Back to Journals » OncoTargets and Therapy » Volume 13
GAS2 Promotes Cell Proliferation and Invasion and Suppresses Apoptosis in Pediatric T-Cell Acute Lymphoblastic Leukemia and Activates Wnt/β-Catenin Pathway
Authors Kong Y, Zhao S, Tian H, Hai Y
Received 31 October 2019
Accepted for publication 10 January 2020
Published 5 February 2020 Volume 2020:13 Pages 1099—1108
DOI https://doi.org/10.2147/OTT.S236854
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leo Jen-Liang Su
Yan Kong, Shouyong Zhao, Hurong Tian, Yang Hai
Clinical Lab, Liaocheng People’s Hospital, Liaocheng City, Shandong Province 252000, People’s Republic of China
Correspondence: Hurong Tian
Clinical Lab, Liaocheng People’s Hospital, No. 67, Dongchang West Road, Dongchangfu District, Liaocheng City, Shandong Province 252000, People’s Republic of China
Tel +86 0635 8276172
Email [email protected]
Purpose: This study aimed to investigate the effect of growth arrest specific 2 (GAS2) on T-cell acute lymphoblastic leukemia (T-ALL) and its potential molecular mechanism.
Methods: The GAS2 expression level was detected by qRT-RCR and Western blot in normal T lymphocytes and T-ALL cells Jurkat and CCRF-CEM. The proliferation and invasion of Jurkat and CCRF-CEM cells were detected by MTT and Transwell assay, respectively. Apoptosis and cell cycle were measured by flow cytometry. In addition, the chemotherapeutic sensitivity of Jurkat and CCRF-CEM cells was measured MTT assay and flow cytometry.
Results: GAS2 was highly expressed in Jurkat and CCRF-CEM cells. GAS2 could promote cell proliferation and invasion, and inhibit apoptosis of Jurkat and CCRF-CEM cells. GAS2 also promoted cell cycle changes from G0/G1 phase to S phase in Jurkat and CCRF-CEM cells. In addition, GAS2 could reduce the chemotherapeutic sensitivity of Jurkat and CCRF-CEM cells. GAS2 overexpression could promote the expression levels of ki67, proliferating cell nuclear antigen (PCNA), Bcl-2, c-myc, cyclin D1 and β-catenin, while GAS2 knockdown could inhibit their expression levels. Meanwhile, GAS2 overexpression could inhibit Bax expression. Moreover, Wnt/β-catenin pathway inhibitor XAV939 could inhibit the expressions of c-myc, cyclin D1 and β-catenin, but activator LiCl could promote their expression.
Conclusion: Our study demonstrated that GAS2 could promote cell proliferation and invasion, and induce cell cycle, as well as inhibit apoptosis and could activate the Wnt/β-catenin pathway in T-ALL cells.
Keywords: T-ALL, GAS2, proliferation, apoptosis, invasion, cell cycle, Wnt/β-catenin pathway
Introduction
T-cell acute lymphoblastic leukemia (T-ALL), is an uncommon hematological malignancy, accounting for 15% and 25% of pediatric and adult ALL.1–3 Although substantial improvements have been made on the treatment of T-ALL in recent years, the clinical outcome remains poor.4 Therefore, it is urgent to find new molecular diagnosis methods and targets for the treatment of T-ALL.
Growth arrest specific 2 (GAS2) is a cytoplasmic protein and first identified by Schneider et al.5 More and more studies have suggested that GAS2 can modulate multiple cell biological behaviors including cell cycle, proliferation, apoptosis and differentiation.6–9 Previous researches indicate that GAS2 is essential in hepatocellular carcinoma10 and colorectal cancer.11 Zhou et al12 have reported that GAS2 is upregulated in chronic myeloid leukemia cells. Sun et al13 have indicated that GAS2 has higher expression in the leukemic cells than that in control cells and promotes the growth of leukemic cells. However, the effects of GAS2 in T-ALL and its potential molecular mechanism have not been fully elucidated.
Wnt/β-catenin pathway is involved in several stages of embryogenesis, lymphocyte development and self-renewal of hematopoietic stem cells.14,15 More and more studies have confirmed that the abnormal activation of the Wnt/β-catenin pathway is implicated in the formation of multiple human malignancies14 and mediating the anti-tumor function of histone deacetylase inhibitors in T-ALL.16 In addition, GAS2 is reported to interact directly with β-calpain and inhibit β-calpain activity.17,18 However, whether GAS2 affects T-ALL through regulating the Wnt/β-catenin pathway is unknown.
In this study, we explored the effects of GAS2 on the biological behaviors of the T-ALL cells and its potential molecular mechanisms. Our results revealed that GAS2 could significantly promote cell proliferation and invasion, and induce cell cycle as well as inhibit cell apoptosis and could activate the Wnt/β-catenin pathway in T-ALL cells. The findings of our study may provide a new theoretical foundation for deeply exploring the treatment of T-ALL.
Materials and Methods
Cell Cultures
Normal T lymphocytes and T-ALL cell lines Jurkat and CCRF-CEM (ATCC, Manassas, VA, USA) were cultured in RPMI-1640 (Gibco, USA) complemented with 10% fetal bovine serum (Gibco, USA) at 37°C in 5% CO2.
Cell Transfection Assay
The lentiviral vectors used for inhibiting GAS2 (phU6-EGFP-shRNA-GAS2) or for inducing GAS2 (pUbi-EGFP-GAS2) were supplied by GenePharma (Shanghai, China). Their corresponding control vectors were also obtained from GenePharma. In short, a total of 5×104 Jurkat and CCRF-CEM cells were added into the 24-plate well containing the media with lentiviral vectors overnight, and divided into NC group (transfected with negative control), GAS2 group (transfected with pUbi-EGFP-GAS2), sh-NC group (transfected with pUbi-EGFP empty control vector) and sh-GAS2 group (transfected with phU6-EGFP-shRNA-GAS2). In addition, cells in the GAS2 group when treated with 10-μM XAV939 (Wnt/β-catenin pathway inhibitor, Tocris Bioscience, USA) were GAS2 + XAV939 group. Cells sh-GAS2 group when treated with 20 μM LiCl (Wnt/β-catenin pathway activator, MedChemExpress, USA) were sh-GAS2 + LiCl group.
Cell Proliferation and Apoptosis Assay
Cell proliferation and apoptosis assay were performed according to a previous study.19 Briefly, after transfection, cells were added with 20 μL 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT, 5 mg/mL, Sigma, USA) at the predetermined time, and absorbance at 495 nm was obtained. For cell apoptosis, Jurkat and CCRF-CEM cells were stained with 5 μL Annexin V-FITC and propidium iodide (PI) in dark, and apoptosis was analyzed by a flow cytometer (FACSCanto™ II, BD Biosciences, CA, USA).
Transwell Assay
Cells invasion was examined by a transwell chamber pre-coated with Matrigel (Invitrogen, USA). Briefly, transfected Jurkat and CCRF-CEM cells were seeded in the upper chamber, and 500-μL medium was added into the lower chamber. After 24 h, the cells on the lower side of the membrane were fixed and stained using 0.1% crystal violet dye (Sigma, USA). Finally, the number of invasive cells was counted under an optical microscope.
Cell Cycle Assay
Jurkat and CCRF-CEM cells cycle were detected using a cell cycle detection kit according to the manufacturer’s instructions (KeyGEN Biotech, Jiangsu, China). Briefly, cells were planted into a 6-well plate, and were stained with PI for 30 min at 4°C. After transfection, the cell cycle was analyzed by using a FACSCanto™ II flow cytometer (BD Biosciences).
Evaluation of Drug Susceptibility
Jurkat and CCRF-CEM cells were added into a 96-well plate. When the cells adhered to the wall, vincristine (VCR, 0.5 μg/mL, Sigma, USA) was added into the well. After 24 h of culture, 20-μL MTT was added to each well and cells were continued to incubate for 4 h. The optical density (OD) was measured at 495 nm by using a microplate reader. The inhibition rate (IR, %) was calculated according to the formula as follows: IR (%) = (1-ODMedicine/ODControl) × 100%.
Real-Time Quantitative PCR (RT-qPCR)
Total RNA was extracted from the normal T lymphocytes, Jurkat and CCRF-CEM cells by using TRIZOL reagent (Invitrogen, USA). Then, RNA was reverse-transcribed into cDNA and measured by using qRT-PCR (Bio-Rad, CA, USA) with SYBR green qPCR Master Mix (Thermo Scientific, USA). Primers used for qRT-PCR analysis were as follows: GAS2 F: 5′-GCAACCCAGAGAAGTGTGTCT-3′, R: 5′-CAGGAGGCTCCACACCAT-3′; β-actin F: 5′-TCAGGTCATCACTATCGGCAAT-3′, R: 5′-AAAGAAAGGGTGTAAAACGCA-3′.
Western Blot Analysis
Protein samples were extracted using RIPA lysis buffer (Beyotime, Shanghai, China). Protein samples with the same amount of protein (50 μg/lane) were subjected to SDS-PAGE and then transferred to the PVDF membrane. After being blocked with 5% non-fat milk, the membrane was incubated at 4°C overnight with the primary antibodies (GAS2, 1:1000, ab175290; ki67, 1:1000, ab92742, Abcam; proliferating cell nuclear antigen (PCNA), 1:1000, #13110; Bax, 1:1000, #5023; Bcl-2, 1:1000, #4223; c-myc, 1:1000, #9402; cyclin D1, 1:1000, #2922; β-Catenin (Ser33/37), 1:1000, #2922 and β-actin, 1:2000, #4967, Cell signaling, USA). Subsequently, HRP-conjugated secondary antibody (1:5000, Sigma, USA) was used to incubate the membranes for 2 h at room temperature. The protein bands were visualized with ECL system (Thermo, USA) and then analyzed by Quantity One 1-D Analysis Software (Bio-Rad).
Statistical Analysis
All statistical analyses were performed using SPSS 22.0 (Chicago, IL). The results were presented in the form of mean ± standard deviation. The differences between various groups were analyzed by a one-way ANOVA followed by the Tukey’s post hoc test, and the data of the two groups were assessed using the Student t test. P < 0.05 was considered to be statistically significant.
Results
The Expression of GAS2 Is Upregulated in Jurkat and CCRF-CEM Cells
After detecting the expression levels of GAS2 using qRT-PCR and Western blot, we found that GAS2 expression in Jurkat and CCRF-CEM cells was significantly higher than that in normal T lymphocytes (P < 0.001) (Figure 1A). As shown in Figure 1B and C, transfection of lentiviral vectors phU6-EGFP-shRNA-GAS2 or pUbi-EGFP-GAS2 could markedly inhibit or increase GAS2 expression in Jurkat and CCRF-CEM cells, suggesting that the cell transfection was successful.
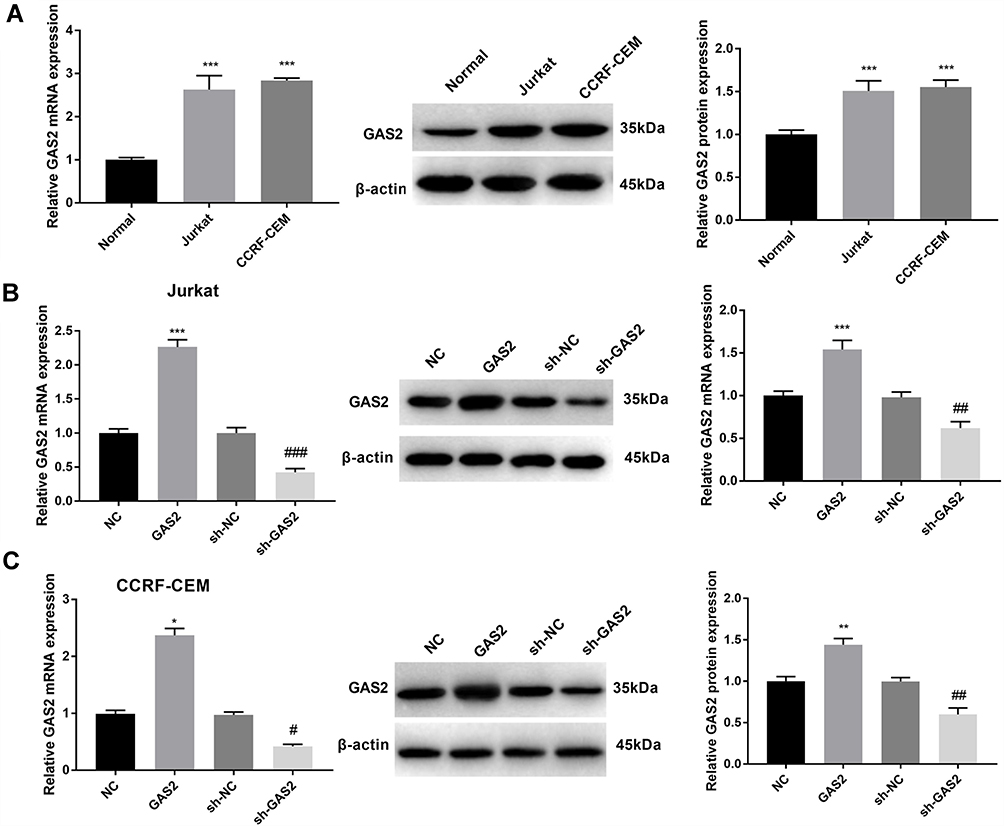 |
Figure 1 The expression of GAS2 was upregulated in Jurkat and CCRF-CEM cells. (A) The mRNA and protein expression of GAS2 was measured by qRT-PCR and Western blot in normal T lymphocytes and acute lymphoblastic leukemia cells Jurkat and CCRF-CEM. (B) The mRNA and protein expression of GAS2 was measured by qRT-PCR and Western blot in the transfected Jurkat cells. (C) The mRNA and protein expression of GAS2 was measured by qRT-PCR and Western blot in the transfected CCRF-CEM cells. Data were presented as mean ± standard deviation with repeated for three times. ***P<0.001, vs Normal group (A). *P<0.05, **P<0.01, ***P<0.001, vs NC group; #P<0.05, ##P<0.01, ###P<0.001, vs sh-NC group (B and C). |
GAS2 Promotes Cell Proliferation
MTT assay revealed that Jurkat and CCRF-CEM cells proliferation was increased at 48 hrs (P < 0.05) and 72 hrs (P < 0.01) in the GAS2 group compared with the NC group (Figure 2A). On the contrary, the cell proliferation was decreased at 48 hrs (P < 0.05) and 72 h (P < 0.01) in the sh-GAS2 group compared with the sh-NC group (Figure 2A). Additionally, compared with the NC and sh-NC group, ki67 and PCNA protein expression was higher in the GAS2 group and lower in the sh-GAS2 group (P < 0.05) (Figure 2B). All those results revealed that GAS2 could promote cell proliferation in Jurkat and CCRF-CEM cells.
 |
Figure 2 GAS2 promoted proliferation of Jurkat and CCRF-CEM cells. (A) The proliferation of transfected Jurkat and CCRF-CEM cells was detected by MTT assay. (B) The expression levels of ki67 and PCNA were measured by Western blot in the transfected Jurkat and CCRF-CEM cells. Data were presented as mean ± standard deviation with repeated for three times. *P<0.05, **P<0.01, vs NC group; #P<0.05, ##P<0.01, vs sh-NC group. |
GAS2 Promotes Cell Cycle Changes from G0/G1 Phase to S Phase
As shown in Figure 3, GAS2 overexpression significantly decreased the proportion of G0/G1 phase in Jurkat and CCRF-CEM cells, and notably increased the proportion of S phase (P < 0.01). On the contrary, knockdown of GAS2 significantly increased the proportion of G0/G1 phase, and markedly decreased the proportion of S phase in Jurkat and CCRF-CEM cells (P < 0.01), indicating that GAS2 could promote cell cycle changes from G0/G1 phase to S phase in Jurkat and CCRF-CEM cells.
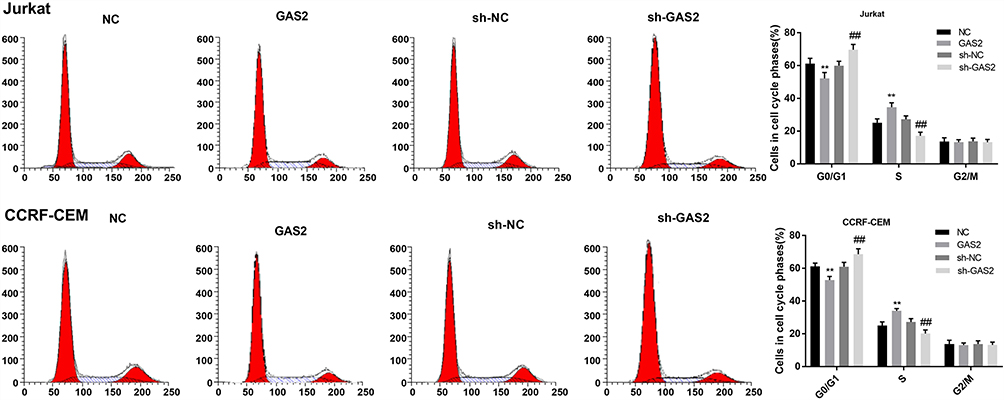 |
Figure 3 GAS2 promoted cell cycle changes from G0/G1 phase to S phase in Jurkat and CCRF-CEM cells. The cell cycle of transfected Jurkat and CCRF-CEM cells was detected by flow cytometry. Data were presented as mean ± standard deviation with repeated for three times. **P<0.01, vs NC group; ##P<0.01, vs sh-NC group. |
GAS2 Inhibits Cell Apoptosis
Cell apoptosis detection showed that the cell apoptosis of Jurkat and CCRF-CEM cells was reduced in the GAS2 group, when compared with the NC group (P < 0.05) (Figure 4A). On the contrary, the cell apoptosis was increased in the sh-GAS2 group compared with the sh-NC group (P < 0.05) (Figure 4A). In addition, the apoptosis-related protein was measured by Western blot (Figure 4B). When compared with the NC group, the expression of Bcl-2 was significantly increased and Bax was decreased in the GAS2 group (P < 0.05). Meanwhile, the opposite expression levels of Bcl-2 and Bax were found in the sh-GAS2 group. The results above revealed that GAS2 could inhibit cell apoptosis in Jurkat and CCRF-CEM cells.
 |
Figure 4 GAS2 inhibited apoptosis of Jurkat and CCRF-CEM cells. (A) The cell apoptosis of transfected Jurkat and CCRF-CEM cells was detected by flow cytometry. (B) The expression levels of Bcl-2 and Bax were measured by Western blot. Data were presented as mean ± standard deviation with repeated for three times. *P<0.05, vs NC group; #P<0.05, vs sh-NC group. |
GAS2 Promotes Cell Invasion in Jurkat and CCRF-CEM Cells
Compared with the NC group, the cell invasion of Jurkat and CCRF-CEM cells was increased in the GAS2 group (P < 0.05) (Figure 5). On the contrary, the cell invasion of Jurkat and CCRF-CEM cells was decreased in the sh-GAS2 group compared with sh-NC group (P < 0.05) (Figure 5), suggesting that GAS2 could promote cell invasion in Jurkat and CCRF-CEM cells.
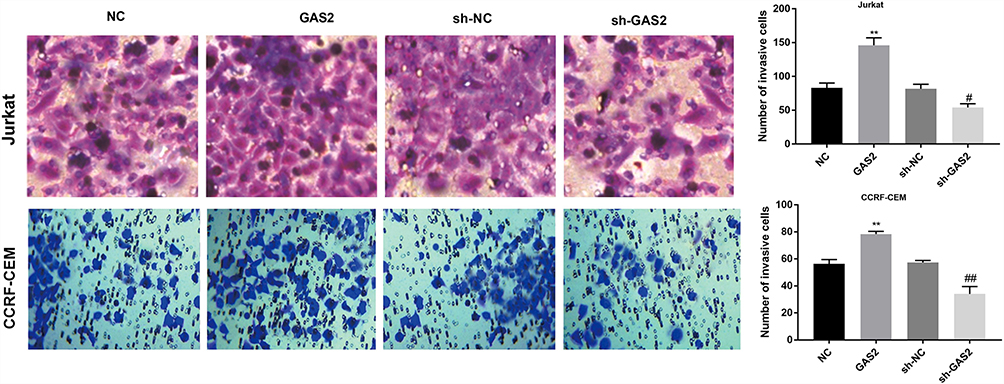 |
Figure 5 GAS2 promoted cell invasion in Jurkat and CCRF-CEM cells. The cell invasion of transfected Jurkat and CCRF-CEM cells was detected by transwell assay. Data were presented as mean ± standard deviation with repeated for three times. **P<0.01, vs NC group; #P<0.05, ##P<0.01, vs sh-NC group. |
GAS2 Reduces the Chemotherapeutic Sensitivity of Jurkat and CCRF-CEM Cells
MTT assays showed that the cell inhibition rate of Jurkat and CCRF-CEM cells in GAS2 +VCR group was lower than that in NC + VCR group (P < 0.05, P < 0.01) (Figure 6A). Meanwhile, the cell inhibition rate in sh-GAS2 + VCR group was increased than that in sh-NC + VCR group (P < 0.05, P < 0.01) (Figure 6A). In addition, as shown in Figure 6B, the cell apoptosis of Jurkat and CCRF-CEM cells was reduced in GAS2 +VCR group compared with the NC + VCR group (P < 0.05), and increased in sh-GAS2 + VCR group compared with sh-NC + VCR group (P < 0.05), indicating that GAS2 reduces the chemotherapeutic sensitivity of Jurkat and CCRF-CEM cells.
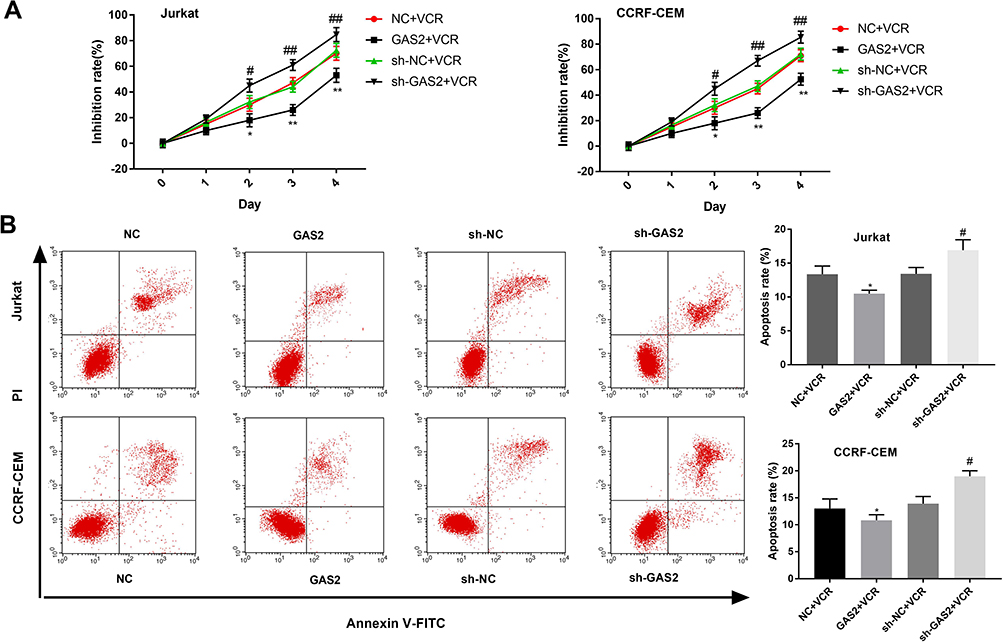 |
Figure 6 GAS2 reduced the chemotherapeutic sensitivity of Jurkat and CCRF-CEM cells. (A) Cell proliferation inhibition rate of Jurkat and CCRF-CEM cells was measured by MTT assay. (B) The cell apoptosis of transfected Jurkat and CCRF-CEM cells was detected by flow cytometry. Data were presented as mean ± standard deviation with repeated for three times. *P<0.05, **P<0.01, vs NC + VCR group; #P<0.05, ##P<0.01, vs sh-NC + VCR group. |
GAS2 Activates Wnt/β-Catenin Pathway
In order to investigate the role of GAS2 on the Wnt/β-catenin pathway, we detected the expression levels of Wnt/β-catenin pathway associated protein using GAS2 overexpression and knockdown. As shown in Figure 7, the expression levels of c-myc, cyclin D1 and β-catenin were increased in GAS2 group and decreased in the sh-GAS2 group compared with the NC group and sh-NC group, respectively (P < 0.01). In addition, to further verify the role of GAS2 on the Wnt/β-catenin pathway, we used the Wnt/β-catenin pathway inhibitor XAV939 and activator LiCl. We found that compared with the GAS2 group, treating with XAV939 could significantly inhibit the expressions of c-myc, cyclin D1 and β-catenin (P < 0.01). Furthermore, LiCl could increase the expressions of c-myc, cyclin D1 and β-catenin compared with sh-GAS2 group (P < 0.01), suggesting that XAV939 and LiCl could eliminate GAS2-induced promotion and GAS2 knockdown-induced inhibition on Wnt/β-catenin pathway. All those results revealed that GAS2 could activate the Wnt/β-catenin pathway in Jurkat and CCRF-CEM cells.
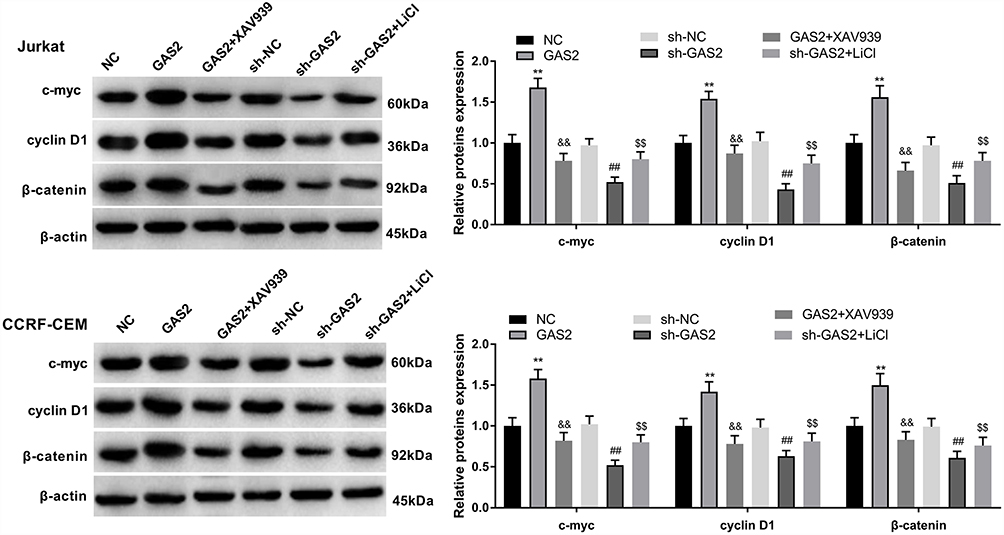 |
Figure 7 GAS2 activated Wnt/β-catenin pathway in Jurkat and CCRF-CEM cells. The expressions of c-myc, cyclin D1 and β-catenin were measured by Western blot in the transfected Jurkat and CCRF-CEM cells. Data were presented as mean ± standard deviation with repeated for three times. **P<0.01, vs NC group; ##P<0.01, vs sh-NC group; &&P<0.01, vs GAS2 group; $$P<0.01, vs sh-GAS2 group. |
Discussion
Although substantial improvements have been made on the treatment of T-ALL, the prognosis of T-ALL patients is still poor due to the drug-resistance and the recurrence and metastasis of disease.20 Therefore, there is an urgent need to find a new molecular approach to identify a novel therapeutic target. Here, we confirmed that GAS2 could promote cell proliferation and invasion, and suppress apoptosis and could activate the Wnt/β-catenin pathway in pediatric T-ALL cells.
GAS2 is a microtubule and microfilament associated protein and the abnormal expression of GAS2 is closely associated with the progress of malignant tumors.21 In addition, GAS2 is upregulated in chronic myeloid leukemia cells12 and leukemic cells.13 In this study, GAS2 expression was upregulated in Jurkat and CCRF-CEM cells relative to normal T lymphocytes, which is consistent with the previous studies. More and more researches have suggested that GAS2 plays a regulatory role in cell biological behaviors including cell cycle, proliferation, apoptosis and differentiation.7,13 Huang et al11 have indicated that GAS2 overexpression is related to cell proliferation, cell cycle and the chemotherapeutic sensitivity in colorectal cancer, and GAS2 contributes to proliferation and inhibits apoptosis of leukemic cells.13 In this study, we demonstrated that GAS2 could promote cell proliferation and invasion, inhibit cell apoptosis and reduce the chemotherapeutic sensitivity of Jurkat and CCRF-CEM cells. Additionally, we further confirmed this result by detecting the expressions of two well-known proliferation markers (PCNA and ki-67 antigen) and the apoptosis-related proteins (Bcl-2 and Bax).
Wnt signaling pathway plays a crucial role in the progress of cell growth and differentiation.22 As the most canonical Wnt pathways, Wnt/β-catenin pathway exerts a crucial regulatory role in multiple tumors, such as hematological malignancies and breast cancer.23,24 More and more researches have indicated that the Wnt/β-catenin pathway is closely related to the hematopoietic cells differentiation and proliferation.25 For example, Zhao et al26 have reported that lncRNA-H19 could promote the proliferation of ALL cells and suppress apoptosis by regulating the Wnt/β-catenin pathway. Xin et al27 have indicated that Dishevelled-Axin domain-containing 1 could promote acute myeloid leukemia cells growth via regulating Wnt/β-catenin pathway. Lyu et al28 suggested that miR-181a-5p could facilitate cell proliferation in ALL by activating Wnt/β-catenin pathway. In addition, cyclin D1 is necessary for the progression of the cell cycle, and its overexpression has been found in ALL cells,29 and c-myc is related to the survival and progression of multiple tumors.30,31 Liu et al32 have also indicated that c-myc and cyclin D1 are generally considered as the downstream targets of the Wnt/β-catenin pathway. In this study, we found that GAS2 overexpression could promote the expression levels of c-myc, cyclin D1 and β-catenin, while GAS2 knockdown could inhibit their expression levels. Moreover, treating with Wnt/β-catenin pathway inhibitor XAV939 or activator LiCl could inhibit or promote the expressions of c-myc, cyclin D1 and β-catenin, suggesting that GAS2 could activate Wnt/β-catenin pathway in Jurkat and CCRF-CEM cells.
Conclusions
In conclusion, our study demonstrated that the expression of GAS2 was upregulated in Jurkat and CCRF-CEM cells. We found that GAS2 could significantly promote cell proliferation and invasion, and induce cell cycle as well as inhibit cell apoptosis, and active Wnt/β-catenin signaling pathway in T-ALL Jurkat and CCRF-CEM cells. Our research provides a novel regulatory mechanism about GAS2 on T-ALL and may provide a reference to the treatment of T-ALL in the future.
Ethics Approval and Consent to Participate
This study was conducted after obtaining Liaocheng People’s Hospital of Shandong Province’s ethical committee approval.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no funding and no conflicts of interest in this work.
References
1. You MJ, Medeiros LJ, Hsi ED. T-lymphoblastic leukemia/lymphoma. Am J Clin Pathol. 2015;144(3):411–422. doi:10.1309/AJCPMF03LVSBLHPJ
2. De Keersmaecker K, Marynen P, Cools J. Genetic insights in the pathogenesis of T-cell acute lymphoblastic leukemia. Haematologica. 2005;90(8):1116–1127.
3. Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med. 2004;350(15):1535–1548.
4. Bleckmann K, Schrappe M. Advances in therapy for Philadelphia-positive acute lymphoblastic leukaemia of childhood and adolescence. Br J Haematol. 2016;172(6):855–869. doi:10.1111/bjh.13896
5. Schneider C, King RM, Philipson L. Genes specifically expressed at growth arrest of mammalian cells. Cell. 1988;54(6):787–793. doi:10.1016/S0092-8674(88)91065-3
6. Brancolini C, Benedetti M, Schneider C. Microfilament reorganization during apoptosis: the role of Gas2, a possible substrate for ICE-like proteases. EMBO J. 1995;14(21):5179–5190. doi:10.1002/embj.1995.14.issue-21
7. Brancolini C, Bottega S, Schneider C. Gas2, a growth arrest-specific protein, is a component of the microfilament network system. J Cell Biol. 1992;117(6):1251–1261. doi:10.1083/jcb.117.6.1251
8. York JP, Ren YA, Zeng J, et al. Growth arrest specific 2 (GAS2) is a critical mediator of germ cell cyst breakdown and folliculogenesis in mice. Sci Rep. 2016;6:34956. doi:10.1038/srep34956
9. Zhang T, Dayanandan B, Rouiller I, Lawrence EJ, Mandato CA. Growth-arrest-specific protein 2 inhibits cell division in Xenopus embryos. PLoS One. 2011;6(9):e24698. doi:10.1371/journal.pone.0024698
10. Zhu R, Mok MT, Kang W, et al. Truncated HBx-dependent silencing of GAS2 promotes hepatocarcinogenesis through deregulation of cell cycle, senescence and p53-mediated apoptosis. J Pathol. 2015;237(1):38–49. doi:10.1002/path.2015.237.issue-1
11. Huang CJ, Lee CL, Yang SH, et al. Upregulation of the growth arrest-specific-2 in recurrent colorectal cancers, and its susceptibility to chemotherapy in a model cell system. Biochim Biophys Acta. 2016;1862(7):1345–1353. doi:10.1016/j.bbadis.2016.04.010
12. Zhou H, Ge Y, Sun L, et al. Growth arrest specific 2 is up-regulated in chronic myeloid leukemia cells and required for their growth. PLoS One. 2014;9(1):e86195. doi:10.1371/journal.pone.0086195
13. Sun L, Zhou H, Liu H, et al. GAS2-Calpain2 axis contributes to the growth of leukemic cells. Acta Biochim Biophys Sin (Shanghai). 2015;47(10):795–804. doi:10.1093/abbs/gmv080
14. Nemeth MJ, Bodine DM. Regulation of hematopoiesis and the hematopoietic stem cell niche by Wnt signaling pathways. Cell Res. 2007;17(9):746–758. doi:10.1038/cr.2007.69
15. Staal FJ, Clevers HC. WNT signalling and haematopoiesis: a WNT-WNT situation. Nat Rev Immunol. 2005;5(1):21–30. doi:10.1038/nri1529
16. Shao N, Zou J, Li J, et al. Hyper-activation of WNT/beta-catenin signaling pathway mediates anti-tumor effects of histone deacetylase inhibitors in acute T lymphoblastic leukemia. Leuk Lymphoma. 2012;53(9):1769–1778. doi:10.3109/10428194.2012.663085
17. Benetti R, Del Sal G, Monte M, Paroni G, Brancolini C, Schneider C. The death substrate Gas2 binds m-calpain and increases susceptibility to p53-dependent apoptosis. EMBO J. 2001;20(11):2702–2714. doi:10.1093/emboj/20.11.2702
18. Benetti R, Copetti T, Dell’Orso S, et al. The calpain system is involved in the constitutive regulation of beta-catenin signaling functions. J Biol Chem. 2005;280(23):22070–22080. doi:10.1074/jbc.M501810200
19. Wang Q, Li Y, Cheng J, et al. Sam68 affects cell proliferation and apoptosis of human adult T-acute lymphoblastic leukemia cells via AKT/mTOR signal pathway. Leuk Res. 2016;46:1–9. doi:10.1016/j.leukres.2016.04.011
20. Graux C, Cools J, Michaux L, Vandenberghe P, Hagemeijer A. Cytogenetics and molecular genetics of T-cell acute lymphoblastic leukemia: from thymocyte to lymphoblast. Leukemia. 2006;20(9):1496–1510. doi:10.1038/sj.leu.2404302
21. Chang CC, Huang CC, Yang SH, Chien CC, Lee CL, Huang CJ. Data on clinical significance of GAS2 in colorectal cancer cells. Data Brief. 2016;8:82–86. doi:10.1016/j.dib.2016.05.010
22. Croce JC, McClay DR. Evolution of the Wnt pathways. Methods Mol Biol. 2008;469:3–18.
23. Ashihara E, Takada T, Maekawa T. Targeting the canonical Wnt/beta-catenin pathway in hematological malignancies. Cancer Sci. 2015;106(6):665–671. doi:10.1111/cas.12655
24. Liu Y, Zhang L, Meng Y, Huang L. Benzyl isothiocyanate inhibits breast cancer cell tumorigenesis via repression of the FoxH1-Mediated Wnt/beta-catenin pathway. Int J Clin Exp Med. 2015;8(10):17601–17611.
25. Fetisov TI, Lesovaya EA, Yakubovskaya MG, Kirsanov KI, Belitsky GA. Alterations in WNT signaling in leukemias. Biochem Biokhimiia. 2018;83(12):1448–1458. doi:10.1134/S0006297918120039
26. Zhao TT, Liu X. LncRNA-H19 inhibits apoptosis of acute myeloid leukemia cells via targeting miR-29a-3p. Eur Rev Med Pharmacol Sci. 2019;23(3 Suppl):224–231.
27. Xin H, Li C, Wang M. DIXDC1 promotes the growth of acute myeloid leukemia cells by upregulating the Wnt/beta-catenin signaling pathway. Biomed Pharmacother. 2018;107:1548–1555. doi:10.1016/j.biopha.2018.08.144
28. Lyu X, Li J, Yun X, et al. miR-181a-5p, an inducer of Wnt-signaling, facilitates cell proliferation in acute lymphoblastic leukemia. Oncol Rep. 2017;37(3):1469–1476. doi:10.3892/or.2017.5425
29. Takahashi-Yanaga F, Sasaguri T. GSK-3beta regulates cyclin D1 expression: a new target for chemotherapy. Cell Signal. 2008;20(4):581–589. doi:10.1016/j.cellsig.2007.10.018
30. Rennoll S, Yochum G. Regulation of MYC gene expression by aberrant Wnt/beta-catenin signaling in colorectal cancer. World J Biol Chem. 2015;6(4):290–300. doi:10.4331/wjbc.v6.i4.290
31. Cairo S, Armengol C, Buendia MA. Activation of Wnt and Myc signaling in hepatoblastoma. Front Biosci (Elite Ed). 2012;4:480–486. doi:10.2741/e393
32. Liu X, Liu S, Chen J, He L, Meng X. Baicalein suppresses the proliferation of acute T-lymphoblastic leukemia Jurkat cells by inhibiting the Wnt/beta-catenin signaling. Ann Hematol. 2016;95(11):1787–1793. doi:10.1007/s00277-016-2766-z
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.