")
Back to Journals » OncoTargets and Therapy » Volume 13
Galangin (GLN) Suppresses Proliferation, Migration, and Invasion of Human Glioblastoma Cells by Targeting Skp2-Induced Epithelial–Mesenchymal Transition (EMT)
Authors Xiong Y, Lai X, Xiang W, Zhou J , Han J, Li H, Deng H, Liu L, Peng J, Chen L
Received 28 May 2020
Accepted for publication 29 July 2020
Published 17 September 2020 Volume 2020:13 Pages 9235—9244
DOI https://doi.org/10.2147/OTT.S264209
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Arseniy Yuzhalin
Yu Xiong,1– 3,* Xue Lai,4,* Wei Xiang,1– 3 Jie Zhou,1– 3 Jizhong Han,1– 3 Hao Li,1– 3 Huajiang Deng,1– 3 Luotong Liu,1– 3 Jianhua Peng,1– 3 Ligang Chen1– 3
1Department of Neurosurgery, Affiliated Hospital of Southwest Medical University, Southwest Medical University, Luzhou, 646000, People’s Republic of China; 2Neurosurgery Clinical Medical Research Center of Sichuan Province, Luzhou 646000 People’s Republic of China ; 3Academician (Expert) Workstation of Sichuan Province; 4Day Surgery Center, Affiliated Hospital of Southwest Medical University, Luzhou 646000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ligang Chen
Department of Neurosurgery, Affiliated Hospital of Southwest Medical University, Southwest Medical University, Luzhou 646000, People’s Republic of China
Tel +86 138 82718881
Email [email protected]
Background: Galangin (GLN), a pure natural flavonoid compound found in plants, has been shown to exert anti-cancer effects against multiple cancer types, including glioma. However, its underlying molecular mechanism remains unclear. Epithelial-to-mesenchymal transition (EMT) performs an important function in the genesis and development of cancer. Skp2, a pivotal component of SCFSkp2 E3 ubiquitin ligase, has been shown to function as an oncogene in GBM invasion that contributes to the EMT process. Thus, we explored whether GLN inhibited Skp2-mediated EMT and the mechanism underlying the Skp2 degradation pathway.
Methods: CCK-8 assay, wound healing assay and transwell assay were used to examine cell proliferation, migration, and invasion after treatment with or without GLN. RT-PCR and Western blotting analysis were performed to evaluate mRNA and protein expression, respectively. Co-immunoprecipitation was conducted to detect ubiquitinated Skp2 levels in vitro and in vivo after GLN treatment. Bioluminescence imaging was performed to examine the intracranial tumor size of U87 xenograft mice. Microscale thermophoresis (MST) experiment was used to detect interactions between Skp2 and GLN.
Results: GLN suppressed GBM cell growth, migration, and invasion, and also downregulated the expression of Skp2 and mesenchymal markers (Zeb1, N-cadherin, snail, vimentin) in vitro. Moreover, the overexpression of Skp2 in GBM cells decreased the effect of GLN on EMT. Furthermore, we demonstrated that GLN degraded skp2 protein through the ubiquitination proteasome pathway and directly interacted with skp2 protein, as shown through the MST assay.
Conclusion: This study is the first to identify Skp2 as a novel target of GLN for the treatment of GBM and report of Skp2 protein degradation in a ubiquitination proteasome pathway. Results from our study indicated the potential of GLN for the treatment of GBM through ubiquitin-mediated degradation of Skp2.
Keywords: galangin, GBM, EMT, Skp2, ubiquitination
Introduction
Glioblastoma multiforme (GBM) is a primary malignancy of the central nervous system. Despite temozolomide (TMZ) treatment, as well as radiotherapy after surgery, tumor recurrence is unavoidable and the median survival of GBM patients is not greater than 1.5 years due to the highly invasive nature of GBM.1–3
Galangin (GLN; 3, 5, 7-trihydroxyflavone) is a pure compound of the flavanol class of flavonoids that is found in shoots of Helichrysum aureonitens plants and is utilized in Asian countries as a medicinal compound. GLN has been shown to exhibit different pharmacological effects, such as anti-oxidative, anti-mutagenic and radical scavenging effects.4–6 Research has indicated that GLN exerts anti-cancer effects against various cancers, such as gastric cancer, melanoma, ovarian cancer, hepatocellular carcinoma, and promyelocytic leukemia,7–10 despite the fact that GLN leads to the growth and apoptosis of certain cancer cells. For example, galangin inhibited proliferation and induced apoptosis of ovarian cancer cells via the p53-dependent pathway.11 Galangin suppressed cell growth and induced HepG2 cell apoptosis through the p38 and AKT signaling pathways.12 In addition, galangin inhibited cell proliferation by downregulating CD44 expression in glioma.13 However, the precise mechanism of action of GLN and its related molecular mechanism in glioma have not been fully elucidated.
Epithelial-to-mesenchymal transition (EMT) has been shown to contribute to the tumorigenic process. Once EMT occurs, cancer cells change their epithelial features into mesenchymal characteristics, thereby promoting cell growth, migration and facilitation of tumor cells to infiltrate nearby tissues and blood vessels. Accumulating evidence has confirmed that EMT causes GBM cells to acquire more malignant biological properties and is negatively associated with GBM prognosis.14,15 Moreover, EMT inhibition is an effective mode of treatment for GBM.16,17 EMT is a complicated process and a variety of molecules have been identified as regulators of EMT.
Skp2 (S-phase-kinase-associated protein 2), an oncogene, has been reported to be expressed in several types of cancers, including GBM. Additionally, multiple studies have reported that Skp2 regulates EMT18 and that a high expression of Skp2 is correlated with poor prognosis.19–21 Therefore, targeting Skp2-mediated EMT may be a novel method for the treatment of GBM.
In this study, we showed that GLN suppressed cell growth and EMT through the inhibition of Skp2 in GBM, both in vitro and in vivo. Moreover, we demonstrated that GLN directly binds to Skp2 protein to promote Skp2 degradation through a ubiquitin-mediated proteasome pathway. In conclusion, our results verified that GLN may act as an anti-cancer agent and provided evidence that GLN exerts its anti-cancer effects through Skp2 suppression.
Materials and Methods
Chemicals, Reagents, and Antibodies
Galangin (Shilan Biology, Tianjin, China) was suspended in DMSO and kept at 4°C. Dulbecco’s modified Eagle medium (DMEM) and fetal bovine serum (FBS) were acquired from Gibco (Grand Island, USA). Skp2, snail, vimentin, N-cadherin, ubiquitin and GAPDH antibodies were bought from Cell Signaling Technology (Beverly, MA). Chloroquine (CQ) and MG132 were purchased from Selleckchem (Houston, USA).
Tissue Culture
GBM cell lines (U87, U251 and U87-luciferase) were provided by the Chinese Academy of Sciences (Beijing, China). The cell lines were maintained in complete medium (high-glucose DMEM) supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin. The cells were cultured at 37°C in a 5% CO2 atmosphere.
Cell Viability Assessment
Cell viability was determined using CCK-8 assay. The cells were plated onto 96-well plates with 4000 cells in 100 μL in each well. Then, the indicated dose of GLN was added and the cells were incubated for 24 h. Thereafter, 10 μL of CCK-8 reagent was added into each well. The plates were maintained at 37°C for 2 hours. The absorbance at a wavelength of 450 nm was quantified utilizing a microplate reader. Data are displayed as viability relative to the controls.
Cell Migration and Invasion Experiments
The migratory and invasive capacities of the GBM cells were determined using transwell assay (Corning, USA). 1 × 104 cells were placed in the upper chamber in 200 µL of serum-free media. The chambers were coated with (invasion) or without (migration) 100 µL of Matrigel (BD Biosciences, CA, USA). The cells were incubated for 24 h at 37°C. Cells that had migrated across the 8 μm polycarbonate basement membrane were stained using 0.1% crystal violet. The number of cells was calculated utilizing five random fields of view in each well using a microscope.
Reverse Transcription Polymerase Chain Reaction (RT-PCR)
Cellular RNA was isolated using TRIzol reagent (Sigma-Aldrich). cDNA was synthesized from the RNA using a PrimeScript RT Reagent kit. A StepOne Plus device (Applied Biosystems) was used to perform RT-PCR at 95°C for 10 s, which was followed by 40 cycles of 95°C for 5 s and 60°C for 20 s using the following primers: human Skp2: GATGTGACTGGTCGGTTGCTGT (forward) and GAGTTCGATAGGTCCATGTGCTG (reverse); and human GAPDH: GTCTCCTCTGACTTCAACAGCG (forward) and ACCACCCTGTTGCTGTAGCCAA (reverse).
Transfection
For the overexpression of Skp2 cells, the cells were transfected with plasmids containing Skp2-flag or flag (OriGene, USA) using Lipofectamine 2000, as per established guidelines.
Western Blotting Analysis
Cell lysates or xenograft glioblastoma tissue homogenates were lysed in 1× RIPA lysis buffer (CWBIO, China). All protein samples underwent separation through electrophoresis on 10% gradient gels in SDS–PAGE and were blotted onto PVDF membranes through electroblotting in a transfer buffer. Then, the membrane was placed in 1× TBST containing 5% BSA for blocking for 1 hour at RT. Thereafter, the membrane was incubated overnight with primary antibodies suspended in 5% BSA (1× TBST) at 4°C. Next, the membranes were incubated with peroxidase–conjugated secondary antibodies and were also diluted in 1× TBST solution. The cells on the membrane were detected using the ECL chemiluminescence method (Merck Millipore, German ECL kit).
Co-Immunoprecipitation
Tissues were lysed in RIPA buffer containing proteinase inhibitors (Thermo Fisher Scientific). The proteins were treated for an hour at 4°C using 20 μL of Protein-A/G agarose beads (Santa Cruz) to decrease non-specific binding. The supernatant was acquired through centrifugation at 3000 rmp for 30 sec, then incubated overnight at 4°C with 2 μg of rabbit anti-Skp2 antibody, followed by addition of Protein A/G Plus-Agarose (Santa Cruz Biotechnology, Santa Cruz, CA) at 4°C overnight, while being slowly swirled. Then, the resulting beads were washed to decrease non-specific binding. After being washed three times with RIPA buffer, the bound proteins were determined using Western blotting analysis.
GLN Treatment of a U87 Xenograft Mouse Model
Approval for experimental procedures using animals was granted by the Institutional Animal Care Committee of the Southwest Medical University. Experiments were performed in accordance with the guideline of the Chinese Animal Welfare Act. Ten weeks old male balb/c nude mice (Vital River Laboratories) were housed at 23–25°C under a 12-hour light/dark cycle with free access to food and water. And the mice were administered 4 μL of 1×106 U87-luciferase cells/mouse at a rate of 0.5μL/min using a micro syringe pump (RWD, China) bound to a Hamilton syringe with a 33-gauge needle (Hamilton, Reno, NV). The following guidelines were used: (mm from the bregma): +0.5 anterior-posterior, +2.2 medio-lateral, and −3.0 dorsoventral. Five days after injection, tumor–bearing mice were divided into two groups and were intraperitoneally injected with GLN (100 mg/kg/day) or the vehicle. The weight of the mice was determined and tumors were quantified once every 7 days. Thereafter, the mice were anesthetized with 3.6% chloral hydrate in 0.9% sterile saline.
Bioluminescence Imaging
The mice were intraperitoneally administered 100 mg/kg body weight D-luciferin (Sigma-Aldrich, UK) and images were captured a minute later using the IVIS Lumina II imaging system (Caliper Life Sciences, MA). Bioluminescence images were acquired using the exposure function. Analysis of signal intensities and images were conducted using Living Image® Software (Caliper Life Sciences).
Microscale Thermophoresis (MST) Experiment
Recombinant Skp2 was marked using the Monolith NT™ Protein Labeling Kit RED as per labeling protocols. Labeled Skp2 was used at a concentration of 50 μM. The samples were incubated for 10 minutes at room temperature and added into MonolithTM standard-treated capillaries. Thermophoresis of the samples was quantified at 25°C after 20 minutes of incubation on a Monolith NT.115 instrument (NanoTemper Technologies, Germany). NT Analysis was used to determine the dissociation constant Kd values.
Statistical Analysis
Two-tailed Student’s t-tests were conducted to assess significant variances among the two groups and one-way ANOVA was used to evaluate multiple comparisons. A P value of <0.05 was considered to indicate statistical significance. The results are shown along with standard errors of the mean.
Results
GLN Inhibited the Growth, Migration and Invasion of GBM Cells
Initially, we detected the anti-proliferative effect of GLN on U87 and U251 cells using CCK-8 and EdU assays. As shown in Figure 1A–D, GLN suppressed proliferation in a concentration-dependent manner. Next, we carried out transwell assays to identify the anti-migratory and invasive abilities of GLN on glioblastoma cells. 10 μM and 20 μM GLN was used to determine the effect of GLN on migration. Compared with the control group, the GLN treatment groups showed lower levels of migration and invasion at the bottom of the membrane (Figure 1E–H). These results indicated that GLN could substantially decrease the migration and invasion abilities of glioblastoma cells.
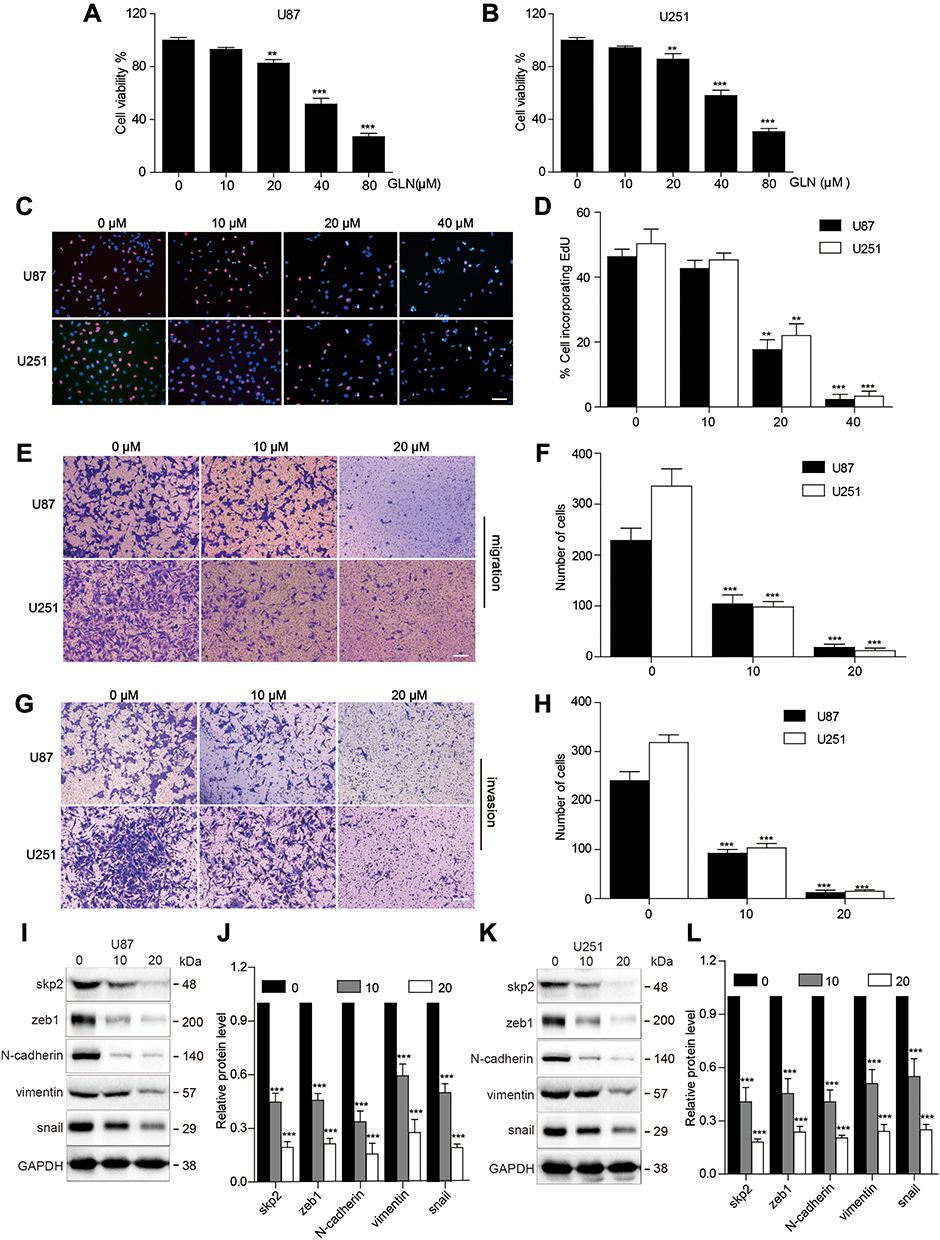 |
Figure 1 GLN suppressed cell growth, migration, invasion and EMT in GBM (A and B) CCK-8 assay was performed to evaluate the proliferation of U87 and U251 cells at the indicated concentrations of GLN. (C and D) U87 and U251 cells were administered different doses of GLN for 24 hours and then EdU assay was used to detect proliferation. (E–H) Transwell assay was conducted to detect the migration and invasion capabilities of U87 and U251 cells after the administration of the indicated concentrations of GLN for 24 hours. (Bar, 100 μm). (I–L) Western blotting analysis was performed to evaluate Skp2 protein and EMT-related marker levels followed by treatment using the indicated concentrations of GLN for 24 hours. **p < 0.01; ***p < 0.001. |
GLN Suppressed Skp2 and EMT-Related Markers in GBM Cells
To evaluate the influence of GLN on Skp2 and EMT in GBM, U87 and U251 cells were treated with 10 μM and 20 μM GLN for 24 hours and examined using Western blotting analysis to assess Skp2 and EMT-related markers (zeb1, N-cadherin, snail, and vimentin). As shown in Figure 1I–L, GLN decreased Skp2 protein level and EMT-related indicators in a concentration-dependent manner in U87 and U251 cells. This finding indicated that Skp2 is a target of GLN and that GLN can suppress EMT in GBM cells.
Skp2 Overexpression Decreased the Influence of GLN on GBM Cells
To validate whether GLN could decrease the effect of Skp2 overexpression, U87 and U251 cells were transfected with a Skp2 overexpressing plasmid or vector, followed by treatment with GLN or vehicle. The results indicated that skp2 overexpression substantially suppressed the influence of GLN on cell growth (Figure 2A and B). In addition, Skp2-induced migration, and invasion abilities of GBM cells were decreased by GLN treatment (Figure 2C–F). Furthermore, Skp2-overexpression in U87 and U251 cells decreased the influence of GLN on Skp2, N-cadherin, snail, and vimentin levels (Figure 2G–J). These results indicated that GLN exerts anti-glioblastoma effects, at least in part, by suppressing Skp2-induced EMT.
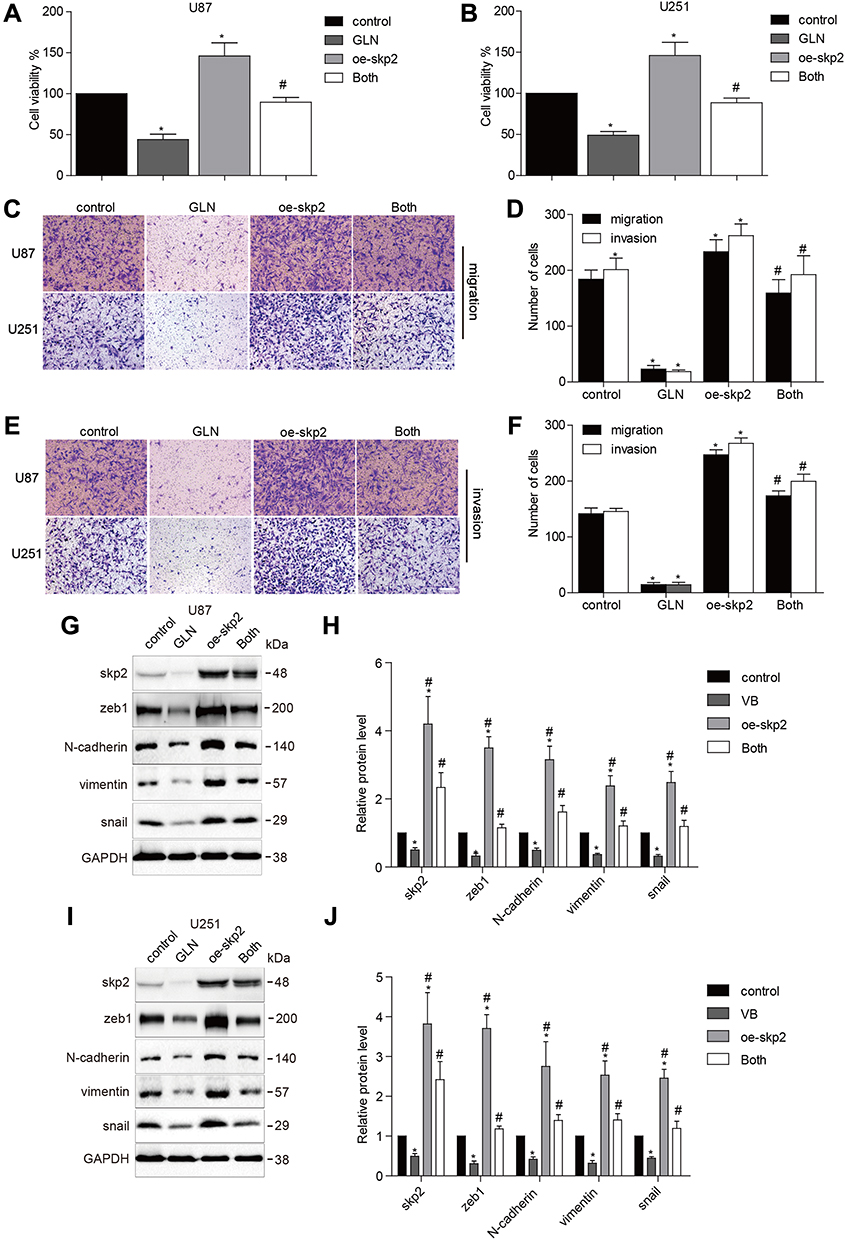 |
Figure 2 Skp2 overexpression abolished the influence of GLN in GBM. U87 and U251 cells were transfected with Skp2 overexpression or vector plasmids for 24 hours (A and B) and were plated in a 96-well plate and were treated with 20 μM GLN or the vehicle for 24 hours. CCK-8 assay was used to detect cell growth. (C–F) Transwell assay was conducted to evaluate the migration and invasion abilities of U87 and U251 cells (Bar, 100 μm). (G–J) Western blotting analysis was used to determine Skp2 and EMT-related marker levels. Control: vector transfection; GLN: vector-transfection and 20 μM GLN; oe-Skp2: Skp2-transfection; Both: Skp2 transfection and 20 μM GLN. *p < 0.05 in comparison to control; #p < 0.05, compared to GLN treatment or Skp2 transfection. |
GLN Induced Skp2 Degradation Through the Ubiquitin-Proteasome-Dependent Pathway in GBM
First, we evaluated Skp2 mRNA levels post-GLN treatment for 24 hours using RT-PCR. The results showed no significant differences between Skp2 levels in U87 and U251 cells (Figure 3A and B). Subsequently, we detected whether Skp2 was degraded via the autophagy-lysosome pathway or ubiquitin-proteasome pathway. The cells were treated with GLN combined with MG132 (a proteasome blocker) or CQ (a lysosome inhibitor). As shown in Figure 3C, MG132, but not CQ, effectively inhibited GLN-induced Skp2 protein degradation. Then, ubiquitination activity assays of U87 and U251 cells were performed to determine the association between ubiquitin and GLN-regulated proteasome breakdown of Skp2 protein. U87 and U251 cells were incubated with MG-132 and treated with GLN. Then, a co-immunoprecipitation assay was performed to examine levels of ubiquitinated Skp2. Compared with control cells, the intensity of Skp2 smeared bands in the GLN treatment group was found to be greater (Figure 3D). These results suggested that GLN enhanced Skp2 degradation through the ubiquitin-proteasome-dependent pathway in GBM.
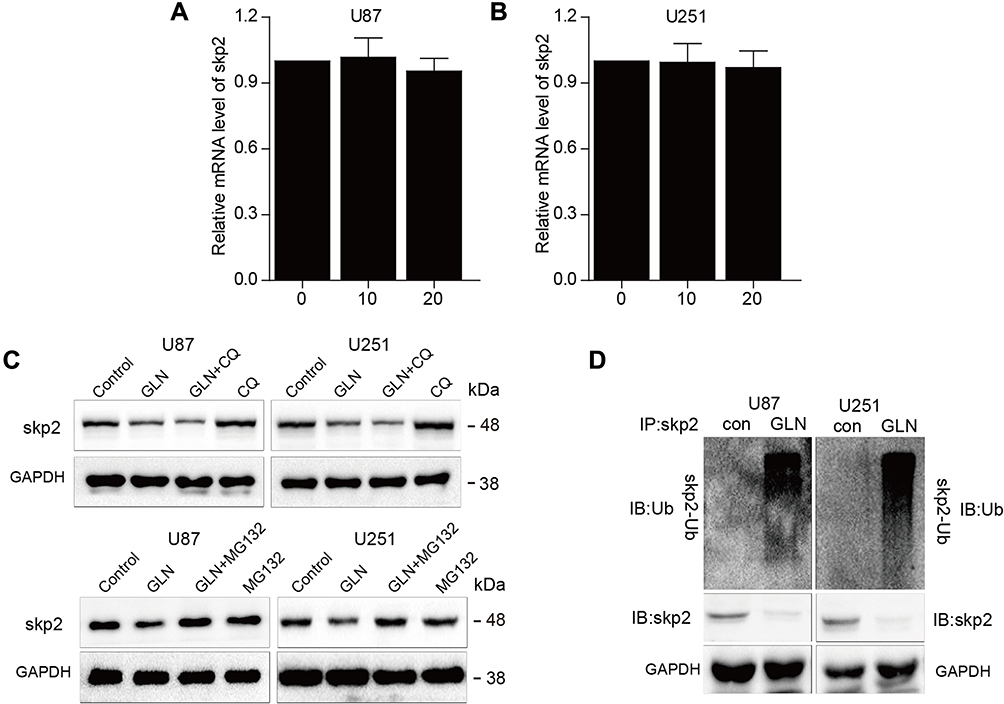 |
Figure 3 GLN strengthened Skp2 degradation through the ubiquitin-proteasome-dependent pathway in GBM. (A and B) Skp2 mRNA expression was evaluated using qRT-PCR in GBM cells incubated with the indicated concentrations of GLN. (C) U87 and U251 cells incubated with 20 μM CQ or 5μM MG132 for 6 hours prior to treatment with 20 μM GLN or vehicle for 18 hours. Skp2 protein levels were evaluated using Western blotting analysis. (D) U87 and U251 cells were pre-treated with MG-132 (5 μM) for 6 hours, and then treated with GLN (20 μm) for 18 hours. Co-immunoprecipitation and Western blotting assays were used to determine levels of Skp2 ubiquitination. |
GLN Inhibited Tumor Proliferation in an Orthotopic Xenograft Mouse Model
To confirm the anti-GBM effect of GLN, we constructed a U87-luciferase orthotopic xenograft mouse model and treated it using GLN or the vehicle. As shown in Figure 4A and B, GLN inhibited intracranial tumor growth. Additionally, the survival rates of mice were higher (Figure 4C) in the 21-day GLN treatment group. Moreover, in the GLN treatment group, the body weight of the mice was higher than that of the vehicle-treatment group (Figure 4D). Similar to the in vitro results, protein levels of Skp2 and EMT-related markers in tumor tissues were downregulated in the GLN-treated group, compared with the vehicle-treated group (Figure 4E and F). Moreover, the co-immunoprecipitation assay confirmed that GLN-enhanced the ubiquitination of Skp2 in the intracranial xenograft model (Figure 4H). These results indicated that GLN treatment is crucially involved in the inhibition of GBM proliferation in a tumor model through inhibition of Skp2-induced EMT.
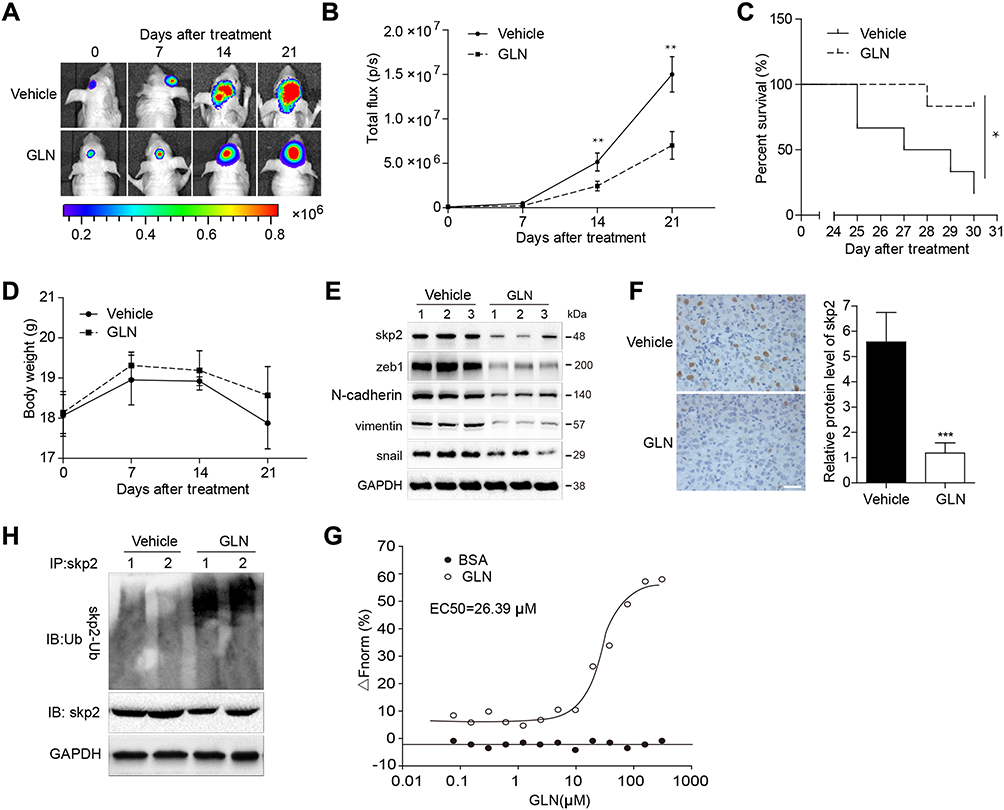 |
Figure 4 Influence of GLN on an orthotopic xenograft mouse model and the specific binding of GLN with Skp2. (A) Representative tumor volume in each group displayed at each time point (n=6). (B) Tumor volumes identified at each time point in each group (n=6). (C) Survival rate of each group (n=10). (D) Body weight differences across the groups (n=6). After the experiment was ended or the mice died, tumor tissues were excised, and the protein lysates (E) were used to evaluate the expression levels of Skp2 and EMT-related markers (F), which were stained using Skp2 antibodies (G) and were immunoprecipitated and immunoblotted to evaluate the ubiquitination of Skp2. (H) MST assay was used to quantify the binding between GLN and Skp2. *p < 0.05; **p < 0.01. |
Specific Binding of GLN to Skp2 Protein
We used the microscale thermophoresis method (MST) system to determine the binding of GLN to Skp2. Compared with the control group (BSA), GLN was found to bind to Skp2 protein and the EC50 was found to be 26.39 μM (Figure 4G), which indicated that Skp2 is a potential direct target of GLN.
Discussion
Increasing evidence has demonstrated that Skp2 is an important EMT regulator that contributes to the commencement and advancement of multiple cancers. GLN is a natural polyphenolic molecule that exerts anti-proliferation and anti-motility effects in several cancers. In this study, we discovered that GLN suppressed Skp2-induced EMT in GBM. Moreover, we also demonstrated that GLN accelerated Skp2 degradation. In addition, GLN was found to have degraded Skp2 in a ubiquitination proteasome dependent manner through its interaction with Skp2. Finally, we confirmed that GLN inhibited GBM in an intracranial GBM model and that the drug plays acts through identical mechanisms in vivo and in vitro.
The invasive property of GBM is due to its aggressive invasion capacity to infiltrate adjacent tissue. Cumulating evidence has demonstrated that EMT is an important mechanism that gives GBM cells an invasive ability.22,23 Based on histopathology, GBM has been regarded as being derived from the neuroepithelium. In addition, similar to carcinoma, GBM contains high expression levels of mesenchymal markers, including zeb1, vimentin, snail, and N-cadherin.24–26 Moreover, GBM patients with increased levels of mesenchymal markers have a worse prognosis.15,26 An increasing number of studies have reported that inhibition of EMT could suppress GBM.27 Cao et al reported that GLN inhibited cell invasion by inhibiting EMT in renal cell carcinoma,28 while Chen et al reported that GLN inhibited EMT by suppressing CD44 in glioma.13 In this study, we showed that GLN could effectively downregulate mesenchymal markers in GBM via downregulating Skp2 in vitro and in vivo. However, the xenograft usually does not grow invasive. Therefore, anti-invasive therapy assessments would be needed in vivo for further investigations.
Skp2 is a part of the F-box family of the specific substrate-recognition subunit of SCF ubiquitin-protein ligase complexes. Many studies have confirmed that Skp2 performs a crucial oncogenic function in glioma pathogenesis. For instance, Skp2 was found to be highly expressed in most glioblastomas and high levels of Skp2 have been observed in about 30% of GBM patients, while high Skp2 expression has been associated with a poor prognosis.19 In addition, inactivation of Skp2 could suppress EMT, as well as glioma growth.28 Thus, inhibition of Skp2 could prevent glioma growth and proliferation. In this study, we reported that GLN could suppress Skp2 expression in GBM. Additionally, we reported that skp2 can be degraded through the ubiquitination-proteasome system. K48-linked Skp2 polyubiquitination is crucial for proteasomal degradation. We also showed that GLN encouraged skp2 degradation through a ubiquitination proteasome pathway.
The Food and Drug Administration (FDA) has reported that 30–40% of currently available anti-cancer therapies originate from plants or their derivatives.29 GLN, a natural compound isolated from the Helichrysum aureonitens plant, has exhibited antitumor properties in a variety of types of cancers.7,10,28 In glioma, the existence of the blood-brain barrier (BBB) makes it difficult for agents to exert their effect in vivo. We confirmed that GLN inhibited GBM using a mouse orthotopic glioma model. Furthermore, we discovered that GLN targeted and enhanced Skp2 degradation, which reduced the side effects of GLN and promotes its anti-glioma ability.
In summary, we demonstrated that GLN blocked GBM proliferation and EMT in vitro and in vivo. Furthermore, we assessed whether GLN enhanced Skp2 degradation in ubiquitination proteasome pathway and found that Skp2 is a potential target of GLN in GBM.
Data Sharing Statement
The data used to support the findings of this study are available from the corresponding author upon reasonable request.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Alexander BM, Cloughesy TF. Adult glioblastoma. J Clin Oncol. 2017;35(21):2402–2409. doi:10.1200/JCO.2017.73.0119
2. Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310(17):1842–1850. doi:10.1001/jama.2013.280319
3. Alifieris C, Trafalis DT. Glioblastoma multiforme: pathogenesis and treatment. Pharmacol Ther. 2015;152:63–82. doi:10.1016/j.pharmthera.2015.05.005
4. Kim ME, Park PR, Na JY, Jung I, Cho JH, Lee JS. Anti-neuroinflammatory effects of galangin in LPS-stimulated BV-2 microglia through regulation of IL-1β production and the NF-κB signaling pathways. Mol Cell Biochem. 2019;451(1–2):145–153. doi:10.1007/s11010-018-3401-1
5. Choi M-J, Lee E-J, Park J-S, Kim S-N, Park E-M, Kim H-S. Anti-inflammatory mechanism of galangin in lipopolysaccharide-stimulated microglia: critical role of PPAR-γ signaling pathway. Biochem Pharmacol. 2017;144:120–131. doi:10.1016/j.bcp.2017.07.021
6. Bacanlı M, Başaran AA, Başaran N. The antioxidant, cytotoxic, and antigenotoxic effects of galangin, puerarin, and ursolic acid in mammalian cells. Drug Chem Toxicol. 2017;40(3):256–262. doi:10.1080/01480545.2016.1209680
7. Chien S-T, Shi M-D, Lee Y-C, Te -C-C, Shih Y-W. Galangin, a novel dietary flavonoid, attenuates metastatic feature via PKC/ERK signaling pathway in TPA-treated liver cancer HepG2 cells. Cancer Cell Int. 2015;15(1):15. doi:10.1186/s12935-015-0168-2
8. Benguedouar L, Lahouel MC, Gangloff S, et al. Ethanolic extract of algerian propolis and galangin decreased murine melanoma T. Anticancer Agents Med Chem. 2016;16(9):1172–1183.
9. Xu YX, Wang B, Zhao XH. In vitro effects and the related molecular mechanism of galangin and quercetin on human gastric cancer cell line (SGC-7901). Pak J Pharm Sci. 2017;30:4.
10. Huang H, Chen AY, Rojanasakul Y, Ye X, Rankin GO, Chen YC. Dietary compounds galangin and myricetin suppress ovarian cancer cell angiogenesis. J Funct Foods. 2015;15:464–475. doi:10.1016/j.jff.2015.03.051
11. Huang H, Chen AY, Ye X, Guan R, Rankin G, Chen YC. Galangin, a flavonoid from lesser Galangal, induced apoptosis via p53-dependent pathway in ovarian cancer cells. Molecules (Basel, Switzerland). 2020;25(7):7. doi:10.3390/molecules25071579
12. Huang H, Chen AY, Ye X, et al. Multifunctional selenium nanoparticles with Galangin-induced HepG2 cell apoptosis through p38 and AKT signalling pathway. Open Biol. 2018;5(11):180509. doi:10.1098/rsos.180509
13. Chen D, Li D, XU X, et al. Galangin inhibits epithelial-mesenchymal transition and angiogenesis by downregulating CD44 in glioma. J Cancer. 2019;10(19):4499–4508. doi:10.7150/jca.31487
14. Noh M-G, Oh S-J, Ahn E-J, et al. Prognostic significance of E-cadherin and N-cadherin expression in Gliomas. BMC cancer. 2017;17(1):583. doi:10.1186/s12885-017-3591-z
15. Iwadate Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol Lett. 2016;11(3):1615–1620. doi:10.3892/ol.2016.4113
16. Wang Z, Liu Z, Yu G, et al. Paeoniflorin inhibits migration and invasion of human glioblastoma cells via suppression transforming growth factor β-induced epithelial–mesenchymal transition. Neurochem Res. 2018;43(3):760–774. doi:10.1007/s11064-018-2478-y
17. Maciaczyk D, Picard D, Zhao L, et al. CBF1 is clinically prognostic and serves as a target to block cellular invasion and chemoresistance of EMT-like glioblastoma cells. Br J Cancer. 2017;117(1):102. doi:10.1038/bjc.2017.157
18. Yang Q, Huang J, Wu Q, et al. Acquisition of epithelial–mesenchymal transition is associated with Skp2 expression in paclitaxel-resistant breast cancer cells. Br J Cancer. 2014;110(8):1958. doi:10.1038/bjc.2014.136
19. Saigusa K, Hashimoto N, Tsuda H, et al. Overexpressed Skp2 within 5p amplification detected by array-based comparative genomic hybridization is associated with poor prognosis of glioblastomas. Cancer Sci. 2005;96(10):676–683. doi:10.1111/j.1349-7006.2005.00099.x
20. Signoretti S, Di Marcotullio L, Richardson A, et al. Oncogenic role of the ubiquitin ligase subunit Skp2 in human breast cancer. J Clin Invest. 2002;110(5):633–641. doi:10.1172/JCI0215795
21. Hershko DD. Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer. 2008;112(7):1415–1424. doi:10.1002/cncr.23317
22. Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer. 2018;18(2):128. doi:10.1038/nrc.2017.118
23. Gherardi E, Birchmeier W, Birchmeier C, Woude GV. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12(2):89. doi:10.1038/nrc3205
24. Iser IC, Pereira MB, Lenz G, Wink MR. The epithelial-to‐mesenchymal transition‐like process in glioblastoma: an updated systematic review and in silico investigation. Med Res Rev. 2017;37(2):271–313. doi:10.1002/med.21408
25. Lee J-K, Joo KM, Lee J, Yoon Y, Nam D-H. Targeting the epithelial to mesenchymal transition in glioblastoma: the emerging role of MET signaling. Onco Targets Ther. 2014;7:1933.
26. Edwards LA, Kim S, Madany M, et al. ZEB1 is a transcription factor that is prognostic and predictive in diffuse gliomas. Front Neurol. 2018;9:1199.
27. Xu B, Jiang C, Han H, et al. Icaritin inhibits the invasion and epithelial‐to‐mesenchymal transition of glioblastoma cells by targeting EMMPRIN via PTEN/AKt/HIF‐1α signalling. Clin Exp Pharmacol Physiol. 2015;42(12):1296–1307. doi:10.1111/1440-1681.12488
28. Cao J, Wang H, Chen F, et al. Galangin inhibits cell invasion by suppressing the epithelial-mesenchymal transition and inducing apoptosis in renal cell carcinoma. Mol Med Rep. 2016;13(5):4238–4244. doi:10.3892/mmr.2016.5042
29. Mans DR, Da Rocha AB, Schwartsmann G. Anti-cancer drug discovery and development in Brazil: targeted plant collection as a rational strategy to acquire candidate anti-cancer compounds. Oncologist. 2000;5(3):185–198. doi:10.1634/theoncologist.5-3-185
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.