liquids, suppositories, suspensions, and cosmetic products.1,2 Polymers are either hydrophilic or hydrophobic in nature.3 Depending on their derivation, they can be categorized as either synthetic or natural. Natural polymers, such as gums and mucilage, are more appealing than synthetic polymers for pharmaceutical applications because they are cheaper, easily available, nontoxic, and biodegradable.4 They are also more acceptable to patients because they have fewer side effects compared to synthetic polymers.5 Gums and mucilage are plant hydrocolloids and polymers of monosaccharides or polysaccharides. Gums are pathological plant products that are formed by dissolving the cell walls caused by injury to the plant or by unfavorable growth conditions,6 while mucilage is a normal physiological product of metabolism.7
Tamarind (Tamarindus indica Linn), from Leguminosae family, is an evergreen tree which grows in South Asia. A tamarind fruit includes up to six seeds. The seed consists of 35%–40% husk and 60%–65% kernel.8 Tamarind seed polysaccharide is a mucilaginous polysaccharide that is obtained from tamarind seed kernels.9,10
Tamarind powder has gelling, creaming, and film forming properties. It is a good source of polysaccharides and forms mucilaginous dispersions with cold water; in addition, the viscosity can be increased when it is heated for 20–30 min at 100°C. Another advantage is that boiling does not affect the properties of polysaccharides.11 Kernel powder contains 46%–48% of gel-forming polysaccharides.12
Oral administration is the most convenient route and carries the lowest cost, which leads to high patient compliance.13,14 Drugs with a short half-life are eliminated from the systemic circulation before complete absorption, requiring frequent dosing to achieve optimal therapeutic effects. However, frequent dosing often leads to increased side effects.15 To overcome this limitation, oral controlled-release formulations are designed to sustain an effective concentration of the drug in the body for an extended period of time by moderating the absorption rate.16
The gastroretentive drug delivery system (GRDDS) is a unique approach in oral drug delivery; its purpose is to improve drug bioavailabity.16,17 Floating drug delivery systems offer a number of applications for local acting drugs or those which absorb from the upper region of the gastrointestinal tract.18,19
Metformin hydrochloric acid (HCl) is an antihyperglycemic agent used to decrease glucose levels to manage type II diabetes. It has very high water solubility (>300 mg/mL at 25°C), poor permeability in the lower area of the gastrointestinal (GI) tract, and is mainly absorbed in the upper GI; therefore, a floating gastroretentive matrix tablet can be helpful in achieving maximum absorption and bioavailability of the drug.20
The current study was aimed to design and improve floating gastroretentive properties of formulations using tamarind powders by a wet granulation process. The primary goal was to prepare matrix tablets with short floating lag time (FLT), required floating and swelling properties, and >90% drug release in the predicted period of time. The effects of combining tamarind seed powder (TSP)/tamarind kernel powder (TKP) and xanthan on gel-forming properties, floating behavior, and the release pattern of metformin HCl were estimated.
Materials and methods
White crystalline powder of metformin HCl BP 98 (batch no 2011MP0244, 99.61% purity) was obtained from Euro Chemo-Pharma Sdn. Bhd (Selangor, Darul Ehsan, Malaysia). Tamarind seeds were supplied by Research Laboratory of Amritum Bio-Botanica Herbs Pvt Ltd (Indore, India). Lactose (C12H22O11·H2O, M =360.31 g/mol), sodium bicarbonate (NaHCO3, M =84.01 g/mol), cellulose microcrystalline (C6H10O5)n, pure xanthan, talc, magnesium stearate, HCl 37% (M =36.46 g/mol), and potassium chloride (KCl, M =74.56 g/mol) were purchased from R&M Marketing (Saffron Walden, UK). Potassium dihydrogen phosphate, perchloric acid 60% solution, orthophosphoric acid, sodium dodecyl (lauryl) sulfate, and acetonitrile were purchased from Thermo Fisher Scientific.
TSP and TKP collection and preparation
TSP and TKP were prepared from tamarind seeds. The seeds obtained from India were a mixture of intact seeds and broken seeds. Intact seeds were cleaned and handpicked to make sure that no traces of pulp existed on the seeds. Both de-husking and grinding processes were performed by a milling cutter machine (Retsch-Allee, Haan, Germany). After running the machine several times, the remaining seeds’ husks were removed from the small particles using a sharp knife. Then, the complete de-husked seed particles were subsequently ground into powder (TKP) using a blender machine and sieved into 0.149 mm using a no 100 sieve. On the other hand, TSP was produced by grinding the whole seed including the husk, and the whole seed was fed into the cutting mill machine to be converted to small particle size and then powdered using a blender and finally passed through a no 100 sieve to standardize the size of particles.21 The TSP and TKP were then stored in an airtight polypropylene bottle and kept in a desiccator until use.21
Matrix tablet preparation using TSP and TKP
The gastroretentive floating tablets were formulated by the wet granulation method using pulverized TSP and TKP, xanthan, sodium bicarbonate, lactose, microcrystalline cellulose, lubricant, and glidant (magnesium stearate and talc). All the ingredients were passed through sieve no 100 to get a uniform size of 0.149 mm and accurately weighed by employing an analytical balance (MS-S/MS-L models; Mettler-Toledo AG Laboratory & Weighing Technologies, Greifensee, Switzerland). Different formulations were prepared using the above ingredients at different concentrations, as shown in Table 1. Eight formulations of MSP2/MKP2, MSP3/MKP3, MSP4/MKP4, and MSP5/MKP5 were designed using tamarind gum:xanthan in the ratios of 4:1, 3:2, 2:3, and 1:4, respectively. MKP1/MSP1 and MKP6/MSP6 were formulated using only TSP/TKP and xanthan, respectively. To prepare the tablets, all ingredients except the lubricant and glidant were initially mixed using the geometrical dilution method. The binder solution (1% w/v xanthan) was used to granulate the powder mixture; then the granules were sieved into 2 mm using a no 10 sieve and dried out at 50°C in a hot air oven to reach a constant moisture level; then sieve no 18 was used to sieve the dried granules to obtain a uniform size of 1 mm. Premixed talc and magnesium stearate were added to them and mixed well to achieve a uniform blend.21 The lubricated granules (783 mg) were manually loaded into the die cavity and compressed into tablet form via a Manual Type Single Punch Tablet Press machine (MTCM-I; GlobePharma, North Brunswick, NJ, USA) using a round, concave 12 mm caplet punch and compressed at 2200 N.21,22
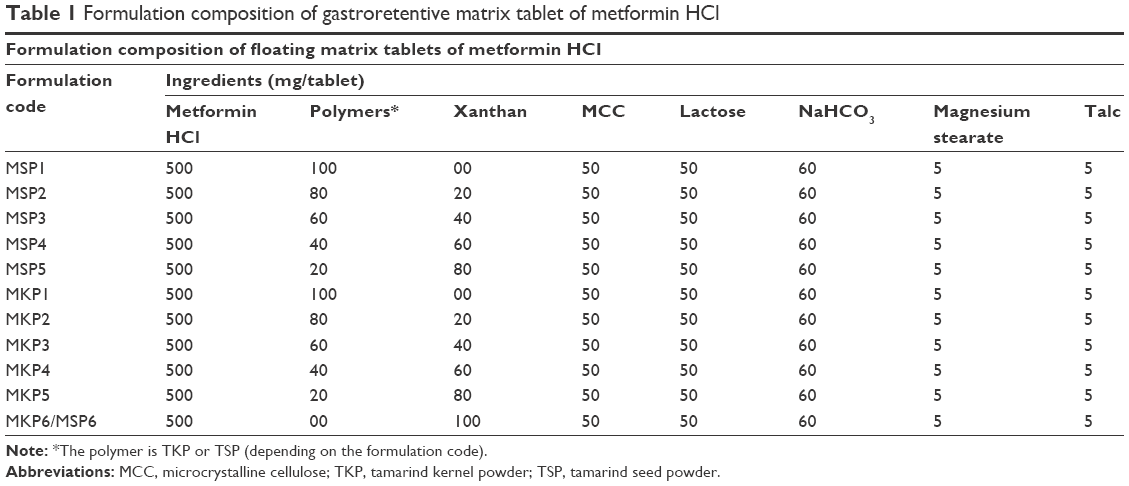 | Table 1 Formulation composition of gastroretentive matrix tablet of metformin HCl |
Evaluation of prepared matrix tablets of metformin HCl
The uniformity of content, weight variation, friability, hardness, FLT, and total floating time (TFT) of all formulations were assessed. The optimal formulation was evaluated by using differential scanning calorimetry (DSC), Fourier transform infrared spectrometry (FTIR), and X-ray diffraction analysis (XRD).
Weight variation of prepared formulations
Twenty tablets were selected randomly, and the average weight was determined by employing an analytical balance (MS-S/MS-L models; Mettler-Toledo AG Laboratory & Weighing Technologies). Prepared tablets were weighed individually to calculate the deviation from individual weight of the average weight.23
Thickness, length, and width of prepared formulations
Randomly ten tablets from each formulation were picked, and a digital harness tester machine (model 6D; Dr Schleuniger Pharmatron, Solothurn, Switzerland) was used to calculate the thickness, width, and length of these tablets.24
Hardness of prepared formulations
Ten randomly selected tablets of each formulation were evaluated for hardness using a hardness test machine (model 6D; Dr Schleuniger Pharmatron).23
Friability of prepared formulations
Ten tablets from each formulation were weighed (W1). A friability tester (model Tar10; Erweka, Ensenstam, Germany) was run at 25 rpm for 4 min, then the tablets were de-dusted and reweighed (W2). The friability was calculated using the formula as shown in Equation 1. According to the United States Pharmacopeia (USP), a friability of <1% is considered acceptable.25
Friability = (W1 − W2)/W1(1) | (1) |
where W1 is the initial weight of the tablets and W2 is the final weight after running the test.
Prepared tablets drug contents estimation
Three randomly selected tablets were crushed; 100 mg of the crushed powder was added to a volumetric flask and 100 mL of distilled water was added.26 The absorbance of the filtered solution was measured using an ultraviolet spectrophotometer (Lambda 35 UV–Vis Spectrometer; PerkinElmer Inc., Waltham, MA, USA) at 233 nm.
FLT and TFT of the floating matrix tablets
The time taken by dosage form to rise to the surface and float on the media is called FLT, whereas TFT is the maximum duration of time that the dosage form stays floating on the medium surface.21 TFT and FLT were measured when the tablets were inserted into a beaker containing 500 mL of simulated gastric fluid (pH 1.2) kept at 37°C. The general duration of tablet float was recorded as TFT, and the period of time between the insertion of the tablet to the medium and its floating to the surface was noted as the FLT.21,27
In vitro dissolution studies
An in vitro drug-release test was conducted in a USP dissolution apparatus (Copley Scientific Ltd, Nottingham, UK), using a paddle speed of 50 rpm in 900 mL of buffer solution (pH 1.2), which was maintained at a constant temperature of 37°C±0.5°C. Samples of 5 mL were withdrawn from the dissolution basket at 0.25, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 9, 10, 11, and 12 h and replaced with an equal volume of fresh medium.21 The sink condition was followed by replacing the amount of sample taken in the dissolution with the dissolution media.28 All the withdrawn samples were analyzed for drug content by measuring the absorbance at 233 nm using a UV–visible spectrophotometer. The release profile of each formulation was compared to the commercial extended-release formulation (Glucophage XR, 500 mg; n=3).
Drug-release kinetics
The data related to the in vitro dissolution test were assigned to mathematical models demonstrating 1) first-order, 2) zero-order, 3) Hixson–Crowell, and 4) Higuchi’s equations adopted from the study performed by Razavi et al.21,22 By using pharmacokinetic parameters, the release of metformin HCl at 0.25, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, and 12 h was calculated. The percentage of metformin HCl release from the effervescent floating tablets was plotted against time.29
Characterization of the optimal formulation using DSC
Pure drug, gastroretentive tablets, and polymers (2.5 mg) were sealed in aluminum pans for DSC scanning using an automatic thermal analyzer system furnace (model DSC 6,000; PerkinElmer). An empty pan sealed in the same way was used as a reference. The entire samples were run at a scanning rate of 10°C/min from 200°C to 300°C. An experiment was performed in a nitrogen atmosphere by purging nitrogen gas at 20 mL/min.30 Pyris software (PerkinElmer, Inc., Waltham, MA, USA) was used to evaluate the thermograms.31
Characterization of the optimal formulation using FTIR
FTIR spectrum of pure drug, polymers, and best formulation was obtained using FTIR (model Nicolet IS10; Thermo Scientific, Waltham, MA, USA) in the range of 400–4,000/cm using the KBr disc preparation method. The data of IR spectral were analyzed for pure drug, polymers, and the best formula using FTIR.32
Characterization of the optimal formulation using XRD
X-ray diffraction spectra were used to analyze samples using an XRD diffractometer (BTX324; Inxitu, Mountain View, CA, USA). Pellets were formed by compressing the powder, and Cu-Kα radiation (40 kV, 60 mA) used to record the diffractograms. The scanning program was scanned at the rate of 2°/min and a speed of 2°/2 cm per 2θ.33
In vivo studies of the optimal formulation in rabbits
All the experimental methods were carried out in accordance with the approved guidelines protocols from the Institutional Animal Care and Use Committee (IACUC) of the Medicine faculty, University of Malaya, Kuala Lumpur, Malaysia, and the IACUC approved this study (2013-12-03/PHAR/R/MR). Male New Zealand White rabbits aged 9–10 weeks weighing 2.5–3.0 kg were purchased from the IACUC center of the University of Malaya. Each rabbit was placed in a clean cage and the temperature was controlled to be 22°C–26°C, relative humidity of 60%±10%, and alternate 12 h of light and 12 h of darkness. They were provided with standard diet ad libitum. Before the experiment, the rabbits were divided into control and test groups, with three rabbits in each group.34
The purpose of the animal work was to evaluate the floating behavior of the selected formulation (MKP3) over a period of 12 h as well as to compare the drug-release patterns acquired from both in vitro and in vivo tests. The test group received the size-reduced formulation of MKP3, and the control group received tablets without polymers and NaHCO3 to investigate the floating ability of these ingredients to determine the release behavior of the tablet (Table 2). The metformin HCl dose was reduced to 250 mg to prevent hypoglycemia and to promote easy oral administration of the tablets. For pharmacokinetic study, the size-reduced tablets were prepared using exactly the same procedures mentioned under matrix tablet preparation; the tablets were given to the test and control groups (n=6) and blood samples of 0.5 mL were collected from the rabbits’ ear veins at time intervals of 0.5, 1, 2, 4, 6, 8, 10, 12, and 14 h; the collected blood was immediately transferred to 2 mL sterile containers containing 10 μL anticoagulant agent (10% w/v sodium citrate). The plasma was separated by centrifuging of the samples at 3,000 rpm for 20 min. Thus gained plasma samples were kept in a refrigerator till further use.
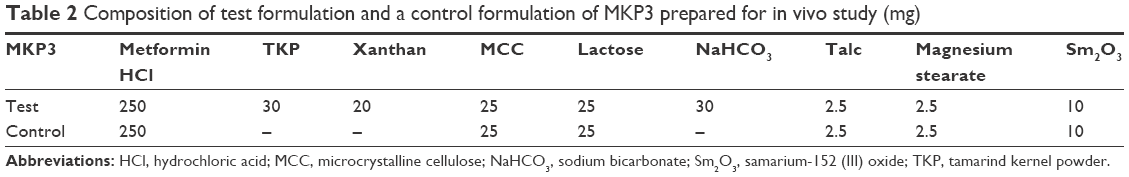 | Table 2 Composition of test formulation and a control formulation of MKP3 prepared for in vivo study (mg) |
The preparation of the tablets for in vivo gamma scintigraphy study followed exactly the same procedures as mentioned in the section of matrix tablet preparation, except that 10 mg of samarium-152 (III) oxide (152Sm2O3) powder was added to the ingredients and mixed well. Prior to the in vivo study, the release profile and floating properties of the 250 mg metformin HCl formulation were tested and compared to the previous result, and it showed that the formulations had a similar release profile and floating time.
Neutron activation of MKP3 tablets for in vivo study
All the test and control tablets used for the in vivo study were neutron activated using a nuclear reactor facility situated at the Malaysian Nuclear Agency (Bangi, Selangor, Malaysia). The facility has a 250 kW open pool-type research reactor (Triga Mark II; General Atomics, San Diego, CA, USA), which operates a uranium zirconium hydride assembly with a low-enriched uranium (20 wt% 235U) fuel source.35 Each tablet was heat-sealed in an individual polyethylene vial and packed into a polyethylene ampoule (commonly known as the “rabbit”). The ampoule was then carried to the reactor core by a pneumatic transport system. The tablet was irradiated in a neutron flux of 1×1013/cm2/s for 5 min to achieve nominal radioactivity of 10 MBq at 48 h after neutron activation.35 Gamma spectroscopy was carried out 24 h after neutron activation using a coaxial, p-type, 40% relative efficiency germanium detector (Canberra Inc., Meriden, CT, USA) and gamma spectrum analysis software (Genie TM 2000 Ver. 3.2; Canberra Inc.) to detect any radioactive impurities.35–37
In vivo gamma scintigraphy of the rabbits
The in vivo study was carried out on two separate occasions; three rabbits were imaged per day. The rabbits had access to only water during the entire day of study.38–40 A pill dispenser was used to administer the tablets orally to the rabbits without crushing; this was followed by 30 mL of drinking water administered to the rabbit with a sterile syringe. Images were taken immediately after drug administration using a dual-detector single-photon emission computed tomography-computed tomography (SPECT-CT) system (BrightView XCT; Philips, Eindhoven, the Netherlands) mounted with a low energy ultra-high-resolution collimator. The CT images were acquired using 120 kVp, 247 mAs, and 1 mm slice thickness at 512×512 matrix size, followed by SPECT imaging (128×128 matrix size, 64 angulations for 360°, 15 s per angulation). The total imaging time for a complete SPECT-CT acquisition was ~16 min. The acquisition was repeated at 1, 2, 4, 6, 8, 10, and 12 h after drug administration. Throughout the scanning procedure, the rabbit movements were controlled and minimized using a towel.
The 3D SPECT-CT images were reassembled using dedicated software (BrightView XCT workstation; Philips) and observed in three different planes, that is, the sagittal, coronal, and axial planes (n=3).
HPLC method for the quantification of metformin HCl in rabbit plasma
Metformin HCl plasma was extracted with protein precipitation method. An amount of 500 μL of plasma and 50 μL of 0.05 M HCl were added and vortex-mixed for 30 s; then 2 mL of acetonitrile was added. The mixture was centrifuged at 1,900 × g for 10 min and filtered. The filtrate was evaporated to dryness at 40°C and under a nitrogen stream. The extracts were reconstituted with 300 μL of mobile phase and 25 μL was injected into the HPLC machine at 30°C under isocratic conditions with a mobile phase of 34% acetonitrile and 66% aqueous phase.41 The aqueous phase contained 10 mM KH2PO4 and 10 mM sodium lauryl sulfate. The pH of the aqueous phase was adjusted to 5.2 by using dilute orthophosphoric acid at a flow rate of 1 mL/min. Eluted metformin HCl was detected by ultraviolet rays at 233 nm.42
Individual plasma concentrations and time curves were plotted. Maximum concentration of metformin HCl in blood (Cmax), the time at which Cmax is observed (Tmax), and half-life (T1/2) were directly calculated from the data and area under curve (AUC) was measured using linear trapezoidal rule.
Results and discussion
This study was aimed to achieve an extended gastric residence time and a prolonged presence of the drug in the GI tract by taking advantage of the immediate floating and swelling properties of the oral dosage form. The development of a GRDDS was based on a combination of floatable and swellable polymers, such as pulverized tamarind seed, xanthan, and NaHCO3 as a gas-generating agent. The physicochemical properties of TSP and TKP have been reported in a previous study,21 which indicated that both TKP and TSP have satisfactory swelling and gelling properties.
Evaluation of the matrix tablets
Figure 1 displays the physical appearance of the prepared formulations; all the tablets were expected to have the same weight and size, but different color due to the difference in the type of polymer used (TKP/TSP) and the percentage of polymer used in their composition. The results of characterization tests are tabulated in Table 3. All the formulations displayed acceptable results and complied with the USP requirements. The tablet thickness (n=10) was within the range of 4.6±0.0 to 4.6±0.9 mm; the average width and length of the prepared formulation were 10.1±0.1 to 10.2±0.4 mm and 18.4±0.0 to 18.4±0.5 mm, respectively. The average weight was between 764±1 and 765±1 mg (n=20). The drug content for three selected tablets from each batch ranged from 97.3%±1% to 98.9%±0.2%, which was within the standard range.43 The hardness of 10 tablets from each batch was assessed, and the values of the MSP and MKP formulations were between the ranges of 120–280 N and 136–280 N, respectively. All formulations had sufficient hardness to withstand abrasion and breakage during storage, transportation, and handling before use. The formulations with higher concentrations of xanthan had greater hardness compared to the others. In addition, it was found that the hardness decreased with increasing TKP and TSP concentration. The friability test for 10 tablets from each formulation was performed; all the tablets passed with friability values <1%, and no obvious cracking was observed in any of the tablets.
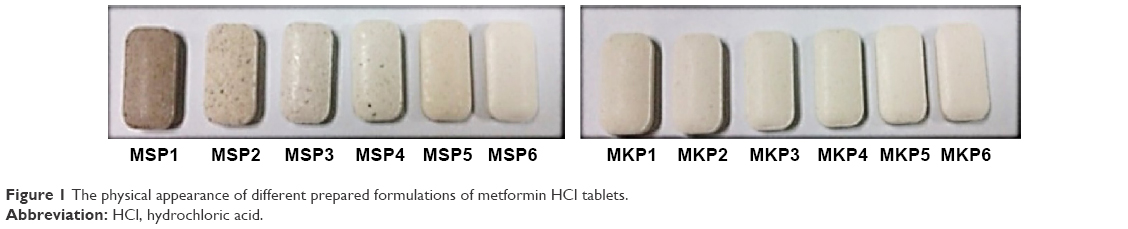 | Figure 1 The physical appearance of different prepared formulations of metformin HCl tablets. |
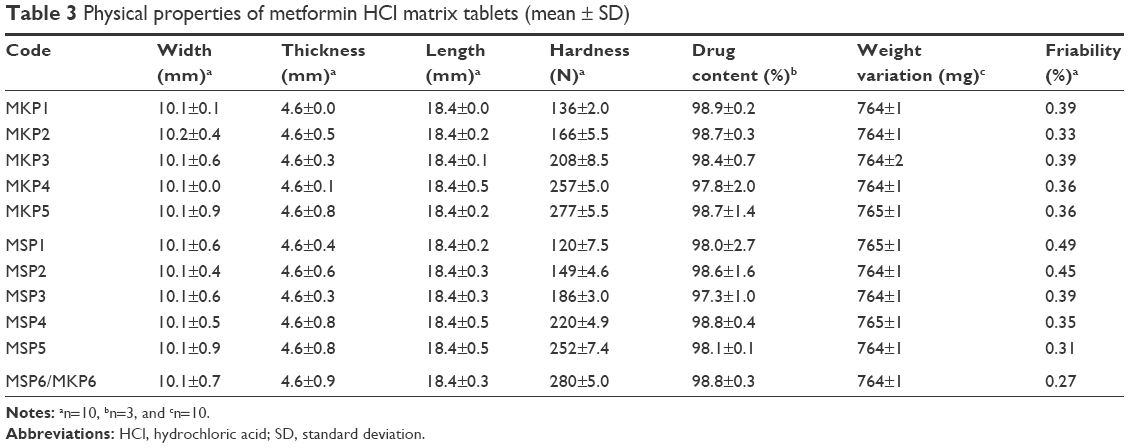 | Table 3 Physical properties of metformin HCl matrix tablets (mean ± SD) |
FLT and TFT
The FLT and TFT behaviors of the tablets are shown in the Figure 2. Based on the literature review, it was evident that the compression force has a significant effect on the physical characteristics, floating, and drug-release profile of the tablets;44 hence, in the present study, all the formulations were compressed with the same force at 2,200 N to minimize the chance of error between the batches. To develop a gastroretentive matrix tablet with the shortest FLT, different concentrations of NaHCO3 were examined. The optimal concentration was found to be 60 mg/tablet, which allowed a tablet to float within a minute.
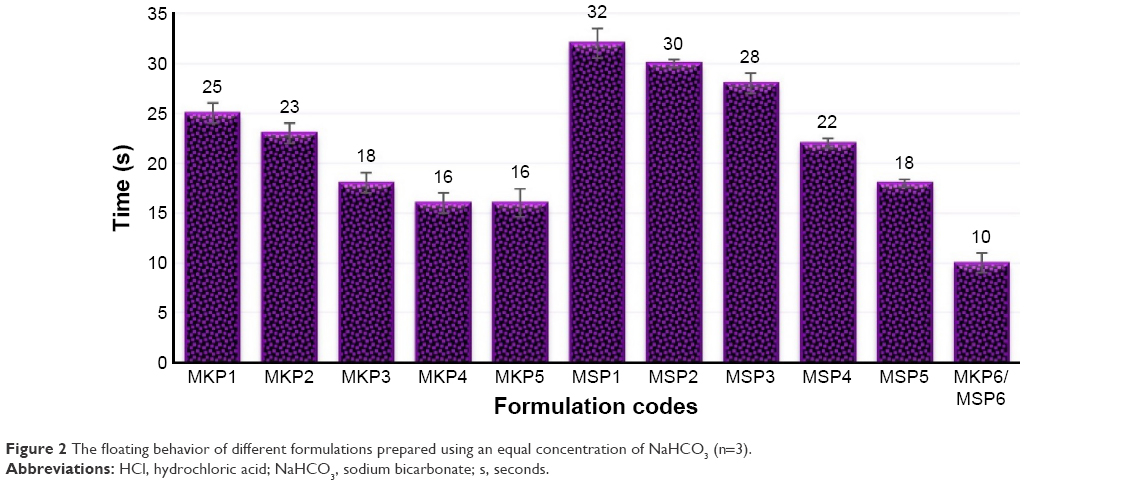 | Figure 2 The floating behavior of different formulations prepared using an equal concentration of NaHCO3 (n=3). |
In fact, the FLT of a tablet depends on two factors, which are critical to reduce the tablet density to <1 g/mL: 1) increases in the bulk volume of the dosage form as a result of the hydrocolloids swelling on the tablet surface when it comes into contact with gastric juice and 2) the existence of voids in the tablet (porosity). The gas-generating agents will generate CO2 in contact with aqueous media, and the gas will be entrapped in the matrix formed by polymers, thus causing the tablet to float.18 Channeling agents, such as lactose and microcrystalline cellulose, were used in the formulations to increase the released drug. These channeling agents fix the tablet’s desired dose and easily dissolve in aqueous media to form fine channels, thus promoting drug release.
In relation to a previous study,21 the viscosity and swelling ability of the polymers were measured. TKP demonstrates better swelling index and viscosity in comparison to TSP, which can be related to the difference in their structural integrity and hydration kinetics.
Immediate floating of the tablets was demonstrated by in vitro buoyancy study of the tablet placed in the simulated gastric fluid. Floating was influenced by the quantity of NaHCO3 and the polymer concentration. Tablets exhibited faster FLT when the gas-generating agent was used. The buoyancy outcomes obviously proved that increase in NaHCO3 concentration causes FLT decline, and the FLT was shorter in formulations containing TKP and xanthan (Figure 2); the TFTs of MKP3, MKP4, MKP5, MKP6, MSP3, MSP4, MSP5, and MSP6 were >24 h, although MSP1, MKP1, MKP2, and MSP2 disintegrated within 1 h.
In vitro drug-release study
The in vitro dissolution study helped in understanding the influence of polymers on the rate of drug release. It was observed that the drug-release profile was greatly dependent on the types of polymers and their concentrations; this information is presented in Figure 3. The presence of TKP/TSP in a formulation improved the rate of drug release in comparison with using xanthan alone. The formulations of MKP1/MSP1 and MSP2 with xanthan:TSP/TKP ratios of 0:100 and 20:80 were not able to control the release for the estimated period of time, and they disintegrated within 1 h. MKP1 and MSP1 achieved 100% release in 2 h, MKP2 and MSP2 completely released the drug in 5 h, and the remaining eight formulations could sustain the release for 12 h. Figure 3 illustrates the percentage of drug release as follows: MKP2 (99.26%), MKP3 (97.87%), MKP4 (96.6%), MKP5 (94.0%), MSP3 (98.4%), MSP4 (97.7%), MSP5 (96.6%), and MSP6/MKP6 (92.8%). Among all the formulations, MKP3 was found to be the optimal formulation; it contained 60 mg of TKP and 40 mg of xanthan; the FLT of this formulation (MKP3) was found to be 18 s and >90% of the drug had been released in 12 h. These in vitro data are consistent with previously published results and demonstrated that the formulation containing TKP could provide superior control release compared to the formulation containing TSP. The rate of drug release was prolonged for a longer time due to better gel-forming ability of the TKP polymer.34,36 This formulation had the highest similarity factor (f2) with commercial formulation (65%). The mechanisms of release for the above formulations were obtained by calculating the R2 value. The first-order, zero-order, Hixson–Crowell, and Higuchi models corresponded to the release data of each formulation. When the maximum value of the correlation coefficient is considered, the drug-release profile for all the prepared formulations appears to fit with the Hixson–Crowell and first-order models. The linearity of the first-order model was higher compared to the zero-order kinetics plot. This indicated that the release of drug from this formulation was dependent on the concentration. Hixson–Crowell was the dominant drug-release mechanism for all the formulations because the Hixson–Crowell plot had higher linearity when compared with the Higuchi model. This showed that the drug was being released by the Hixson–Crowell method from the matrix surface instead of diffusion. This occurred due to the strong matrix formed in the high proportion of polymer which contains a narrow gap space, resulting in limited and slow diffusion of the drug.
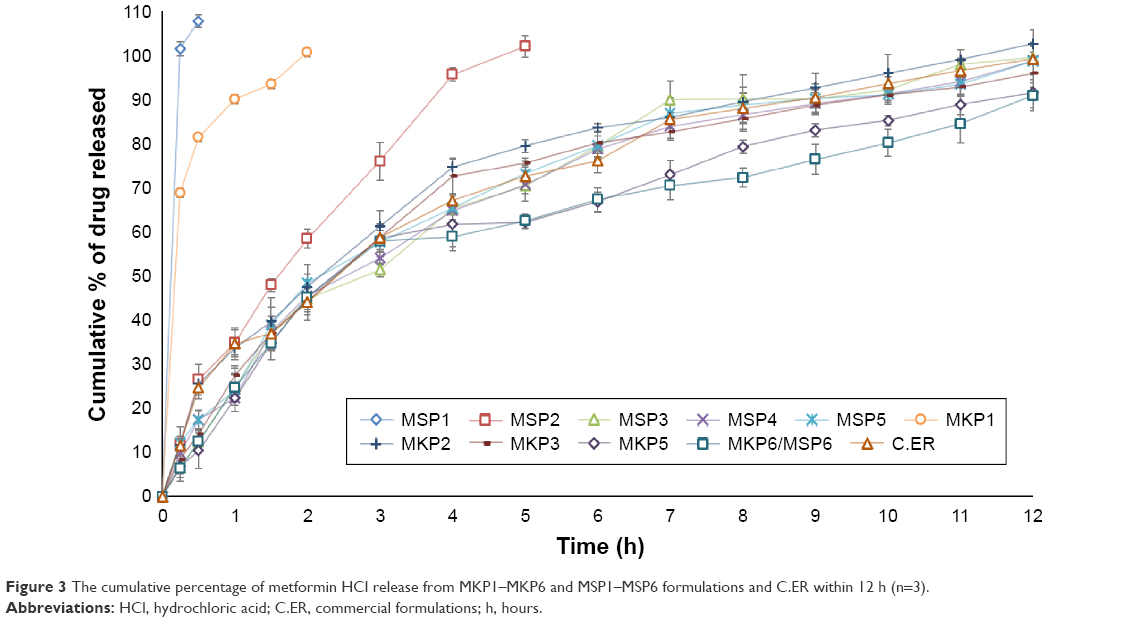 | Figure 3 The cumulative percentage of metformin HCl release from MKP1–MKP6 and MSP1–MSP6 formulations and C.ER within 12 h (n=3). |
Characterization of the optimal formulation using DSC
Pure metformin HCl displayed an endothermic peak at 233.09°C for instance of drug melting point. The presence of the sharp peak indicated high purity of an anhydrous crystalline molecule.45 The thermogram detected broad peaks at 115°C and 121.15°C of pure xanthan and pure TKP, respectively. These findings demonstrated that the polymers melting transition happened at these temperatures. The broad peaks in melting endotherm also indicated the multiple thermal characteristics of TKP and xanthan. From the DSC thermograms of the crushed tablets, the represented peaks correlated to the melting endotherm of pure metformin HCl, xanthan, and TKP. At the melting point of the crushed tablet, no considerable new peak or variation was noticed. This specified that no solid-state interaction occurred; in addition, the drug and polymers were compatible. However, there were some changes in the melting transition of the ingredients in the DSC thermogram of the crushed tablets. The peak’s height was shorter when compared with the thermograms of the pure forms, which may be due to differences in the concentrations of the ingredients in the samples, as other excipients in the sample act as impurities to the pure polymers; the peaks became broader and less pronounced than the peaks of the pure polymer (Figure 4).
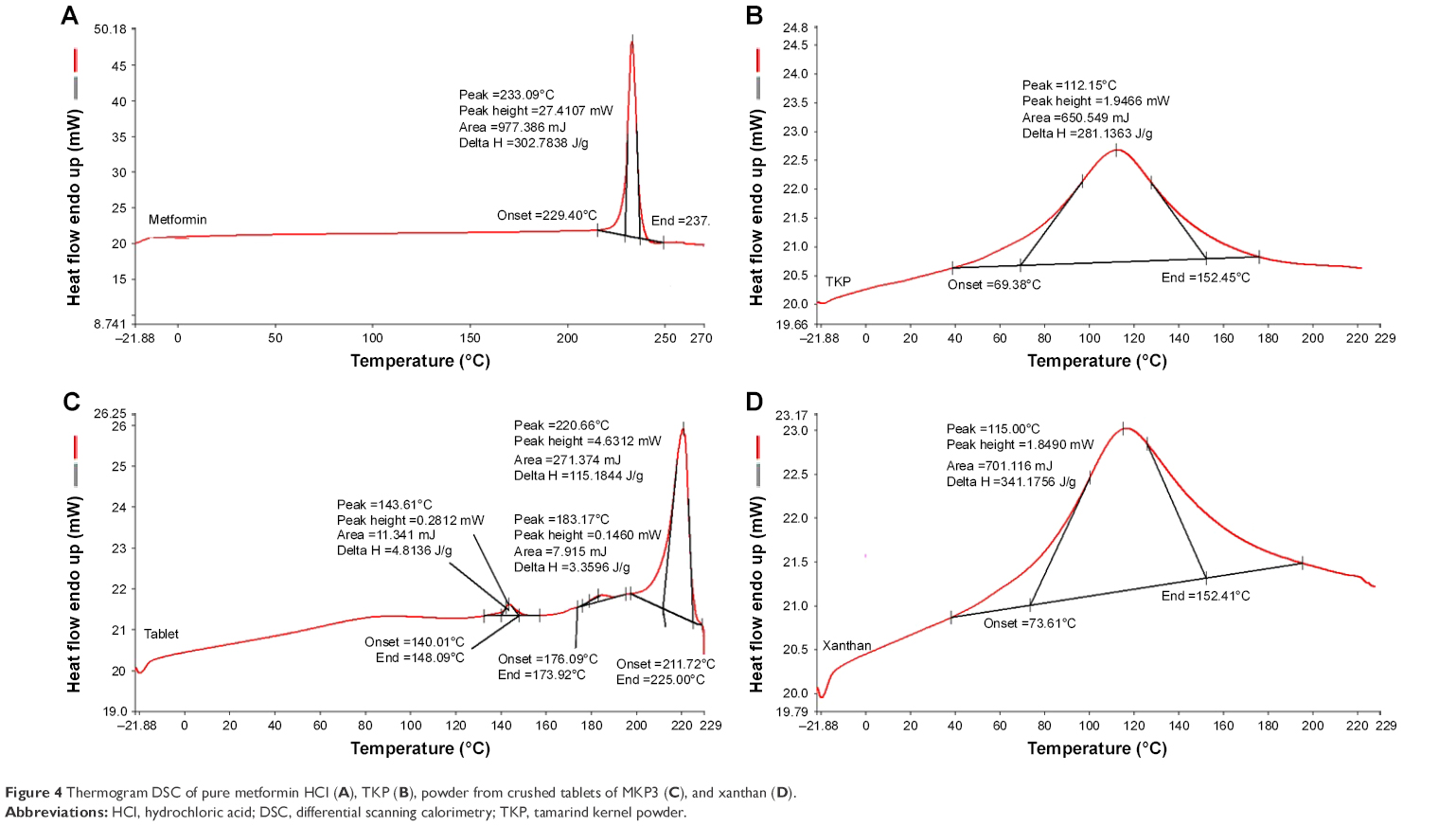 | Figure 4 Thermogram DSC of pure metformin HCl (A), TKP (B), powder from crushed tablets of MKP3 (C), and xanthan (D). |
Characterization of the optimal formulation using FTIR
FTIR is widely used for molecular structural investigations, and a specific range of the wavelengths represents different functional groups present in the tested sample. As seen in Figure 5, major peaks were identified at 3,568.37, 3,402.80, 1,935.40, 1,834.4, 1,077.3, and 963.3/cm for the formulated matrix tablets. The peaks of metformin HCl were detected at 3,369.4, 3,295.75, 3,157.01, 1,565.01, 1,064.23, and 938.16/cm. The TKP and xanthan main peaks were spotted at 1,040.64, 1,645.26, 2,925.27, 3,301.86/cm, and 3,374.48, 2,919.59, and 1,053.84/cm, respectively. The tablet spectrum showed no new peaks, indicative of no chemical alterations between the ingredients through the tablet preparation process.
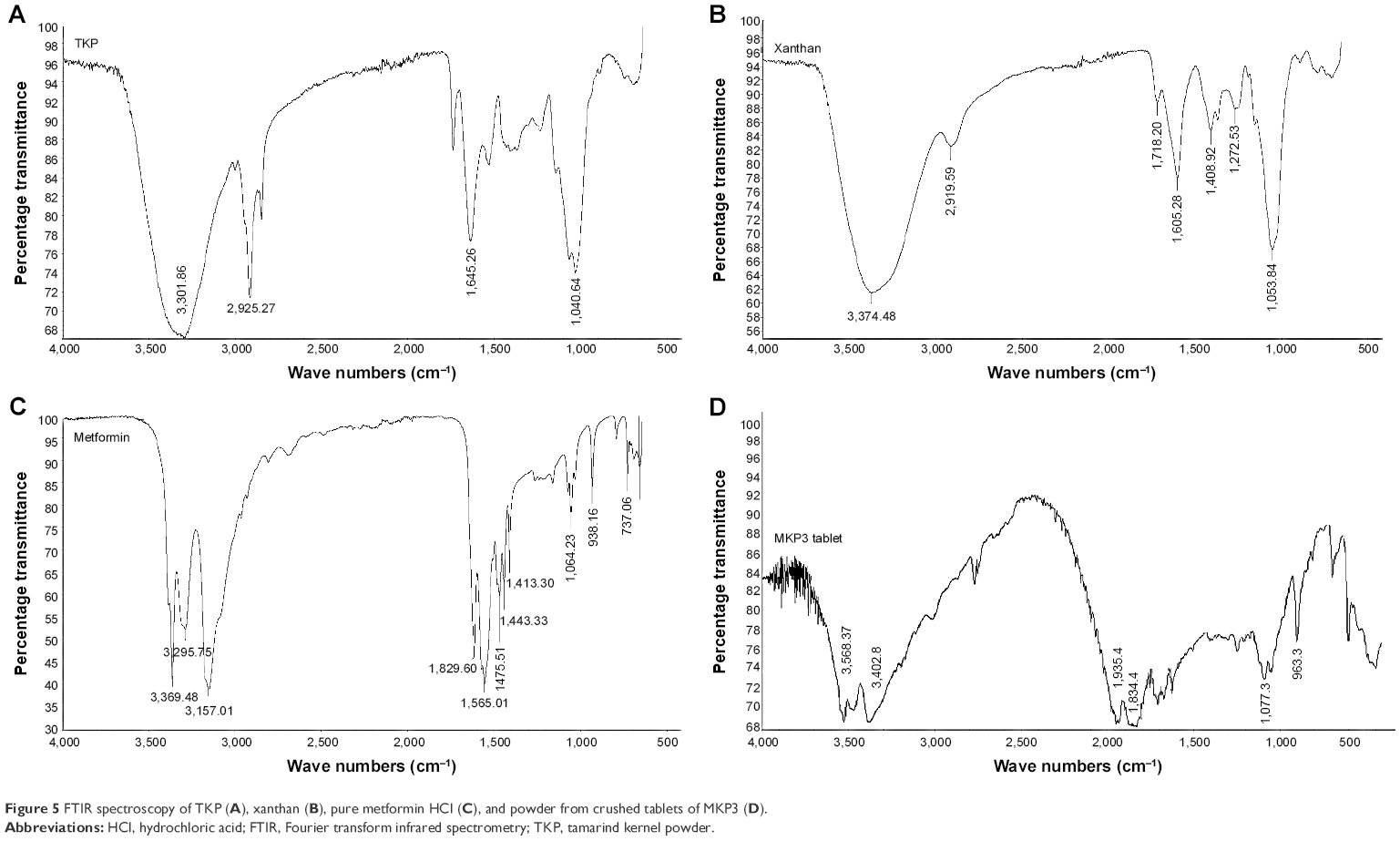 | Figure 5 FTIR spectroscopy of TKP (A), xanthan (B), pure metformin HCl (C), and powder from crushed tablets of MKP3 (D). |
Characterization of the optimal formulation using XRD
Phase identification of a crystalline material is mostly used by XRD which is a quick analytical tool, and it offers cell dimension information as well. Figure 6 displays the X-ray diffractograms of TKP, pure drug, xanthan, and their relevant formulations (MKP3). The sharp distinguishing peaks of metformin HCl were observed at angles (2θ°) of 13, 17.58, 22.5, 23.25, 25.25, 27.5, 28.6, 29.55, and 30.85, which indicates drug crystallinity. Extensive peak of TKP was displayed at 19.7, whereas a peak by xanthan appeared at an angle of 20.4 (2θ°), demonstrating both polymers natures are amorphous. Lesser significant peaks were exhibited by xanthan than TKP, possibly because of its greater amorphous nature. XRD of MKP3 formulation shows a decline in the pure drug crystallinity as reflected in the diffractogram. There was no remarkable change in the formulation peaks except the polymers peaks. No noticeable alteration was detected in the pattern of XRD, which might be due to the physical interactions that happened during tablet compression or polymers dispersion during the mixing.
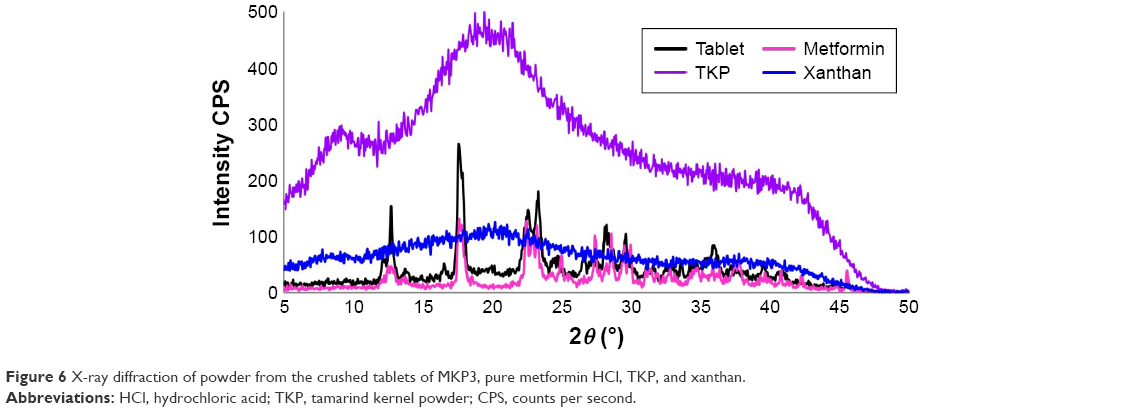 | Figure 6 X-ray diffraction of powder from the crushed tablets of MKP3, pure metformin HCl, TKP, and xanthan. |
In vivo imaging study of the rabbits
The in vitro dissolution study revealed that size reduction and the presence of samarium oxide (Sm2O3) in the formulation did not show any significant effect on the FLT or drug-release profile of the tablets. Furthermore, gamma spectroscopy data confirmed that neutron activation of the tablets produced no unwanted radioactive by-products.34,36 The in vitro dissolution result of the optimal formulation (MKP3) was compared with the MKP3-Sm formulation which contained 152Sm. The drug-release profile of samarium in this formulation was compared with metformin HCl in MKP3 formulation; the similarity factor was calculated and 68% similarity was achieved; so the release profile of 153Sm was considered as the release profile of metformin HCl (Figure 7).
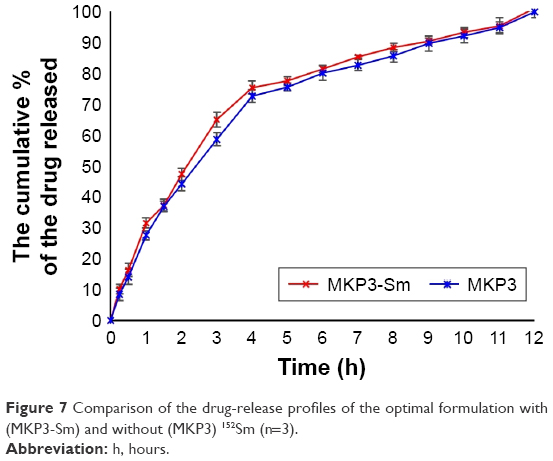 | Figure 7 Comparison of the drug-release profiles of the optimal formulation with (MKP3-Sm) and without (MKP3) 152Sm (n=3). |
Radionuclide 153Sm2O3 has an effective half-life of 1.93 days and emits γ and β radiations. For the last 20 years, 153Sm2O3 has been used as a radiotracer in numerous pharmacoscintigraphy research studies for reasons such as its availability, proper gamma energy, easy production, and minimal radiation exposure.46 153Sm2O3 is chemically stable and water insoluble; therefore, it will not be absorbed into the blood plasma during its transit in the GI tract; it is excreted through feces from the body and eventually decays to 153Eu as a stable nuclide.37
The objective of in vivo gamma scintigraphy study was to validate the in vitro test results of the optimal formulation. Hybrid imaging of SPECT-CT was chosen because it provides excellent information on both anatomy and physiology. The activity of the radiotracer (153Sm) was clearly seen on the 3D images with anatomical details. Patterns of the SPECT-CT images of rabbit’s stomach at different times are shown in Figures 8 and 9. The images display the kinetic of the 153Sm activity, which represents the kinetics of the drug, throughout the GI system of the rabbit over a period of 12 h. The results represent a prolonged gastric retention time and the release of the drug in all the rabbits administered with MKP3 formulation (Figure 8). In contrast, the control formulation showed shorter gastric retention time, and the drug was released rapidly throughout the period of imaging (Figure 9). This study revealed that gastroretentive properties of the MKP3 formulation were not altered by gastric emptying. The drug-release profiles for MKP3 (tested both in vivo and in vitro) and the control formulation are shown in Figure 10. The MKP3 release at 1, 2, 4, 6, 8, 10, and 12 h was found to be 25%, 40.3%, 65.8%, 75.9%, 79.9%, 88.2%, and 97.4%, and for the control formulation the values were 55.2%, 80.3%, 85.0%, 88.5%, 91.5%, 94.8%, and 95.0%, respectively. The obtained in vivo gamma scintigraphy results were similar to those of previously published studies, and it confirmed and proved that using a gel-forming agent such as TKP could sustain the drug release and that tablets will remain in the stomach for longer period of the time with optimum release rate.34,36
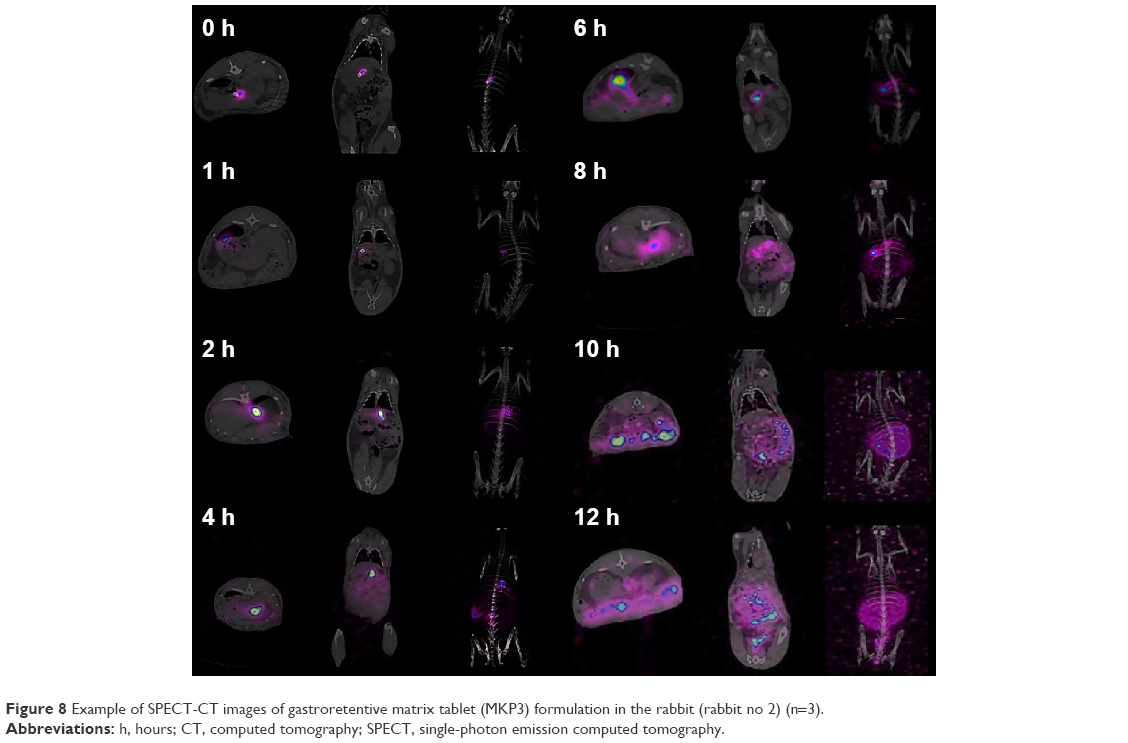 | Figure 8 Example of SPECT-CT images of gastroretentive matrix tablet (MKP3) formulation in the rabbit (rabbit no 2) (n=3). |
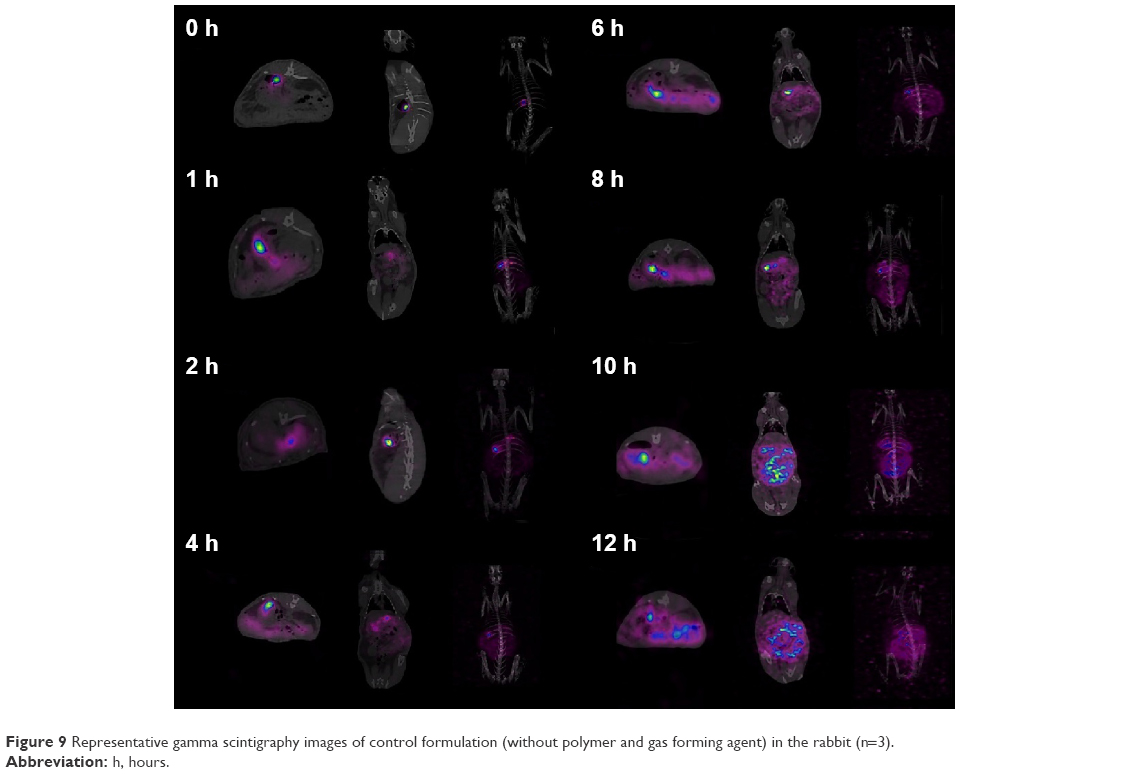 | Figure 9 Representative gamma scintigraphy images of control formulation (without polymer and gas forming agent) in the rabbit (n=3). |
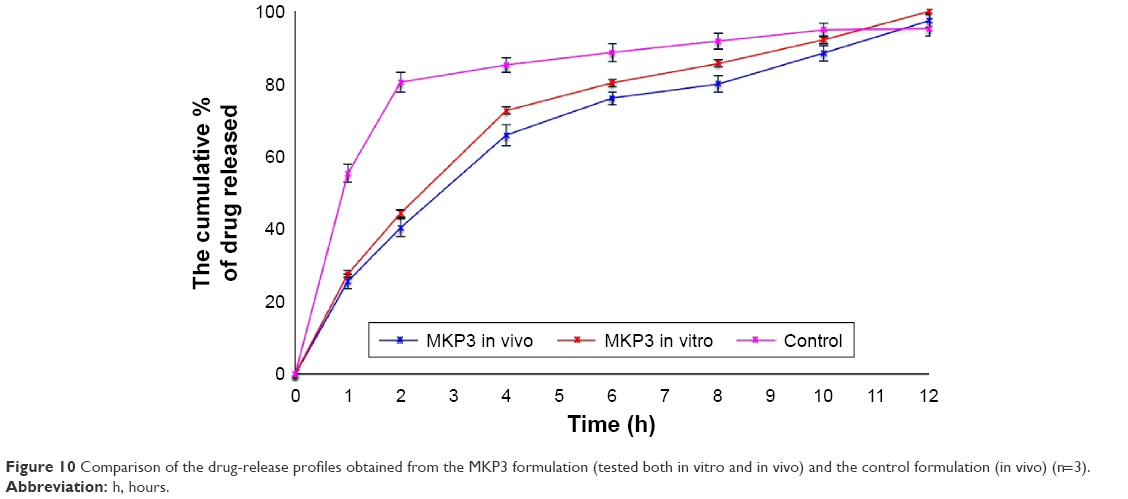 | Figure 10 Comparison of the drug-release profiles obtained from the MKP3 formulation (tested both in vitro and in vivo) and the control formulation (in vivo) (n=3). |
Pharmacokinetic study
The calibration curve was obtained by plotting the ratio of the chromatographic peak area (metformin HCl) versus concentration of metformin HCl. Samples were prepared and injected on the same day. The calibration curve was linear in the given range (R2=0.9983). Estimated pharmacokinetic parameters are presented in Table 4 and Figure 11.
 | Table 4 Pharmacokinetic parameters of metformin HCl in plasma samples (n=6, mean ± SD) |
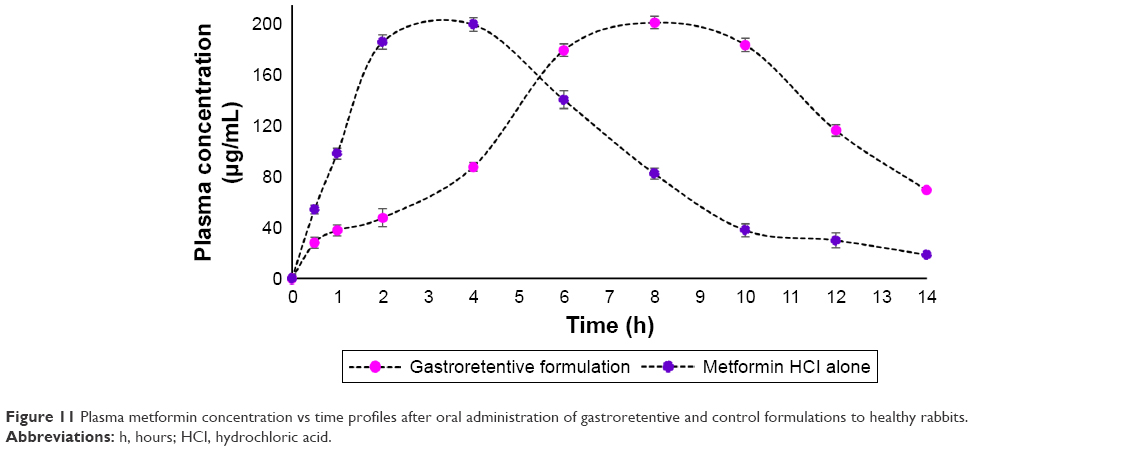 | Figure 11 Plasma metformin concentration vs time profiles after oral administration of gastroretentive and control formulations to healthy rabbits. |
Pharmacokinetic study of data shows increase in AUC and Tmax of the sample administered with gastroretentive formulation of metformin HCl compared to the control formulation containing metformin HCl alone without polymers. A 4 h delay was obvious in Tmax, which can be justified due to the sustaining release property of the gastroretentive formulation. The bioavailability was increased because of the polymer’s ability to form a strong matrix tablet and delay the release; so a greater amount of the drug reaches systemic circulation (this is depicted by the higher AUC for the gastroretentive formulation compared to the control formulation [P<0.05]). The aim of the therapy is to achieve a steady state blood level that is therapeutically effective and nontoxic for an extended period of time. The present study concurs with this aim where the Cmax values between the control formulation and the gastroretentive formulation are significantly the same (P>0.05); Figure 11 shows that the maintenance of Cmax for the optimized preparation is extended. The objective of gastroretentive formulation is to maintain a therapeutic blood level over an extended period, so the drug must enter the circulation at approximately the same rate at which it is eliminated. The elimination rate is quantitatively described by the half-life (T1/2). This study clearly shows that the half-life of a normal tablet and the optimized tablet exhibit significantly the same (P>0.05) half-life for both preparations (Table 4).
Conclusion
The in vitro data revealed that tamarind and xanthan are good candidates for sustained-release formulation. Among all the formulations, MKP3 with a TKP:xanthan ratio of 3:2 showed the best sustained-release properties with relatively short FLT; hence, it was selected as the optimal formulation in this study. The in vitro data were validated by in vivo scintigraphy imaging and further confirmed that the MKP3 formulation successfully prolongs the gastric retention time by its swelling and floating behavior; in addition, a sustained release of >12 h was achieved. This formulation has potential to be developed further for clinical trials in humans, particularly for diabetes II mellitus patients. The in vitro data indicated that low concentrations of TKP and TSP were insufficient for controlling drug release in the dosage form. In addition, this formulation had 61% similarity with the in vivo result obtained from the gamma scintigraphy study of the rabbits and may be a potential candidate for use as an excipient in sustained-release formulations.
Acknowledgments
The research leading to these results has received funding from the University of Malaya, UMRG- Frontier Science (RP021D-14AFR), and University Malaya Research Grant (RG044/11BIO) are greatly appreciated.
Disclosure
The authors report no conflicts of interest in this work.
References
Van Krevelen DW, Te Nijenhuis K. Properties of Polymers: Their Correlation with Chemical Structure; Their Numerical Estimation and Prediction from Additive Group Contributions. Oxford: Elsevier; 2009. | ||
Malviya R, Srivastava P, Kulkarni G. Applications of mucilages in drug delivery – a review. Adv Biol Res. 2011;5(1):1–7. | ||
Tiwari SB, Murthy TK, Pai MR, Mehta PR, Chowdary PB. Controlled release formulation of tramadol hydrochloride using hydrophilic and hydrophobic matrix system. AAPS PharmSciTech. 2003;4(3):18–23. | ||
Prajapati VD, Jani GK, Moradiya NG, Randeria NP. Pharmaceutical applications of various natural gums, mucilages and their modified forms. Carbohydr Polym. 2013;92(2):1685–1699. | ||
Jani GK, Shah DP, Prajapati VD, Jain VC. Gums and mucilages: versatile excipients for pharmaceutical formulations. Asian J Pharm Sci. 2009;4(5):309–323. | ||
Nussinovitch A. Plant Gum Exudates of the World: Sources, Distribution, Properties, and Applications. Taylor & Francis Group, Boca Raton, London, New York: CRC Press; 2009. | ||
Jani GK, Shah DP, Prajapati V, Jain V. Gums and mucilages: versatile excipients for pharmaceutical formulations. Asian J Pharm Sci. 2009;4(5):309–323. | ||
Bhatta R, Krishnamoorthy U, Mohammed F. Effect of tamarind (Tamarindus indica) seed husk tannins on in vitro rumen fermentation. Anim Feed Sci Techn. 2001;90(3):143–152. | ||
Kaur H, Yadav S, Ahuja M, Dilbaghi N. Synthesis, characterization and evaluation of thiolated tamarind seed polysaccharide as a mucoadhesive polymer. Carbohydr Polym. 2012;90(4):1543–1549. | ||
Gupta V, Puri R, Gupta S, Jain S, Rao G. Tamarind kernel gum: an upcoming natural polysaccharide. Syst Rev Pharm. 2010;1(1):50. | ||
Mishra A, Malhotra AV. Tamarind xyloglucan: a polysaccharide with versatile application potential. J Mater Chem. 2009;19(45):8528–8536. | ||
Ho C-T. African natural plant products: new discoveries and challenges in chemistry and quality. OUP Catalogue. 2010. Available from: https://www.amazon.com/African-Natural-Plant-Products-Discoveries/dp/0841269874. Accessed October 28, 2016. | ||
Weh FH, Razavi M, Erh CH, et al. Formulation and in vitro evaluation of hydrodynamically balanced matrix tablets of famotidine using pectin as controlled release polymer. Lat Am J Pharm. 2014;33(3):420–431. | ||
Khan AD, Bajpai M. Floating drug delivery system: an overview. Int J PharmTech Res. 2010;2(4):2497–2505. | ||
Deshpande AA, Shah NH, Rhodes CT, Malick W. Evaluation of films used in development of a novel controlled-release system for gastric retention. Int J Pharm. 1997;159(2):255–258. | ||
Prajapati VD, Jani GK, Khutliwala TA, Zala BS. Raft forming system – an upcoming approach of gastroretentive drug delivery system. J Control Release. 2013;168(2):151–165. | ||
Streubel A, Siepmann J, Bodmeier R. Floating matrix tablets based on low density foam powder: effects of formulation and processing parameters on drug release. Eur J Pharm Sci. 2003;18(1):37–45. | ||
Singh BN, Kim KH. Floating drug delivery systems: an approach to oral controlled drug delivery via gastric retention. J Control Release. 2000;63(3):235–259. | ||
Narang N. An updated review on: floating drug delivery system (FDDS). Int J Appl Pharm. 2011;3(1):1–7. | ||
Boldhane SP, Kuchekar BS. Gastroretentive drug delivery of metformin hydrochloride: formulation and in vitro evaluation using 32 full factorial design. Curr Drug Deliv. 2009;6(5):477–485. | ||
Razavi M, Nyamathulla S, Karimian H, Moghadamtousi SZ, Noordin MI. Hydrogel polysaccharides of tamarind and xanthan to formulate hydrodynamically balanced matrix tablets of famotidine. Molecules. 2014;19(9):13909–13931. | ||
Razavi M, Nyamathulla S, Karimian H, Noordin MI. Novel swellable polymer of orchidaceae family for gastroretentive drug delivery of famotidine. Drug Design Dev Ther. 2014;8:1315. | ||
Zade P, Kawtikwar P, Sakarkar D. Formulation, evaluation and optimization of fast dissolving tablet containing tizanidine hydrochloride. Int J Pharm Tech Res. 2009;1(1):34–42. | ||
Shinde AJ, Patil MS, More HN. Formulation and evaluation of an oral floating tablet of cephalexin. Indian J Pharm Educ Res. 2010;44(3):243–252. | ||
Savaşer A, Özkan Y, Işimer A. Preparation and in vitro evaluation of sustained release tablet formulations of diclofenac sodium. Il Farmaco. 2005;60(2):171–177. | ||
Kulkarni A, Bhatia M. Development and evaluation of regioselective bilayer floating tablets of Atenolol and Lovastatin for biphasic release profile. Iran J Pharm Res. 2010;8(1):15–25. | ||
Baumgartner S, Kristl J, Vrečer F, Vodopivec P, Zorko B. Optimisation of floating matrix tablets and evaluation of their gastric residence time. Int J Pharm. 2000;195(1):125–135. | ||
Desai D, Wong B, Huang Y, et al. Surfactant-mediated dissolution of metformin hydrochloride tablets: wetting effects versus ion pairs diffusivity. J Pharm Sci. 2014;103(3):920–926. | ||
Costa P, Lobo JMS. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13(2):123–133. | ||
Chopra S, Patil GV, Motwani SK. Release modulating hydrophilic matrix systems of losartan potassium: optimization of formulation using statistical experimental design. Eur J Pharm Biopharm. 2007;66(1):73–82. | ||
Darapu B, Sundaramoorthy K, Vetrichelvan T. Formulation and in-vitro evaluation of gastroretentive floating microspheres of ranitidine hydrochloride. Res J Pharm Dosage Forms Technol. 2011;3(1):24. | ||
Gupta NV, Shivakumar H. Development of a gastroretentive drug delivery system based on superporous hydrogel. Trop J Pharm Res. 2010;9(3):257–264. | ||
Wang X, Du Y, Luo J. Biopolymer/montmorillonite nanocomposite: preparation, drug-controlled release property and cytotoxicity. Nanotechnology. 2008;19(6):065707. | ||
Razavi M, Karimian H, Yeong CH, Chung LY, Nyamathulla S, Noordin MI. Gamma scintigraphic evaluation of floating gastroretentive tablets of metformin HCl using a combination of three natural polymers in rabbits. Drug Design Dev Ther. 2015;9:4373. | ||
Yeong C-H, Abdullah BJJ, Ng K-H, et al. Production and first use of 153 SmCl 3-ion exchange resin capsule formulation for assessing gastrointestinal motility. Appl Radiat Isot. 2012;70(3):450–455. | ||
Razavi M, Karimian H, Yeong CH, et al. Gamma scintigraphic study of the hydrodynamically balanced matrix tablets of metformin HCl in rabbits. Drug Design Dev Ther. 2015;9:3125. | ||
Yeong C-H, Abdullah BJJ, Ng K-H, et al. Neutron-activated 153Sm-ion-exchange resin as a tracer for gastrointestinal scintigraphy. Nucl Med Commun. 2011;32(12):1256–1260. | ||
Hu L-D, Liu Y, Tang X, Zhang Q. Preparation and in vitro/in vivo evaluation of sustained-release metformin hydrochloride pellets. Eur J Pharm Biopharm. 2006;64(2):185–192. | ||
Gusler G, Gorsline J, Levy G, et al. Pharmacokinetics of metformin gastric-retentive tablets in healthy volunteers. J Clin Pharm. 2001;41(6):655–661. | ||
Marathe P, Arnold M, Meeker J, Greene D, Barbhaiya R. Pharmacokinetics and bioavailability of a metformin/glyburide tablet administered alone and with food. J Clin Pharm. 2000;40(12):1494–1502. | ||
Porta V, Schramm SG, Kano EK, et al. HPLC-UV determination of metformin in human plasma for application in pharmacokinetics and bioequivalence studies. J Pharm Biomed Anal. 2008;46(1):143–147. | ||
Chhetri HP, Thapa P, Van Schepdael A. Simple HPLC-UV method for the quantification of metformin in human plasma with one step protein precipitation. Saudi Pharm J. 2014;22(5):483–487. | ||
Kumar R, Patil S, Patil M, Patil SR, Paschapur MS. Design and in vitro evaluation of oral floating matrix tablets of aceclofenac. Int J Chem Tech Res. 2009;1(4):815–825. | ||
Velasco M, Ford JL, Rowe P, Rajabi-Siahboomi AR. Influence of drug: hydroxypropylmethylcellulose ratio, drug and polymer particle size and compression force on the release of diclofenac sodium from HPMC tablets. J Control Release. 1999;57(1):75–85. | ||
Singh AN, Pathak K. Development and evaluation of dual controlled release microballoons containing riboflavin and citric acid: in vitro and in vivo evaluation. J Microencapsul. 2011;28(5):442–454. | ||
Yeong C-H, Abdullah BJJ, Ng K-H, et al. Production and first use of 153SmCl3-ion exchange resin capsule formulation for assessing gastrointestinal motility. Appl Radiat Isot. 2012;70(3):450–455. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.