Back to Journals » Drug Design, Development and Therapy » Volume 11
Gancidin W, a potential low-toxicity antimalarial agent isolated from an endophytic Streptomyces SUK10
Authors Zin NM, Baba MS, Zainal-Abidin AH, Latip J, Mazlan NW, Edrada-Ebel R
Received 1 September 2016
Accepted for publication 14 November 2016
Published 8 February 2017 Volume 2017:11 Pages 351—363
DOI https://doi.org/10.2147/DDDT.S121283
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Noraziah Mohamad Zin,1 Mohd Shukri Baba,2 Abu Hassan Zainal-Abidin,3 Jalifah Latip,4 Noor Wini Mazlan,5 RuAngelie Edrada-Ebel6
1Programme of Biomedical Science, School of Diagnostic and Applied Health Sciences, Faculty of Health Sciences, Universiti Kebangsaan Malaysia, Kuala Lumpur, 2Department of Biomedical Sciences, Kulliyyah of Allied Health Sciences, International Islamic University Malaysia, Kuantan, 3Department of Parasitology, Faculty of Medicine, Universiti Teknologi MARA, Shah Alam, 4School of Chemical Sciences and Food Technology, Faculty of Science and Technology, Universiti Kebangsaan Malaysia, Bangi, 5Analytical and Environmental Chemistry, School of Marine and Environmental Sciences, Universiti Malaysia Terengganu, Kuala Terengganu, Malaysia; 6Strathclyde Institute of Pharmacy and Biomedical Sciences, University of Strathclyde, Glasgow, UK
Abstract: Endophytic Streptomyces strains are potential sources for novel bioactive molecules. In this study, the diketopiperazine gancidin W (GW) was isolated from the endophytic actinobacterial genus Streptomyces, SUK10, obtained from the bark of Shorea ovalis tree, and it was tested in vivo against Plasmodium berghei PZZ1/100. GW exhibited an inhibition rate of nearly 80% at 6.25 and 3.125 µg kg-1 body weight on day four using the 4-day suppression test method on male ICR strain mice. Comparing GW at both concentrations with quinine hydrochloride and normal saline as positive and negative controls, respectively, 50% of the mice treated with 3.125 µg kg-1 body weight managed to survive for more than 11 months after infection, which almost reached the life span of normal mice. Biochemical tests of selected enzymes and proteins in blood samples of mice treated with GW were also within normal levels; in addition, no abnormalities or injuries were found on internal vital organs. These findings indicated that this isolated bioactive compound from Streptomyces SUK10 exhibits very low toxicity and is a good candidate for potential use as an antimalarial agent in an animal model.
Keywords: antimalarial, Shorea ovalis, in vivo, endophytic, Streptomyces, gancidin W
Introduction
Malaria is a leading human parasitic disease that increasingly threatens much of the world’s population. The emergence of resistance by malarial parasites to currently commercially available antimalarial drugs is one of the major factors that lead to infection of more than 3 billion people living in high-incidence areas, particularly children and the elderly.1,2 Chloroquine resistance is most common in tropical and subtropical regions, particularly in Southeast Asia,3,4 and there has also been clinical and laboratory evidence of resistance to sulfadoxine, pyrimethamine, artemisinin, primaquine, mefloquine and quinine.3,5 Plasmodium falciparum, the most dominant causative agent of malaria, has high adaptability, brought about through many types of mutations, and has become resistant to nearly all antimalarial drugs.3 Therefore, new families of bioactive compounds are needed in order to combat these problems and reduce the risk of resistance.
Malaysian tropical forests have not been fully scientifically explored even though they are among the oldest on the planet and as many as 1,300 plants are claimed to have medicinal properties.6 Previous findings have shown that plants used by the Malaysian native population as alternative medicines are actually produced by endophytic microorganisms and may have the potential to treat malaria and many other parasitic diseases.7,8 Some of these antibiotic derivatives include alkaloid congeners as potential anticancer and antimicrobial agents;9 nonadecene compounds as antifungals10 and antioxidants;11 as well as biologically active diketopiperazine (DKP) derivatives.12,13 DKP derivatives have demonstrated a variety of good biological activities, such as antibacterial, antiviral, antitumor, fungicidal and many more.14,15
Streptomyces, a well-studied genus of gram-positive actinobacteria, shows outstanding chemical and morphological diversity with a distinct evolutionary line.15 As these filamentous bacteria have a similar physiology as fungi,16 most Streptomyces spp. have the ability to synthesize various secondary metabolites that have numerous medical applications, as antibiotic, herbicidal, antiparasitic, antitumor, antifungal and enzyme inhibiting agents.17 Producing more than two-thirds of clinically useful antibiotics of natural origin,18 endophytic Streptomyces spp. have the ability to inhibit a variety of human pathogens, including bacteria and fungi.9,19–22
In this work, the DKP gancidin W (GW)23 was isolated from the crude extract of an endophytic Streptomyces strain designated as SUK10, which was obtained from the bark of the Shorea ovalis tree. Screening of GW for antimalarial activity against Plasmodium berghei PZZ1/100 strain was carried out in order to assess the optimum in vivo antimalarial concentration of GW. Assessment of toxicity in terms of activity of enzymes in mice blood and total protein determination, as well as histopathology evaluation of the selected vital organs treated with GW, was also carried out.
Materials and methods
Production of crude extract
Nutrient broth (NB) (Sigma-Aldrich, Kuala Lumpur, Malaysia) was prepared at pH 7.0. To produce sufficient amount of isolates for extraction, five blocks of 5×5 mm International Streptomyces Project 2 agar enriched with Streptomyces SUK10 were cut and inoculated into 400 mL of autoclaved NB at pH 7.0 in a 1,000 mL conical flask. Fermentation was carried out using an orbital shaker at 28°C at rotation rate of 200 rpm for 21 days. By modifying a previous method,22 the supernatant was then extracted three times using ethyl acetate to obtain 5.2 g of Streptomyces SUK10 crude extract.
Isolation and purification of bioactive compound
Approximately 2.8 g of SUK10 crude extract was fractionated using flash chromatography (Reveleris®; Grace & Co, London, UK). The extract was chromatographed over 40 g of generic silica and eluted with hexane and ethyl acetate as the mobile phases A and B, respectively. The peaks were detected under UV light at absorption wavelengths of 245 and 310 nm coupled to an electron light scattering detector (ELSD). A gradient elution was done at 35 mL min−1, commencing with 100% hexane:100% ethyl acetate for 80.0 minutes, yielding 22 fractions (V1–V22), which were collected according to peak response. The purity of the fractions was further validated by normal phase thin-layer chromatography. Fractions 1–7 were monitored using 80:20 (v/v) of hexane/ethyl acetate as eluent, fractions 8–13 with 60:40 (v/v) hexane/ethyl acetate and fractions 14–22 with 98:2 (v/v) dichloromethane/methanol.
Identification of bioactive compound
All fractions were subjected to nuclear magnetic resonance (NMR) spectroscopy in deuterated chloroform (CDCl3) for one-dimensional (1D) and 2D 1H-NMR analyses (400 MHz). Correlation spectroscopy (COSY) (12 scans), heteronuclear multiple-quantum coherence (HMQC) (24 scans) and heteronuclear multiple-bond coherence (HMBC) (32 scans) were also further implemented for the more purified fractions 11, 15 and 20. Fraction 15 was also subjected to 13C-NMR and distortionless enhancement by polarization transfer (DEPT) studies (both 4,000 scans). In addition to the NMR studies, liquid chromatography/high-resolution mass spectrometry (LC/HRMS) analysis (Accela autosampler; Grace & Co.) was also carried out, and the obtained spectral data were then compared with AntiBase database.
In vivo antimalarial screening of bioactive compound
ICR strain male mice (n=120, 25–30 g, 6–8 weeks old) were used in all animal experiments and were divided into 20 groups (n=6). All groups were housed in stainless steel cages under 12:12 hours with and without light conditions at 28°C with daily ad libitum feed. To initiate the infection, 0.1 mL of 1.0×106 P. berghei PZZ1/100 parasitized red blood cell (RBC) solution was intraperitoneously administered into the host. To determine the best concentration, 50, 25, 12.5, 6.25 and 3.125 μg kg−1 bw of GW solutions were prepared by dissolving the obtained fractions of GW in 1.0 mL dimethyl sulfoxide (DMSO) as the stock compound solution before they were serially diluted with sterile distilled water to achieve the targeted concentrations. The 4-day suppression test4 was chosen to implement the antimalarial screening, whereby day zero of the treatment was the day when the mice were treated with 0.1 mL of GW at all concentrations, immediately within 2 hours postinfection. In parallel, 0.1 mL of 10 mg kg−1 bw of dH2O-diluted quinine hydrochloride (QH) and 0.9% normal saline were respectively used as positive control (PC) and negative control (NC) solutions.
Similarly, successively, day one, day two and day three were referred when the same treatment was repeated for the next 3 days respectively without any infection. On day four, the parasitemia density (%) and inhibition rate (%) were calculated by a Giemsa blood smear from the treated and control mice using the following expressions:
|

and
|
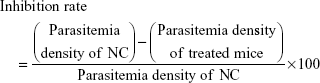
After 20 days postinfection, the mice were observed daily for their survival period (days). Treatment regime with inhibition rate of >65% was considered as having antimalarial activity, and the mice group with the longest survival time was considered as receiving the best treatment.
In vivo toxicity assessment of the compound
Mice with the same characteristics as during antimalarial screening (ICR strain, male, 25–30 g, 6–8 weeks old, n=36) were used for in vivo toxicity assessment. All animal experiments for toxicity assessment were conducted under the same Universiti Kebangsaan Malaysia Animal Ethics Committee (UKMAEC) approval code for antimalarial screening. The mice were divided into 12 groups (n=6), and all groups were subjected to the same conditions (28°C room temperature, stainless steel cage, 12:12 hours with and without light and daily ad libitum feed). At the best-detected concentration during antimalarial screening, toxicity tests were carried out on blood samples of mice treated with GW according to two types of toxicity regime: acute exposure (daily treatment for 7 days) and subacute exposure (daily treatment for 28 days). Each toxicity regime was divided into two groups of treatment: without infection and immediately within 2 hours after infection on day zero. For labeling purposes, all the mice groups were respectively labeled as TA for acute exposure without infection, TB (acute exposure immediately within 2 hours after infection), TC (subacute exposure without infection) and TD (subacute exposure immediately within 2 hours after infection). Data from the two control regimens, namely, normal mice without any infection and treatment (CN) and single-dose infected mice (CI), were also obtained for comparison. The animals were sacrificed under diethyl ether anesthesia, 0.8–1.0 mL of the blood was collected from each mouse by cardiac puncture on day eight and day 29 postexposure and tested for serum total protein (STP), alanine aminotransferase (ALT), aspartate aminotransferase (AST) and alkaline phosphatase (ALP) levels; their normal ranges (NRs) were compared with the data supplied by Research Animal Resources, University of Minnesota, MN, USA.
From the same diethyl ether-anesthetized mice used for biochemical toxicity tests, three vital organs, namely, liver, kidney and spleen, were individually collected for organ toxicity and histology studies. The infiltration procedure was done automatically by a tissue processor (Leica EG1169, Leica Biosystems [SEA] Pte. Ltd., Singapore). The tissues were next embedded in hot paraffin wax using an embedding machine (Shandon HistoCentre 2, GMI-Advance, Ramsey, MN, USA). These tissues were then cut (0.4–0.6 μm thick) by a microtome (Leica, RM2235). Histologic tissue preparation was done using hematoxylin-and-eosin (H–E) staining method. The H–E-stained slides for organ histology were observed using a computerized light microscopic camera (Zeiss Primo Star, New York Microscope Co., New York, NY, USA, and Dell, Dell Global Business Sdn. Bhd., Selangor, Malaysia) at 100× magnification by placing a drop of immersion oil on the surface of the slide or without the immersion oil at 40× magnification; this method was vital to give a clear picture of the internal structure of the targeted tissues, as well as for assessing and identifying abnormalities, toxicity and injuries in the tissues.
Ethics statement
The animal protocols used in this study were evaluated and approved by the UKMAEC of Universiti Kebangsaan Malaysia (UKM) under the given Animal Use Protocol code FSK/Biomed/341 and were in accordance with the UKM Laboratory Animal Care and Management Guidelines.
Statistical analysis
All results were expressed as the mean ± standard deviation for every six mice per group (n=6). The Shapiro–Wilk test was used to determine all of the antimalarial screening and toxicity test parameters. Statistical significance was declared when the P-value was equal to or less than 0.05 (P≤0.05).
Results and discussion
After culture optimization, the Streptomyces SUK10 isolate produced 10–12 mg of crude dried extract after 21 days of fermentation in NB at pH 7.0 and an agitation rate of 200 rpm. As shown in most previous studies,8,20,24–28 biomass production was greater at higher agitation speeds. Higher rotating speeds during the fermentation process gave better aeration and oxygen could be efficiently supplied to the organisms for their growth and metabolism.21,25,27 The fermentation process was scaled up to obtain a crude extract with a sufficient yield of 5.2 g. The crude extract was fractionated, and all 22 fractions were subjected to 1H-NMR experiments to determine purity and chemical structure. Based on the 1H-NMR analyses, fraction V15 was found to contain interesting signals that corresponded to the major constituent. V15 at 98% purity was isolated at 16.2 mg (0.31% yield) as a light brownish solid material with a molecular weight of 210.275 g/mol and molecular formula of C11H18N2O2, established by LC-high-resolution electrospray ionization mass spectrometry at a retention time of 7.17 minutes. Literature search revealed that the 1H- and 13C-NMR data for V15 (Table 1) were in accordance to the cyclodipeptide structure for S-prolyl-R-leucyl-diketopiperazine (Figure 1), previously isolated from the sponges Calyx cf. podatypa29 and Callyspongia.30 A similar compound, named as gancidin W (GW) and elucidated as cyclo-(L-leu-L-pro) but with unknown absolute stereochemistry, was earlier isolated from strains of Streptomyces gancidus.31,32 In this study, the obtained natural product gave an optical rotation of [a]D =−138.2° (c=1.0, EtOH), which was compatible with those isolated earlier from other Streptomyces strains.31–34 Therefore, in this study, the nomenclature GW was assigned for the isolated compound V15.
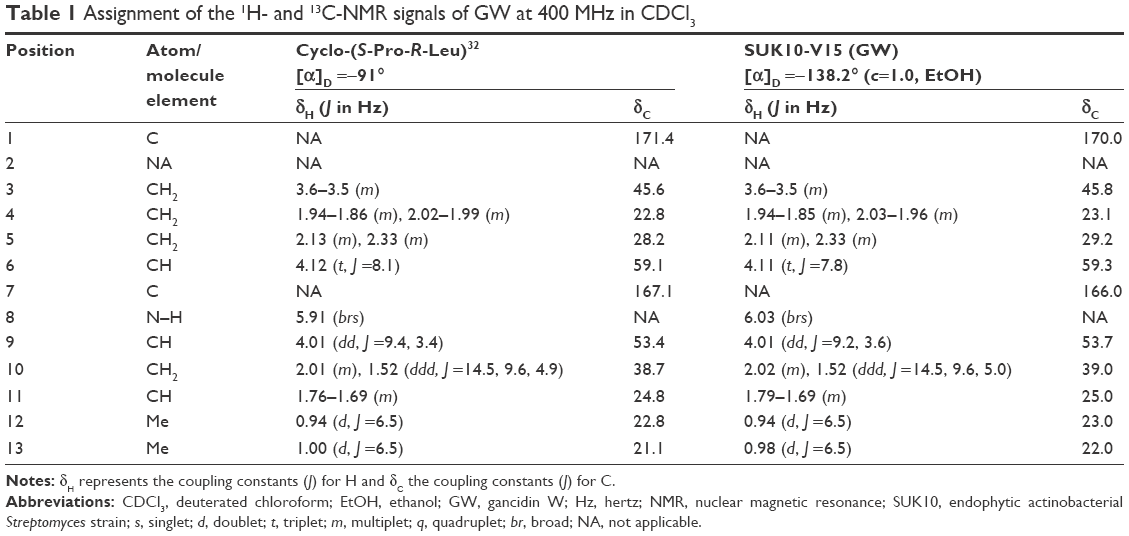 | Table 1 Assignment of the 1H- and 13C-NMR signals of GW at 400 MHz in CDCl3 |
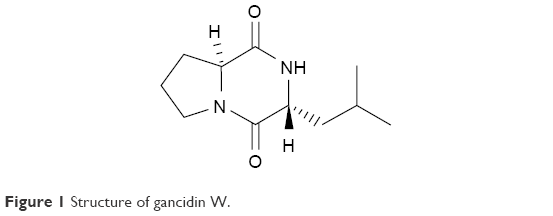 | Figure 1 Structure of gancidin W. |
With less than 2.0% of parasitemia density, there was a significant difference in this parameter (P<0.05, n=6) between 3.125 μg kg−1 bw and the other four concentrations of GW. Nevertheless, in animals given a dose of 6.25 μg kg−1 bw, 1.43% of parasitemia density was observed, which was still far lower (Table 2) than that in animals given 3.125 μg kg−1 bw concentration. Figure 2 shows the microscopic blood smear from mice treated with 0.1 mL of GW at five different concentrations. At 0.1 mL of 10 mg kg−1 bw of QH administered to the PC group, no infected RBCs were found in any of the mice on day four postinfection regardless of the tested concentrations. Previous studies4,33,35,36 demonstrated that no physical side effects were observed in the host treated with 10% DMSO. Therefore, it is hypothesized that there is no crucial requirement to have 10% DMSO as one of our control treatments.
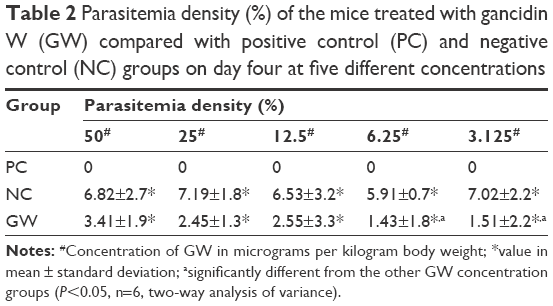 | Table 2 Parasitemia density (%) of the mice treated with gancidin W (GW) compared with positive control (PC) and negative control (NC) groups on day four at five different concentrations |
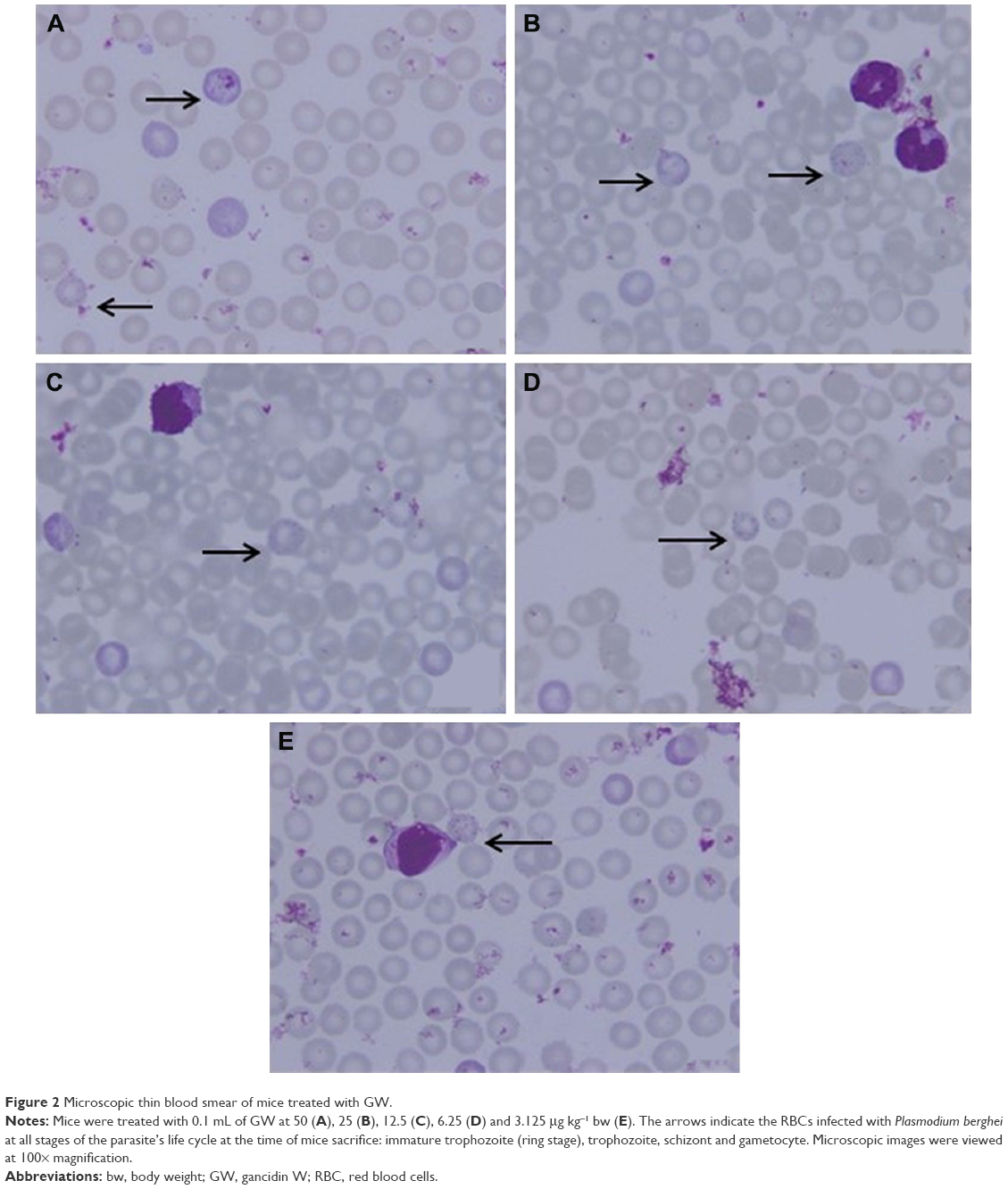 | Figure 2 Microscopic thin blood smear of mice treated with GW. |
P. berghei PZZ1/100 strain, which is the most established and reliable rodent malaria parasite, was used as the infectious agent in this study. This strain was a quinine-sensitive and chloroquine-resistant strain,37–39 making QH application as a control drug in this study valid and significant. Moreover, P. berghei is a rodent malaria parasite that was used to test the antimalarial property of the bioactive compound before this compound can be introduced in clinical trials.40 Stepniewska et al37 documented that in terms of molecular structure, there was a similarity between the monomeric structure of lactate dehydrogenase from all strains of P. berghei (PbLDH) and P. falciparum lactate dehydrogenase (PfLDH). These two forms of the enzyme are important in glycolysis because of the generation of ATP as an energy source, and they are persistent in the erythrocytes of the host.41–43 This fact proves that the mouse model is valid to test in vivo antimalarial activity using P. berghei as it was also comparable with results from infection with P. falciparum in a human study.
A group of scientists discussed the relative merits of a range of animal models and their overlap with the complex clinical syndromes of human malaria.44,45 These researchers agreed that malaria research using male rodents was acceptable and understandable and that rodents were 95% systemic pathologically, endocrinologically and immunologically reflective of experimental human P. falciparum malaria. This was based on the results from a study showing favorable exploitation by the female hormonal system when an infectious agent was administered.43 The interferences from any female hormones in microbiological and pathological studies should be considered a bias because the results obtained by research would be difficult to analyze. Among the research efforts was a study on the new antibiotic azithromycin, which has been shown to be effective in male mice and monkeys and has undergone a successful trial to prevent malaria in humans.43
GW was subjected to antimalarial screening across a narrow range of five different concentrations in order to determine which dose gave the best antimalarial activity. The value for parasitemia density directly reflected the value of percentage inhibition, where the higher the percentage inhibition – the more effective the treatment was.4 The inhibition rate at 3.125 μg kg−1 bw dose was found to exhibit the greatest value. Reaching almost 80% inhibition rate, there was a significant difference (P<0.05, n=6) between the 3.125 μg kg−1 bw group and the groups given the other four concentrations. Inhibition rate higher than 65%, which was considered a benchmark for in vivo antimalarial activity, was also reached at 6.25 μg kg−1 bw dose, whereby this value was far better than the other three dose concentrations (Table 3).
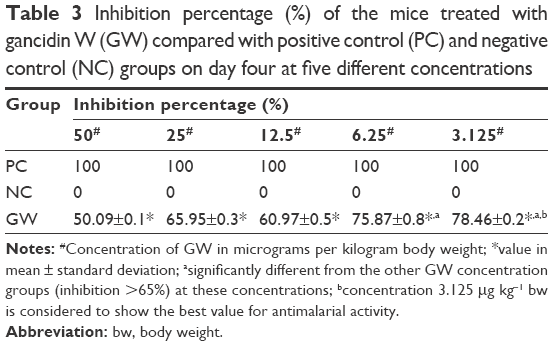 | Table 3 Inhibition percentage (%) of the mice treated with gancidin W (GW) compared with positive control (PC) and negative control (NC) groups on day four at five different concentrations |
The survival time of mice treated with 3.125 μg kg−1 bw dose of GW was steadily almost twofold longer than that of mice given 12.5 μg kg−1 bw and was proportionately higher than that for mice given a dose of 6.25 μg kg−1 bw (Figure 3). As previously demonstrated, both in vivo and in vitro analyses4,14,46–48 predicted that the survival times of the treated mice would be longer at a higher parasite inhibition rate. The longest period of mouse survival (235.53± 2.20 days) was achieved by treatment with GW at a dose concentration of 3.125 μg kg−1 bw, which was significantly higher than that with other dose concentrations (P<0.05, n=6). In conjunction with this, surprisingly, the remaining 50% (n=3) of the group treated with GW at this concentration individually managed to survive until 291.13±0.5 days postinfection. Based on previous documentations,49–53 this period of survival time was considered to almost reach the life span of normal male mice at approximately 12–18 months. Until 411 postinfection days, all PC mice survived and this observation was in parallel with results of previous in vivo antimalarial studies,4,14,47,48,52 while the NC mice survived to 7–9 days postinfection, as also recorded in this study.
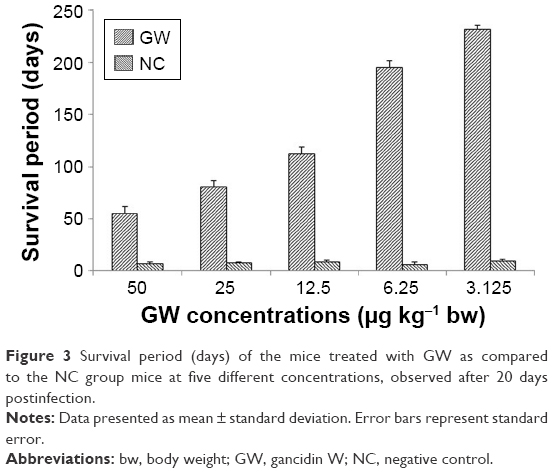 | Figure 3 Survival period (days) of the mice treated with GW as compared to the NC group mice at five different concentrations, observed after 20 days postinfection. |
There was no comparable benchmark found in the literature for the animals’ survival period in in vivo studies when test groups partially had their respective control groups.53 Therefore, it was not surprising that the survival time at 12.5 μg kg−1 bw was longer than that at 25 μg kg−1 bw, although the inhibition rate at 25 μg kg−1 bw was slightly higher. As recommended by the World Health Organization (WHO), for every single dose of an antimalarial drug, regardless of the type of drug and malaria etiological agent, the daily therapeutic dosage in treating malaria was 5–25 mg kg−1 bw for the first month of medication.54,55 This dosage was significantly higher than the best concentration recorded in this study.
Except for the mice in the TD regime (those immediately subjected to subacute exposure within 2 hours after the 28-day postinfection period), ALT and AST levels slightly increased. At the treatment dose of 3.125 μg kg−1 bw, all tested enzymes and total protein levels were in their NRs, regardless of the type of exposure regime (Table 4). The biochemical toxicity test results in this study revealed that all of the enzymes and STPs levels for all the regime groups were within the NR. However, there was a slightly higher average recorded for ALT and AST levels in the mice group that was treated daily with 3.125 μg kg−1 bw of GW for 28 days. However, it is too early to conclude that the higher levels of ALT and AST in this study for this group was due to damaged liver or oversecretion of these enzymes into the blood stream. The Department of Research Animal Resources, University of Minnesota (MN, USA), circulated the guidelines that the values of ALT, AST, ALP and STP can vary and be beyond the NR depending on the age, bw, gender and the animal model.56 In the mice group receiving a daily dose of 3.125 μg kg−1 bw of GW for 4 weeks, the values of ALT and AST exceeded the maximum normal values at 3 and 5 IU L−1, respectively. These values were already considered significant because blood was sampled after 28 days posttreatment, at which time point, the mice were 4 weeks older from day 0 of their treatment regime. Most enzymes will be oversecreted as the animals mature in age.45 Therefore, the normal levels of these enzymes were basically set up according to the animal’s age together with other factors mentioned herein. Another factor to consider in this study was the animals’ experimental environment.57,58 Animals (n=6) of the same gender were housed together in a transparent walled box (45×25×25 cm). The animals were situated in a stress condition, which induced oversecretion and elevation of ALT and AST levels from their NRs. In addition, the mice in this group received the treatment daily by the intraperitoneal route for 28 days. In an earlier study,57 under similar stress conditions, the AST and ALT levels significantly reached a twofold increase from the NR.
 | Table 4 ALT, AST and ALP levels (IU L−1), as well as the STP level (g dL−1), in the mice treated with gancidin W (GW) at 3.125 μg kg−1 bw under four toxicity regimens |
The selected tissues from all mice in the groups were stained with H–E. None of the tissues showed any abnormality, including the organs taken from the mice in the TD regime group (treated daily with 3.125 μg kg−1 bw of GW for 28 days); prior to this, their recorded ALT and AST levels were slightly higher than normal. Figures 4–6 show the microscopic H–E histology slides for the toxicity assessment of the kidney, spleen and liver tissues, respectively, on selected mice treated with 3.125 μg kg−1 bw of GW from each of the regime groups. Histologically, there was no morphological difference between the control and the treatment groups, regardless of whether they were administered acute or subacute regimens. At the same time, no physical injury or swelling, morphological abnormalities or color changes of the targeted organs were spotted during the isolation process from the sacrificed mice.
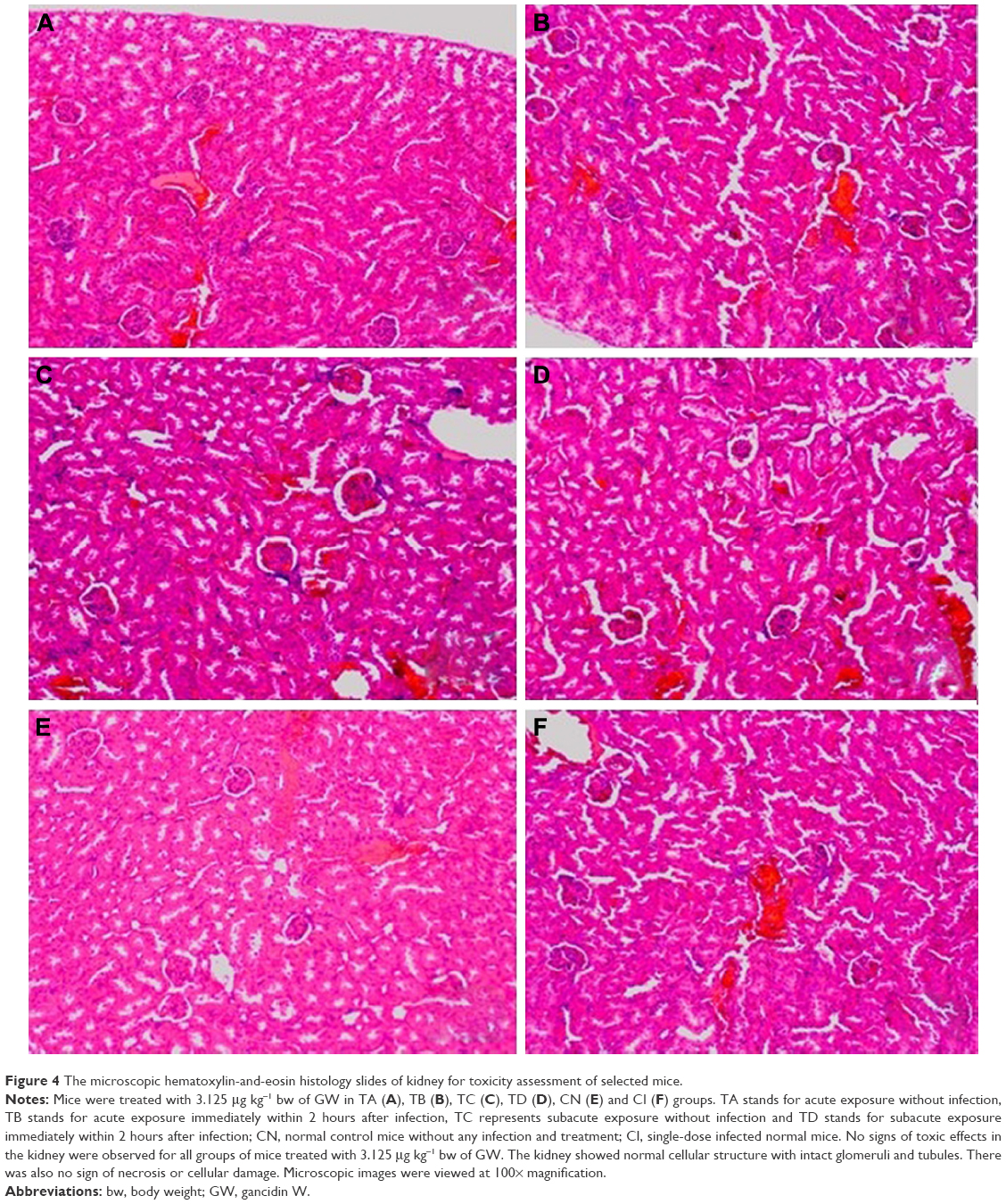 | Figure 4 The microscopic hematoxylin-and-eosin histology slides of kidney for toxicity assessment of selected mice. |
 | Figure 5 The microscopic hematoxylin-and-eosin histology slides of spleen for toxicity assessment of selected mice. |
 | Figure 6 The microscopic hematoxylin-and-eosin histology slides of liver for toxicity assessment of selected mice. |
In this study, the histopathology assessment was observed for three vital organs of the mice treated with 3.125 μg kg−1 bw of GW. Neither in the acute (7 days) nor in the subacute (28 days) treatment regime, or in the control treatment groups, were activated kupffer cells, cytoplasmic vacuolation and sinusoidal dilatation observed by H–E staining of liver, which could basically indicate their association with the presence of abnormal morphology or toxicity characteristics.59 There was no evidence of hepatic injuries in any of the treatment groups as they exhibited defined histological structures in their liver tissue without any signs of cytoplasmic condensation, mild vascular inflammatory changes, congested nuclear changes and necrosis.60 Similarly, no presence of granular and cellular cast was noticed in the kidney tissue of all treated and control mice groups regardless of their regime. There was also a normal histological structure of the kidney glomerulus and tubules, with no glomerular atrophy and tubular necrosis, as well as no vascular shrinkage being observed in the renal tubules at the cortical zone, which could directly indicate a degenerative phenomenon.61,62
The histopathology assessment in the sectional spleen tissue in all treatment regimens did not reveal any evidence of vascular changes and periarterial hyperplasia of the red pulp and white pulp or malpighian follicle nodules. Positively, there was also no proliferation of the mononuclear cells around the sinusoid cord structure, which generally could have activated the accumulation of organic compounds in the blood stream.63,64 No shrinkage or deformed size and shape of splenic cord that could indicate any spleen degenerative phenomenon was evidenced.52 Onkar and Govardhan65 and Rungruang et al66 claimed that in the same sacrificed mice, any abnormalities spotted and visualized on the four vital organs, namely, liver, spleen, lungs and kidney, were also histologically recorded by the H–E staining method. In this study, concurrently, the selected organs of the mice were extracted for histopathology assessment after their blood samples were taken for biochemical tests. Because there were no ultimate evidences of blood toxicity in all the mice groups, in parallel, similar observations were also expected from the histopathology evidences of these selected organ tissues of all mice groups. These findings showed that GW at 3.125 μg kg−1 bw left no undesired effects on the treated host.
Conclusion
Administration of antimalarial drugs totally depends on the regime combination, stage of infection, the side effects experienced by the infected host and the type of plasmodial species.47,52,67,68 Quinine has been proven to kill all Plasmodium cells, regardless of the species. However, it is only effective in the early stages of malarial infection, during which generally, the host will experience asymptomatic signs.69,70 All the untreated control mice in this study died between 7 and 9 days postinfection, as described in previous in vivo antimalarial studies,4,18,51,60,71 regardless of the type of animal model used.
Although GW has been previously reported from other Streptomyces species,31,32,34 its antimalarial properties have not yet been revealed. In fact, our results indicate that GW is one of the metabolites responsible for the in vivo antimalarial activity of the ethyl acetate crude extract from SUK10. The outcomes of the presented study indicate that Streptomyces SUK10 living endophytically in S. ovalis tree is a good source of a potential antimalarial agent with relatively very low toxicity. Apart from the fact that the progression of the erythrocytic cycle in the Plasmodium-infected host is still poorly understood,36 there is also a tendency to obtain more significant values for certain antimalarial parameters in particular cycles or stages of the Plasmodium life cycle,43,44,46,54 which have not been considered in this study.
Acknowledgments
The authors appreciate the financial assistance provided by the Ministry of Higher Education, Malaysia, through the grant UKM-NN03-FRGS 0042-2009, and the Universiti Kebangsaan Malaysia, for the grant UKM_GUP-TKP-08-22-074. We would also like to thank and express our appreciation to the International Islamic University, Malaysia (http://www.iium.edu.my), and the Tunku Naquiyuddin Foundation, Malaysia (http://ewarga4.ukm.my/ewarga/pdf/082011/19-83.pdf), for their financial support and for making this study possible, as well as expressing a special appreciation to the laboratory management and staff of Strathclyde Institute of Pharmacy and Biomedical Sciences (SIPBS), University of Strathclyde, Glasgow, UK, for their technical support and permissions for us to use their laboratory instruments and materials.
Disclosure
The authors report no conflicts of interest in this work.
References
Joy D, Feng X, Mu J, et al. Early origin and recent expansion of Plasmodium falciparum. Science. 2003;300(5617):318–321. | ||
Yotoko KSC, Elisei C. Malaria parasites (Apicomplexa, Haematozoea) and their relationships with their hosts: is there an evolutionary cost for the specialization. J Zool Syst Evol Res. 2006;44(4):265–273. | ||
Wellems TE. Plasmodium chloroquine resistance and the search for a replacement antimalarial drug. Science. 2002;298(5591):124–126. | ||
Abdulelah HA, Zainal-Abidin BAH. In vivo anti-malarial test of Nigella sativa (black seed) different extracts. Am J Pharmacol Toxicol. 2007;2(2):46–50. | ||
Alecrim MGC, Alecrim W, Macedo V. Plasmodium vivax resistance to chloroquine (R2) and mefloquine (R3) in Brazilian Amazon region. Rev Soc Bras Med Trop. 1999;32(1):67–68. | ||
Ibrahim J, Mat-Ali R, Goh SH. Toxic and antifungal properties of the essential oils of Cinnamomum species from Peninsular Malaysia. J Trop For Sci. 1994;6(3):286–292. | ||
Strobel G, Daisy B. Bioprospecting for microbial endophytes and their natural products microbial. Microbiol Mol Biol Rev. 2003;67(4):491–502. | ||
Zin NM, Ng KT, Sarmin NM, et al. Anti-trypanosoma activity of endophytic Streptomycete. Curr Res Bacteriol. 2011;4:1–8. | ||
Smaoui S, Mathieu F, Elleuch L, et al. Taxonomy, purification and chemical characterization of four bioactive compounds from new Streptomyces sp. TN256 strain. World J Microbiol Biotechnol. 2012;28(3):793–804. | ||
El-Sakhawy FS, El-Tantawy ME, Ross SA, et al. Composition and antimicrobial activity of the essential oil of Murraya exotica. Flavour Fragrance J. 1998;13(1):59–62. | ||
Yassa N, Masoomi F, Rohani-Rankouhi SE, et al. Chemical composition and antioxidant activity of the extract and essential oil of Rosa damascene from Iran, population of Guilan. DARU J Pharm Sci. 2009;17(3):175–180. | ||
Ben-Ameur-Mehdi R, Mellouli L, Chabchoub F, et al. Purification and structure elucidation of two biologically active molecules from a new isolated Streptomyces sp. US24 strain. Chem Nat Compd. 2004;40(5):510–513. | ||
Ben-Ameur-Mehdi R, Sioud S, Fourati-Ben-Fguira L, et al. Purification and structure determination of four bioactive molecules from a newly isolated Streptomyces sp. TN97 strain. Process Biochem. 2006;41(7):1506–1513. | ||
Magyar A, Zhang X, Abdi F, Kohn H, Widger WR. Identifying the bicyclomycin binding domain through biochemical analysis of antibiotic-resistant Rho proteins. J Biol Chem. 1999;274(11):7316–7324. | ||
Vining LC. Secondary metabolism, inventive evolution and biochemical diversity: a review. Gene. 1992;115(2):135–140. | ||
Hopwood DA. Streptomyces in Nature and Medicine: The Antibiotic Markers. Vol. 62. 1st ed. New York City, NY: Oxford University Press; 2007:435–467. | ||
Olano C, Mendaz C, Salar JA, et al. Antitumor compounds from marine Streptomyces. Mar Drugs. 2009;7(2):210–248. | ||
Kurosawa Y, Dorn A, Kitsuji-Shirane M, et al. Hematin polymerization assay as a high-throughput screen for identification of new antimalarial pharmacophores. Antimicrob Agents Chemother. 2000;44(10):2638–2644. | ||
Rajendra PM, Elisabeth H, Oliver K, et al. Anti-cancer and antibacterial trioxacarcins with high anti-malarial activity from a marine Streptomycete and their absolute stereochemistry. J Antibiot (Tokyo). 2004;57(12):771–779. | ||
Ghadin N, Zin NM, Sabaratnam V, et al. Isolation and characterization of a novel endophytic Streptomyces SUK 06 with antimicrobial activity from Malaysian plants. Asian J Plant Sci. 2008;7(2):189–194. | ||
Zin NM, Loi CS, Sarmin NM, et al. Cultivation-dependent characterization of endophytic actinomycetes. Res Microbiol. 2010;5(8):717–724. | ||
Castillo UF, Strobel GA, Ford EJ, et al. Munumbicins, wide spectrum antibiotics produced by Streptomyces (NRRL 30562) endophytic on Kennedia nigriscans. Microbiology. 2002;148(9):2675–2685. | ||
Mohd-Shukri BB, Zainal-Abidin BAH, Latip J, et al. In vivo antimalarial activity of the endophytic actinobacterium, Streptomyces SUK10. J Microbiol. 2015;53(12):847–855. | ||
Foley M, Tilley L. Quinoline antimalarials: mechanisms of action and resistance and prospects for new agents. Pharmacol Ther. 1998;79(1):55–87. | ||
Castillo UF, Harper JK, Strobel GA, et al. Novel antibiotics from Streptomyces sp. NRRL 30566, an endophyte on Grevillea pteridifolia. Microbiology. 2003;244(2):183–190. | ||
Conn VM, Franco MM. Analysis of the endophytic actinobacterial population in the roots of wheat (Triticum aestivum L.) by terminal restriction fragment length polymorphism and sequencing of 16S rRNA clones. Appl Environ Microbiol. 2004;70(3):1787–1794. | ||
Coombs JT, Franco MM. Isolation and identification of actinobacteria from surface-sterilized wheat roots. Appl Environ Microbiol. 2003;69(9):5603–5608. | ||
Zin NM, Sarmin MN, Ghadin N, et al. Bioactive endophytic Streptomycetes from the Malay Peninsula. FEMS Microbiol Lett. 2007;274(1):83–88. | ||
Adamczeski M, Reed AR, Crews P. New and known diketopiperazines from the Caribbean sponge, Calyx cf. podatypa. J Nat Prod. 1995;58(2):201–208. | ||
Bin Y, Junde D, Xuefeng Z, et al. Proline-containing dipeptides from a marine sponge of a Callyspongia species. Helv Chim Acta. 2009;92(6):1112–1117. | ||
Wakaki S, Marumo H, Tomioka K, et al. Purification and isolation study on gancidins. J Antibiot (Tokyo). 1958;11(4):150–155. | ||
Aiso K, Arai T, Suzuki M, Takamizawa T. Gancidin, an antitumor substance derived from Streptomyces sp. J Antibiot (Tokyo). 1956;9(3):97–101. | ||
Ata-ur-Rehman S, Malik H, Cun H, Clardy J. Isolation and structure determination of nigellicine, a novel alkaloid from seeds of Nigella sativa. Tetrahedron Lett. 1995;26(2):2759–2762. | ||
Jain TC, Dingerdissen JJ, Weisbach JA, et al. Isolation and structure elucidation of Gancidin. Heterocycles. 1977;7(1):341–346. | ||
Peters W, Robinson BL. The chemotherapy of rodent malaria XLVII: studies on pyronaridine and other mannich base antimalarials. Ann Trop Med Parasitol. 1992;86(5):455–465. | ||
Anthony PJ, Fyfe L, Smith H. Plant active components – a source for anti-parasitic agents. Trend Parasitol. 2005;21(10):462–468. | ||
Stepniewska K, Taylor WR, Mayxay M, et al. In vivo assessment of drug efficacy against Plasmodium falciparum malaria: duration of follow-up. Antimicrob Agents Chemother. 2004;48(3):4271–4280. | ||
Wellems TE, Plowe CV. Chloroquine-resistant malaria. J Infect Dis. 2001;184(3):770–776. | ||
White NJ, Krishna S, Waller D, Craddock C, Kwiatkowski D, Brewster D. Open comparison of intramuscular chloroquine and quinine in children with severe chloroquine-sensitive falciparum malaria. Lancet. 1989;2(8675):1313–1326. | ||
Fidock DA. Drug discovery – priming the antimalarial pipeline. Nature. 2010;465(8):297–298. | ||
Epiphanio S, Campos MG, Pamplona A, et al. VEGF promotes malaria-associated acute lung injury in mice. PLOS Pathog. 2010;6(5):e1000916. | ||
Lovegrove FE, Gharib SA, Pena-Castillo L, et al. Parasite burden and CD36-mediated sequestration are determinants of acute lung injury in an experimental malaria model. PLOS Pathog. 2008;4(5):e1000068. | ||
Marinho CR, Neres R, Epiphanio S, Gonçalves LA, Catarino MB, Penha-Gonçalves C. Recrudescent Plasmodium berghei from pregnant mice displays enhanced binding to the placenta and induces protection in multigravida. PLoS One. 2009;4(4):e5630. | ||
Hee L, Dinudom A, Mitchell AJ, et al. Reduced activity of the epithelial sodium channel in malaria-induced pulmonary oedema in mice. Int J Parasitol. 2011;41(2):81–88. | ||
Craig AG, Grau GE, Janse C, et al. The role of animal models for research on severe malaria. PLOS Pathog. 2012;8(2):e1002401. | ||
Zainal-Abidin BAH, Noorakmal Z, Othman O, et al. Profile of blood glucose levels in mice infected with Trypanosoma evansi. Malays Appl Biol. 1985;32(2):22–27. | ||
Isaka M, Jaturapat A, Kramyu J, Tanticharoen M, Thebtaranonth Y. Potent in vitro antimalarial activity of metacycloprodigiosin isolated from Streptomyces spectabilis BCC4785. Antimicrob Agents Chemother. 2001;46(4):1112–1113. | ||
Prudhomme J, McDaniel E, Ponts N, et al. Marine actinomycete: a new source of compounds against the human malarial parasite. PLoS One. 2008;3(6):e2335. | ||
Sage R, Atchley W, Capanna E, et al. House mice as models in systemic biology. Syst Biol. 1993;42(4):523–561. | ||
Szenczi P, Bánszegi O, Groó Z, Altbäcker V. Development of the social behavior of two mice species with contrasting social systems. Aggress Behav. 2012;38(4):288–297. | ||
Gartner K. Life expectancy, its relation to sexual activity and body weight in male inbred mice. J Exp Anim Sci. 1992;35(3):132–135. | ||
Wiesner J, Henschker D, Hutchinson DB, Beck E, Jomaa H. In vitro and in vivo synergy of fosmidomycin, a novel antimalarial drug, with clindamycin. Antimicrob Agents Chemother. 2002;46(9):2889–2894. | ||
Ernest EMC. Advantages and limits of in vivo screening test. Ann Occup Hyg. 1995;39(5):727–735. | ||
Premji Z, Umeh RE, Owusu-Agyei S, et al. Chlorproguanil-dapsone-artesunate versus artemether-lumefantrine: a randomized, double-blind phase III trial in African children and adolescents with uncomplicated Plasmodium falciparum malaria. PLoS One. 2009;4(8):e6682. | ||
Van-Vugt M, Brockman A, Gemperli B, et al. Randomised comparison of artemether-benflumetol and artesunate-mefloquine in the treatment of multidrug-resistant falciparum malaria. Antimicrob Agents Chemother. 1998;42(1):135–139. | ||
Guidelines. Research Animal Resources, University of Minnesota. Available from: http://www.ahc.umn.edu/rar/refvalues.html. Accessed January 8, 2017. | ||
Nagaraja HS, Anupama BK, Jeganathan PS, et al. Stress responses in albino rats. Thai J Physiol Sci. 2006;19(2):8–15. | ||
Nalpas B, Vassault A, Charpin S, Lacour B, Berthelot P. Serum mitochondrial aspartate aminotransferase as a marker of chronic alcoholism: diagnostic value and interpretation in a liver unit. Hepatology. 1986;6(4):608–614. | ||
Arsad SS, Esa NM, Hamzah H, et al. Histopathologic changes in liver and kidney tissues from male sprague dawley rats treated with Rhaphidophora decursiva (Roxb.) schott extract. J Cytol Histol. 2014;4(1):10–18. | ||
Lim AY, Segarra I, Chakravarthi S, Akram S, Judson JP. Histopathology and biochemistry analysis of the interaction between sunitinib and paracetamol in mice. BMC Pharmacol. 2010;10(14):14–30. | ||
Mitsumari K, Shibutani S, Sato S, et al. Relationship between the development of hepatorenal toxicity and cadmium accumulation in rats. Arch Toxicol. 1998;72(9):545–552. | ||
Finn WF, Portr GA. Urinary biomarkers and nephrotoxicity. Miner Electrolyte Metab. 2008;20(1):92–130. | ||
Machalinska A, Wiszniewska B, Tarasiuk J, et al. Morphological effects of sodium fluoride on hematopoietic organs in mice. Fluoride. 2002;32(231):231–238. | ||
Podder S, Chattopadhyay A, Bhattacharya S, et al. Histopathology and cell cycle alteration in the spleen of mice from low and high doses of sodium fluoride. Fluoride. 2010;439(4):237–245. | ||
Onkar DP, Govardhan SA. Comparative histology of human and dog spleen. J Morphol Sci. 2013;30(1):16–20. | ||
Rungruang T, Chaweeborisuit P, Klosek SK. Effect of malaria infection and dexamethasone on spleen morphology and histology. Southeast Asian J Trop Med Public Health. 2010;41(6):1290–1296. | ||
Folarin OA, Gbotosho GO, Sowunmi A, Olorunsogo OO, Oduola AM, Happi TC. Chloroquine resistant Plasmodium falciparum in Nigeria: relationship between pfcrt and pfmdr1 polymorphisms, in-vitro resistance and treatment outcome. Open Trop Med J. 2008;1(1):74–82. | ||
Schellenberg D, Kahigwa E, Drakeley C, et al. The safety and efficacy of sulfadoxine-pyrimethamine, amodiaquine, and their combination in the treatment of uncomplicated Plasmodium falciparum malaria. Am J Trop Med Hyg. 2002;67(1):17–23. | ||
Nkrumah LJ, Riegelhaupt PM, Moura P, et al. Probing the multifactorial basis of Plasmodium falciparum quinine resistance: evidence for a strain-specific contribution of the sodium–proton exchanger PfNHE. Mol Biochem Parasitol. 2009;165(2):122–131. | ||
Pelleau S, Bertaux L, Briolant S, et al. Differential association of Plasmodium falciparum Na+/H+ exchanger polymorphism and quinine responses in field- and culture-adapted isolates of Plasmodium falciparum. Antimicrob Agents Chemother. 2011;55(12):5834–5841. | ||
Angulo-Barturen I, Jiménez-Díaz MB, Mulet T, et al. A murine model of falciparum malaria by in vivo selection of competent strains in non-myelodepleted mice engrafted with human erythrocytes. PLoS One. 2008;3(5):e2252. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.