Back to Journals » Drug Design, Development and Therapy » Volume 17
Galangin Attenuates Myocardial Ischemic Reperfusion-Induced Ferroptosis by Targeting Nrf2/Gpx4 Signaling Pathway
Authors Yang T ![]() , Liu H, Yang C, Mo H, Wang X, Song X, Jiang L, Deng P, Chen R, Wu P, Chen A, Yan J
, Liu H, Yang C, Mo H, Wang X, Song X, Jiang L, Deng P, Chen R, Wu P, Chen A, Yan J
Received 19 February 2023
Accepted for publication 17 August 2023
Published 22 August 2023 Volume 2023:17 Pages 2495—2511
DOI https://doi.org/10.2147/DDDT.S409232
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Tao Yang,1– 4 Haiqiong Liu,1,3,4 Chaobo Yang,1,3,4 Huaqiang Mo,1,3,4 Xianbao Wang,1,3,4 Xudong Song,1,3,4 Luping Jiang,2 Ping Deng,2 Ran Chen,2 Pengcui Wu,2 Aihua Chen,1,3,4 Jing Yan1,3,4
1Department of Cardiology, Heart Center, Zhujiang Hospital, Southern Medical University, Guangzhou, Guangdong, People’s Republic of China; 2Department of Cardiovascular Medicine, The Affiliated Changsha Central Hospital, Hengyang Medical School, University of South China, Changsha, People’s Republic of China; 3Laboratory of Heart Center, Zhujiang Hospital, Southern Medical University, Guangzhou, Guangdong, People’s Republic of China; 4Guangdong Provincial Key Laboratory of Shock and Microcirculation, Southern Medical University, Guangzhou, Guangdong, People’s Republic of China
Correspondence: Jing Yan; Aihua Chen, Department of Cardiology, Heart Center, Zhujiang Hospital, Southern Medical University, NO. 253, Gongye Avenue, Guangzhou, 510282, People’s Republic of China, Tel/Fax +86-2061643686, Email [email protected]; [email protected]
Purpose: Myocardial ischemic reperfusion injury (MIRI) is a crucial clinical problem globally. The molecular mechanisms of MIRI need to be fully explored to develop new therapeutic methods. Galangin (Gal), which is a natural flavonoid extracted from Alpinia Officinarum Hance and Propolis, possesses a wide range of pharmacological activities, but its effects on MIRI remain unclear. This study aimed to determine the pharmacological effects of Gal on MIRI.
Methods: C57BL/6 mice underwent reperfusion for 3 h after 45 min of ischemia, and neonatal rat cardiomyocytes (NRCs) subjected to hypoxia and reoxygenation (HR) were cultured as in vivo and in vitro models. Echocardiography and TTC-Evans Blue staining were performed to evaluate the myocardial injury. Transmission electron microscope and JC-1 staining were used to validate the mitochondrial function. Additionally, Western blot detected ferroptosis markers, including Gpx4, FTH, and xCT.
Results: Gal treatment alleviated cardiac myofibril damage, reduced infarction size, improved cardiac function, and prevented mitochondrial injury in mice with MIRI. Gal significantly alleviated HR-induced cell death and mitigated mitochondrial membrane potential reduction in NRCs. Furthermore, Gal significantly inhibited ferroptosis by preventing iron overload and lipid peroxidation, as well as regulating Gpx4, FTH, and xCT expression levels. Moreover, Gal up-regulated nuclear transcriptive factor Nrf2 in HR-treated NRCs. Nrf2 inhibition by Brusatol abolished the protective effect of Gal against ferroptosis.
Conclusion: This study revealed that Gal alleviates myocardial ischemic reperfusion-induced ferroptosis by targeting Nrf2/Gpx4 signaling pathway.
Keywords: galangin, ferroptosis, myocardial ischemia/reperfusion injury, Nrf2, Gpx4
Introduction
Acute myocardial infarction has become one of the main causes of mortality and morbidity in humans.1 Reperfusion therapies, including thrombolysis and percutaneous coronary intervention, can immediately alleviate cardiac injury by reopening the occluded culprit artery.2 However, it usually causes arrhythmias, slow flow, and acute heart failure, which is defined as myocardial ischemic reperfusion injury (MIRI).3 So far, methods to curb MIRI remain limited,4 thus finding novel strategies that can efficiently treat MIRI is an urgent need.
Cell death can occur in regulated and non-regulated forms following cardiac ischemic reperfusion injury. Previous studies have indicated that programmed cell death, including apoptosis,5 necroptosis,6 pyroptosis,7 and autophagic cell death,8 are involved in MIRI development. Ferroptosis is a newly found programmed cell death type, distinct from apoptosis, necroptosis, and autophagy in morphology, biochemistry, and genetics.9,10 The main characteristic of ferroptosis is iron overload, which causes lipid peroxidation and induces cell death. Recently, ferroptosis inhibitors have shown cardioprotective effects against MIRI. Moreover, Wang et al11 and Jiang et al12 demonstrated that ferrostatin-1 and iron chelator ameliorated myocardial ischemic/reperfusion (IR) injury in mice. Ferroptosis regulation may be an effective therapeutic strategy for attenuating MIRI.
Galangin (Gal) is a natural flavonoids compound isolated from traditional Chinese medicines-Alpinia Officinarum Hance and Propolis, which has multiple biological effects, such as antioxidant, modulating endothelial dysfunction, anti-inflammation, and inhibiting apoptosis.13–15 As previously reported, Gal might prevent the pathological progression of cardiovascular diseases. Gal ameliorates pressure overload or angiotensin II–induced cardiac remodeling via MEK1/2-ERK1/2 and PI3K-AKT pathways.16 Further, Gal attenuated diabetic cardiomyopathy by regulating oxidative stress, apoptosis, and inflammation in rats.17
Additionally, Gal is a potential substance for inhibiting ferroptosis, as it reduces liver iron levels and alleviates iron-evoked oxidative stress.18 Thus, we hypothesize that Gal might alleviate myocardial injury after ischemic reperfusion through ferroptosis inhibition. Nuclear factor erythroid 2-related factor 2 (Nrf2) is an antioxidant transcription factor that up-regulates the transcription of a series of antioxidant and anti-ferroptosis enzymes.19–21 Glutathione peroxidases 4 (Gpx4) plays a crucial role in reducing phospholipid peroxides, thereby protecting cells from the pro-death effects of ferroptosis.22,23 Hence, we have investigated the possibility that Gal might suppress ferroptosis through the Nrf2/Gpx4 signaling pathway after myocardial ischemic reperfusion (MIR).
Materials and Methods
IR Mouse Model
All the animal care and experimental procedures follow the Guidelines for the Care and Use of Laboratory Animals developed by the Ministry of Science and Technology of China, and the Ethics Committee of the Southern Medical University approved this study (Permit Number: LAEC-2021-007). All animals were raised in standard conditions with a 12-h light/dark cycle and free access to water and food. Adult male wild-type C57BL/6 mice (6–8 weeks; 20–25 g) from the Laboratory Animal Center of Guangdong Province underwent left anterior descending artery ligation to establish the IR model as previously described.24 Briefly, mice were anesthetized with 50 mg/kg of pentobarbital sodium by intraperitoneal injection (IP). Surgical procedures were started and the left anterior descending coronary (LAD) was ligated with a 6–0 suture (no ligation for sham) after intratracheal incubation and ventilation using a mini-ventilator with air. After 45 min of ischemia, 3 h of reperfusion was allowed by opening the knot (echocardiography was performed after reperfusion for 7 days).24
We set the groups as 1) Sham group, IR group, and IR + Ferrostatin-1 (Fer-1) group and 2) Sham group, Gal group, IR group, and IR + Gal group. Fer-1 at 2 mg/kg or Gal at 15 mg/kg was dissolved in a mixture of dimethyl sulfoxide (DMSO), polyethylene glycol 300 (PEG 300), Tween-80, and normal saline following the manufacturer’s instructions. Fer-1 or Gal was given by IP 15 min or 4 days preoperatively, respectively, as elucidated elsewhere.25–27 The sham group was administered with the same dose of solvent mixture. Then, the mice were euthanized and hearts were collected at 3 h following reperfusion (except 7 days in the echocardiography test). The present study purchased Gal and solvent mixture from MedChemExpress (MCE) Inc (Shanghai, China).
Cardiomyocyte Isolation and Culture
Neonatal rat cardiomyocytes (NRCs) were separated from Sprague–Dawley rats for 1–3 days and cultured as previously described.24 NRCs were incubated in a 5% CO2 atmosphere at 37°C. First, cells were cultured in Dulbecco’s Modified Eagle Medium containing 10% fetal bovine serum for 72 h. Then, hypoxia/reoxygenation (HR) treatment was initiated. Briefly, cells were incubated with phosphate-buffered saline (PBS) in a hypoxic chamber (Modular Incubator Chamber) for 2 h at 37°C equilibrated with 95% N2 and 5% CO2. Then, reperfusion was initiated by returning to normal air conditions and normoxic complete medium for 2 h. Gal was added to NRCs 24 h before the HR injury. At the beginning of hypoxia, 30 μM of apoptosis inhibitor Z-VAD-FMK (FMK), 20 μM of necroptosis inhibitor necrostatin-1 (Nec-1), 5 mM of autophagy inhibitor 3-MA, and 1 μM of Nrf2 inhibitor Brusatol (MCE, NJ, USA) were added into NRCs, 10 μM of ferroptosis inhibitor Fer-1 was given 24 h before hypoxia. Thus, NRCs were randomly divided into the following groups: 1) (a) control group; (b) HR group, NRCs subjected to HR injury; (c) HR + Fer-1 group, treated with Fer-1 in HR-treated NRCs; (d) HR + FMK group, treated with FMK in HR-treated NRCs; (e) HR + Nec-1 group, treated with Nec-1 in HR-treated NRCs; (f) HR + 3-MA group, treated with 3-MA in HR-treated NRCs; 2) (a) control group; (b) HR group, NRCs subjected to HR injury; (c) HR + Gal group treated with Gal (25 μM) in HR-treated NRCs; (d) HR + Gal + Brusatol group treated with Gal (25 μM) and Brusatol (1 μM) in HR-treated NRCs; (e) HR + Brusatol group treated with Brusatol (1 μM) in HR-treated NRCs. NRCs were collected at the end of reoxygenation and used for later analysis.
Cell Viability Test
Cell Counting Kit-8 (CCK‐8) assay kit (MCE, NJ, USA) was used to measure the viability of NRCs following the manufacturer’s instructions. NRCs were seeded in 96‐well plates and then treated by different administrations. Then, the NRCs were incubated in the dark for 4 h with 10 μL of CCK‐8 reagent. A microplate reader (Thermo Scientific, NY, USA) tested the absorbance at 450 nm, and the readings were normalized with vehicle control.
Propidium Iodide Staining
A propidium iodide (PI) staining kit (Yeasen, Shanghai, China) was used to evaluate the necrosis of NRCs in different groups. NRCs were incubated in laser confocal dishes. Then, cells were collected and washed three times with PBS. The cells were incubated with PI staining and Hoechst 33342 solution for 25 min. Then, A fluorescence microscope (Olympus, Japan) captured the images.
Cardiac Injury Biomarker Measurement
The serum samples were collected from the different groups of mice at the indicated time points. The levels of cardiac injury biomarkers, including creatine kinase-MB (CK-MB) and cardiac troponin T (TnT), were measured by enzyme-linked immunosorbent assay kits (HUAYUN, Guangzhou, China) following the manufacturer’s protocol. The absorbance was measured at 450 nm using a microplate reader (Thermo Scientific, NY, USA). A lactate dehydrogenase (LDH) cytotoxicity assay kit (Beyotime, Shanghai, China) was used to test the LDH level in NRCs following the manufacturer’s instructions. A microplate reader tested the absorbance at 490 nm.
Western Blot
The collected mice hearts and NRCs were lysed in radioimmunoprecipitation assay (RIPA) buffer. Bicinchoninic acid protein assay kits (Thermo Scientific) were used to measure the protein concentrations. Protein was isolated by Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) and then transferred into a polyvinylidene fluoride membrane and incubated at room temperature for 2 h with a 5% non‐fat milk-blocking buffer. Afterward, membranes were incubated with the primary antibodies at 4°C overnight, including anti-Clearved Caspase-3 (1:1000; cat#: 9664, Cell Signaling Technology), anti-RIPK3 (1:1000; cat#: 10188, Cell Signaling Technology), anti-LC3 I/II (1:1000; cat#: 4108, Cell Signaling Technology), anti‐Gpx4 (1:1000; cat#: 59735, Cell Signaling Technology), anti‐xCT (1:1000; cat#: ab300667, Abcam), anti‐FTH (1:1000; cat#: 4393, Cell Signaling Technology), anti‐Nrf2 (1:1000; cat#: ab62352, Abcam), and anti‐β actin (1:10000, cat#: 4970, Cell Signaling Technology). The membranes were incubated at room temperature for 2 h with secondary antibodies (1:10000, Boster, Shanghai, China) and visualized by ECL kits (Engreen, Beijing, China). The protein expression was quantified by ImageJ software and was normalized to control.
Transmission Electron Microscopy
Small fragments of myocardium sized ∼1 mm3 were fixed overnight in 2.5% glutaraldehyde and then immersed in 1% osmium tetroxide in 0.1 M of cacodylate buffer for 1 h and incubated with 2% aqueous uranyl acetate for 2 h. Samples were dehydrated by a graded series of ethanol and cut into small slices. A Philips CM 10 electron microscope checked the slices.
Mitochondrial Membrane Potential Measurement
JC-1 staining probe (Beyotime, Shanghai, China) was used to measure the mitochondrial membrane potential following the manufacturer’s instructions. NRCs were incubated at 37°C for 20 min with JC-1 staining solution. Then, NRCs were washed three times with buffer solution. Images were captured with a fluorescence microscope. Green and red fluorescence indicates decreased and normal mitochondrial membrane potential, respectively. The fluorescence density was calculated as red/green ratio and analyzed with ImageJ software.
TTC-Evans Blue Staining
Evans Blue and 2,3,5-Triphenyltetrazolium chloride (TTC) staining were used to determine the area at risk (AAR) and infarct area (IA) as previously described.28 LAD was re-ligated after reperfusion for 3 h, and 2 mL of 2% Evans Blue dye was injected via the jugular vein. Then, the heart was collected immediately and frozen at −80°C for 10 min. Subsequently, the heart was sliced into 1–2 mm sections and placed into 2% TTC in a dark incubator at 37°C for 20 min. Afterward, the heart slices were fixed in 4% paraformaldehyde for 24 h. Finally, the ImageJ software was used to photograph and analyze the slices. The percentage of viable myocardium (dark blue), AAR (red and white), and IA (white) of the left ventricle (LV) were calculated.
Echocardiography
A Vevo 2100 System equipped with a 30‐MHz transducer (Fujifilm Visual Sonics, Inc., Toronto, Canada) was used for echocardiography in mice 7 days after IR injury. M‐mode echocardiographic was performed after anesthetization with 2% isoflurane, as previously described.29 The left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) were calculated.
Measurement of Malondialdehyde (MDA) Level
MDA, which is the end product of lipid peroxidation, was assessed to evaluate the lipid peroxidation level. The procedure was performed following the manufacturer’s instructions (Beyotime, Shanghai, China). The heart tissues or NRCs were lysed in RIPA buffer. Then, the lysates were centrifuged to separate the supernatant. MDA levels were detected by testing the absorbance at 532 nm with a microplate reader (Thermo Scientific, NY, USA).
C11BODIPY Fluorescence Probe
NRCs were seeded into the confocal dish and incubated with a 10-μM C11BODIPY fluorescence probe (Cayman, Michigan, USA) at 37°C in a dark incubator for 25 min and then washed with PBS three times to assess lipid reactive oxygen species (ROS) production in vitro. A fluorescence microscope captured the images.
FerroOrange Fluorescence Probe
FerroOrange fluorescence probe (F374, Dojindo, Japan) at 1 µM dissolved in serum-free medium was added to NRCs and incubated for 30 min to detect Fe2+ level in cells. Finally, a fluorescence microscope captured the images.
Prussian Blue Staining
Prussian blue staining was performed following the previous study to determine the iron deposited in cardiac tissue.30 Heart sections were deparaffinized at 60°C for 1 h and hydrated in distilled water. Then, the heart tissues were incubated with the Prussian blue solution for 3 min. Finally, a light microscope (Olympus, Japan) was used to observe the samples.
Data and Statistical Analysis
GraphPad Prism 8 software was used for data analysis. All data are indicated as mean ± standard deviation. Comparisons between multiple groups were used with a one‐way analysis of variance followed by Bonferroni’s or Dunnett’s T3 tests. P-values of <0.05 were defined as statistically significant. Data normalization was performed to control for sources of variation of baseline parameters.
Results
The Ferroptosis Inhibition Attenuated IR-Induced Myocardial Injury
We detected the hallmarks of different cell deaths by immunoblotting to clarify the presence of different programmed cell deaths in the MIRI. We found an increased expression level of Cleaved Caspase-3, RIPK3, and LC3II/I and a reduced expression level of Gpx4 in the HR-treated NRCs compared with those in the control group, indicating the presence of apoptosis, necroptosis, autophagy, and ferroptosis in the HR-treated NRCs (Figure 1A and B). Furthermore, we used different cell death inhibitors to confirm the important role of ferroptosis in MIRI, including Fer-1 for ferroptosis, FMK for apoptosis, Nec-1 for necroptosis, and 3-MA for autophagy. The CCK-8 (Figure 1C) and LDH assay (Figure 1D), as well as PI staining (Figure 1E and F), demonstrated that the cell injury and death were all attenuated after the treatment of these inhibitors, indicating the involvement of ferroptosis, apoptosis, necroptosis, and autophagy in NRCs exposed to HR treatment. Of note, the protective effect of ferroptosis inhibitor Fer-1 was better than the apoptosis inhibitor FMK, necroptosis inhibitor Nec-1, and autophagy inhibitor 3-MA. Therefore, ferroptosis inhibition seemed to be a critical strategy to improve MIRI. Fer-1 was administered to mice with MIRI to further verify the role of ferroptosis in MIRI, and we revealed that cardiac injury biomarkers, CK-MB and TnT, were decreased compared to those in the IR group (Figure 1G and H). Taken together, these results revealed the crucial role of ferroptosis in MIRI.
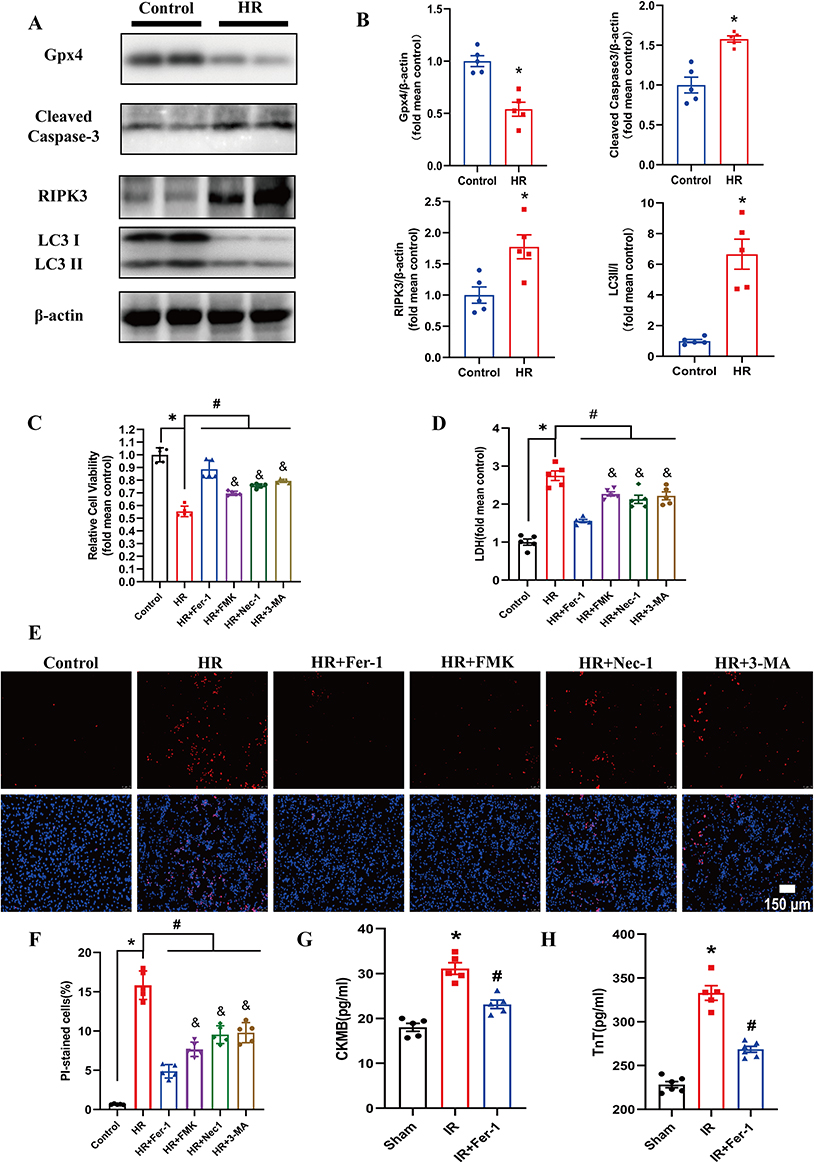 |
Figure 1 Multiple regulated cell death modes were involved in MIRI. (A and B) Representative pictures of Western Blot and quantitative analysis of Gpx4, Cleaved Caspase-3, RIPK3, and LC3II/I in different groups. Values are expressed as means ± SD; n = 5. *P < 0.05 vs control group. (C) The effect of different cell death inhibitors on NRCs exposed to HR was determined. CCK-8 assay was used to measure cell vitality. Values are expressed as means ± SD; n = 5. *P < 0.05 vs control group. #P < 0.05 vs HR group. &P < 0.05 vs HR+Fer-1 group. (D) Effect of different cell death inhibitors on cell injury in HR-exposed NRCs. LDH assay was used to measure cell injury. Values are expressed as means ± SD; n = 5. *P < 0.05 vs control group. #P < 0.05 vs HR group. &P < 0.05 vs HR + Fer-1 group. (E) PI staining was performed to detect the cell death of NRCs. The higher panels indicated the PI‐positive nuclei which were stained red. The lower panels indicated that the PI‐positive nuclei were merged with total nuclei which were stained with Hoechst 33342. The scale bar represents 150 μm. (F) The quantification of the percentage of PI-stained cells. Values are expressed as means ± SD; n = 5. *P < 0.05 vs control group. #P < 0.05 vs HR group. &P < 0.05 vs HR + Fer-1 group. (G and H) CK-MB and TnT levels of mice after IR surgery were detected. Fer-1 at 15 mg/kg was intraperitoneally injected into mice 15 min before IR surgery. Values are expressed as means ± SD; n = 5. *P < 0.05 vs sham group. #P < 0.05 vs IR group. Abbreviations: MIRI, myocardial ischemic reperfusion injury; HR, hypoxia-reoxygenation; NRCs, neonatal rat cardiomyocytes; LDH, lactate dehydrogenase; SD, standard deviation; CCK-8, Cell Counting Kit-8; CK-MB, creatine kinase-MB; TnT, troponin T. |
Gal Alleviated Myocardial Injury in Mice After IR Surgery
Initially, TTC-Evans Blue staining was conducted to assess the effects of Gal on myocardial injury after IR. The results revealed a markedly reduced IA/AAR in the IR + Gal group compared with that in the IR group (Figure 2A). No significant differences in AAR/LV were found between IR and IR + Gal groups. Additionally, TEM was performed to investigate the protective effects of Gal treatment on the mitochondria and myocardial fibrils because mitochondrial damage is one of the major characteristics in cardiomyocytes after IR.31 Dense and tight mitochondria and orderly arranged and intact myocardial fibrils could be seen in the cytoplasm in the sham group. In contrast, the IR-treated group demonstrated loose and swollen mitochondria with ruptured myocardial fibrils, but this damage was alleviated after Gal treatment (Figure 2B), indicating that Gal attenuated the mitochondrial and myocardial fibril injury during IR. Furthermore, we performed echocardiography to evaluate the cardiac function in mice. The results revealed that LVFS and LVEF were lower in the IR group than those in the sham group; however, these diminishments were partially reversed by Gal treatment (Figure 2C). These results revealed that Gal alleviated myocardial injury in mice after IR injury.
 |
Figure 2 Galangin (Gal) attenuated myocardial injury after IR in in vivo study. (A) Representative TTC-Evans Blue stained sections of hearts from each group. The non-risk area stained by Evans Blue is indicated in blue, the area at risk (AAR) stained by TTC is indicated in red and white, whereas the unstained (white) area represents the infarcted area (IA). The ratio of ARR/LV and IA/ARR are shown. Values are expressed as means ± SD; n = 5. #P < 0.05 compared with IR group. (B) Representative images of cardiomyocyte mitochondria and myofibril captured by TEM in different groups. Mitochondria (Mi) and Myofibril (My) were marked in red. The scale bar represents 1 μm. (C) M‐mode echocardiography was used to measure left ventricular EF and FS in the different groups. Representative echocardiograms and the quantification of EF and FS were shown. Values are expressed as means ± SD; n = 5. *P < 0.05 compared with Sham group, #P < 0.05 compared with IR group. Abbreviations: IR, ischemic reperfusion; LV, left ventricle; TEM, transmission electron microscopy; EF, ejection fraction; FS, fractional shortening; SD, standard deviation. |
Gal Alleviated Myocardial Injury in HR-Treated NRCs
Different Gal concentrations (10, 25, 50, and 100 μM) were added to NRCs, and the cell vitality was investigated by the CCK‐8 assay. First, we added Gal concentrations of 5–100 μM into NRCs without exposure to HR to exclude the toxicity of Gal, which revealed the constant cell vitality, indicating that Gal was nontoxic to NRCs with the above concentrations. Further, we revealed that cell viability was diminished in HR-treated NRCs, but Gal administration of 25, 50, and 100 μM significantly promoted cell viability. Gal at 25 μM demonstrated the optimal cell viability (Figure 3A). Thus, Gal at 25 μM was selected for the following in vitro experiments. Consistently, PI staining demonstrated that Gal prevented HR‐induced cell death in NRCs (Figure 3B). Moreover, we used a JC-1 fluorescence probe to further identify the effects of Gal on mitochondrial potential in vitro study. The mitochondrial potential was reduced in NRCs subjected to HR, but this reduction was improved by Gal, as shown in Figure 3C. In conclusion, these data demonstrated that Gal ameliorated cell damage in HR-treated NRCs.
 |
Figure 3 Galangin (Gal) attenuated myocardial injury after HR in in vitro study. (A) The effect of different Gal doses on NRCs exposed or not exposed to HR was determined. Cell vitality was measured by CCK-8 assay. (B) PI staining was performed to detect the cell death of NRCs. The higher panels indicated that the PI‐positive nuclei were stained red. The lower panels indicated that the PI‐positive nuclei were merged with total nuclei which were stained with Hoechst 33342. The scale bar represents 50 μm. The quantification of the percentage of PI-stained cells is shown on the right. (C) Mitochondrial membrane potential was measured with JC‐1 staining and captured by the microscope. Red fluorescence represented intact mitochondrial membrane potential. Green fluorescence represented reduced mitochondrial membrane potential. The scale bar represents 50 μm. The ratio of red/green fluorescence intensity is shown on the right. Values are expressed as means ± SD; n = 5. *P < 0.05 vs control group, #P < 0.05 compared with HR group. Abbreviations: HR, hypoxia-reoxygenation; NRCs, neonatal rat cardiomyocytes; SD, standard deviation; CCK-8, Cell Counting Kit-8. |
Gal Attenuates Myocardial IR Injury Through Ferroptosis Inhibition in Mice
We investigated the effects of Gal on ferroptosis, including two major factors, including Fe2+ overload and lipid peroxidation after IR, because ferroptosis is the important mechanism involved in cardiac IR injury. Prussian blue iron staining was performed and revealed that the iron ions deposited in the cardiac tissue after IR injury, but Gal treatment could eliminate the iron overload induced by IR (Figure 4A). Additionally, the end product of lipid peroxidation MDA was elevated in the IR group than in the sham group. Gal evidently reduced MDA accumulation in the cardiac tissue of IR-treated mice (Figure 4B). Moreover, the expression level of anti-ferroptosis proteins, including Gpx4 and FTH, was decreased in the IR group compared with that in the sham group, but this reduction was restored by Gal administration (Figure 4C). These data concluded that Gal inhibited IR-induced ferroptosis.
 |
Figure 4 Gal inhibited ferroptosis in mice subjected to IR surgery. (A) Representative pictures of Prussian blue staining in different groups. The blue stained area pointed with the black arrows represents the deposition of iron. The scale bar represents 50 μm. (B) MDA levels in cardiac tissue in mice in each group. (C) Representative pictures of Western Blot and quantitative analysis of Gpx4, FTH in different groups. Summary data are presented as means ± SD; n = 5. *P < 0.05 vs sham group, #P < 0.05 vs IR group. Abbreviations: IR, ischemic reperfusion; MDA, malondialdehyde; SD, standard deviation. |
Gal Inhibits Ferroptosis in HR‐treated NRCs
Initially, we revealed that HR significantly increased MDA levels in NRCs, but Gal inhibited MDA accumulation compared with that in the HR group (Figure 5A). Additionally, a C11BODIPY probe was performed to detect the lipid oxidative stress in vitro. HR treatment demonstrated increased lipid ROS in comparison with those in NRCs. Whereas, the lipid ROS production markedly decreased compared with those in the HR group in the presence of Gal (Figure 5B and C).
 |
Figure 5 Galangin (Gal) suppressed ferroptosis in HR-treated NRCs. (A) MDA levels in NRCs in different groups. (B and C) Representative images and quantitative data of lipid ROS level estimated with C11BODIPY fluorescence probe in different groups. The scale bar represents 50 μm. (D) Representative images (Left) and quantitative data (Right) of Fe2+ quantified with FerroOrange fluorescence probe in different groups. The scale bar represents 25 μm. (E) Representative pictures of Western Blot and quantitative analysis of Gpx4, xCT, and FTH in different groups. Summary data are presented as means ± SD; n = 5–6. *P < 0.05 vs control group, #P < 0.05 vs HR group. Abbreviations: HR, hypoxia-reoxygenation; NRCs, neonatal rat cardiomyocytes; MDA, malondialdehyde; Fe2+, ferrous ion; SD, standard deviation. |
Next, we verified the effects of Gal on Fe2+ level in NRCs with the FerroOrange fluorescence probe application. NRCs subjected to HR exhibited enhanced Fe2+ levels in comparison with that in the control group. On the contrary, the Fe2+ level was significantly reduced in the HR + Gal group (Figure 5D). The three crucial markers in the ferroptosis included xCT, Gpx4, and FTH,32–34 thus we evaluated these proteins in NRCs. The Gpx4, xCT, and FTH expression were markedly reduced in NRCs after being subjected to HR injury, but Gal significantly elevated the xCT, Gpx4, and FTH expressions in HR-treated NRCs (Figure 5E). Taken together, Gal suppressed HR-induced ferroptosis in NRCs.
Gal Suppresses Erastin-Induced Ferroptosis in vitro
Erastin is a recognized ferroptosis stimulator applied in numerous cell models.35 We induced ferroptosis by erastin in vitro to further determine whether Gal inhibits ferroptosis in NRCs. The CCK8 assay demonstrated that Gal could improve the decreased cell vitality induced by erastin (Supplementary Figure S1A). Furthermore, erasin treatment markedly increase the MDA level in NRCs, whereas Gal inhibited MDA accumulation compared with that in the erastin group (Supplementary Figure S1B). Moreover, erastin treatment markedly reduced Gpx4, xCT, and FTH expressions in NRCs, and Gal significantly reversed the expression of these proteins compared to those in the erastin group (Supplementary Figure S1C). Thus, these data further confirmed that Gal inhibited ferroptosis.
Gal Suppresses Ferroptosis via Nrf2/Gpx4 Pathway in vitro
Gpx4 is regulated by the antioxidative transcription factor Nrf2 (Nuclear factor erythroid 2-related factor 2), as previously reported.23 We investigated the Nrf2 expression in NRCs under HR to identify the involvement of Nrf2 in Gal-inhibited ferroptosis and revealed a markedly decreased Nrf2 compared with that in the control group. However, Gal treatment up-regulated the Nrf2 expression in HR-treated NRCs (Figure 6A). Additionally, we downregulated the Nrf2 expression level with the inhibitor Brusatol (Bru) to explore whether Gal modulates ferroptosis via the Nrf2/Gpx4 pathway. We revealed that HR treatment markedly reduced the Gpx4 and Nrf2 expression levels in the untreated NRCs, but Gal significantly promoted these proteins’ expression in the HR-treated NRCs. However, the effects of Gal on Gpx4 and Nrf2 expression were abolished by Bru, indicating that Gal activated Nrf2/GPX4 axis (Figure 6B). CCK8 assay demonstrated that Gal improved cell vitality after HR. Whereas co-treatment with Gal and Bru decreased the elevated cell vitality in the HR + Gal group (Figure 6C). Furthermore, we revealed that Gal reduced the content of MDA in HR-treated NRCs and co-treatment with Gal and Bru eliminated the effects of Gal (Figure 6D). Altogether, these results indicated that Gal suppresses ferroptosis via the Nrf2/Gpx4 pathway.
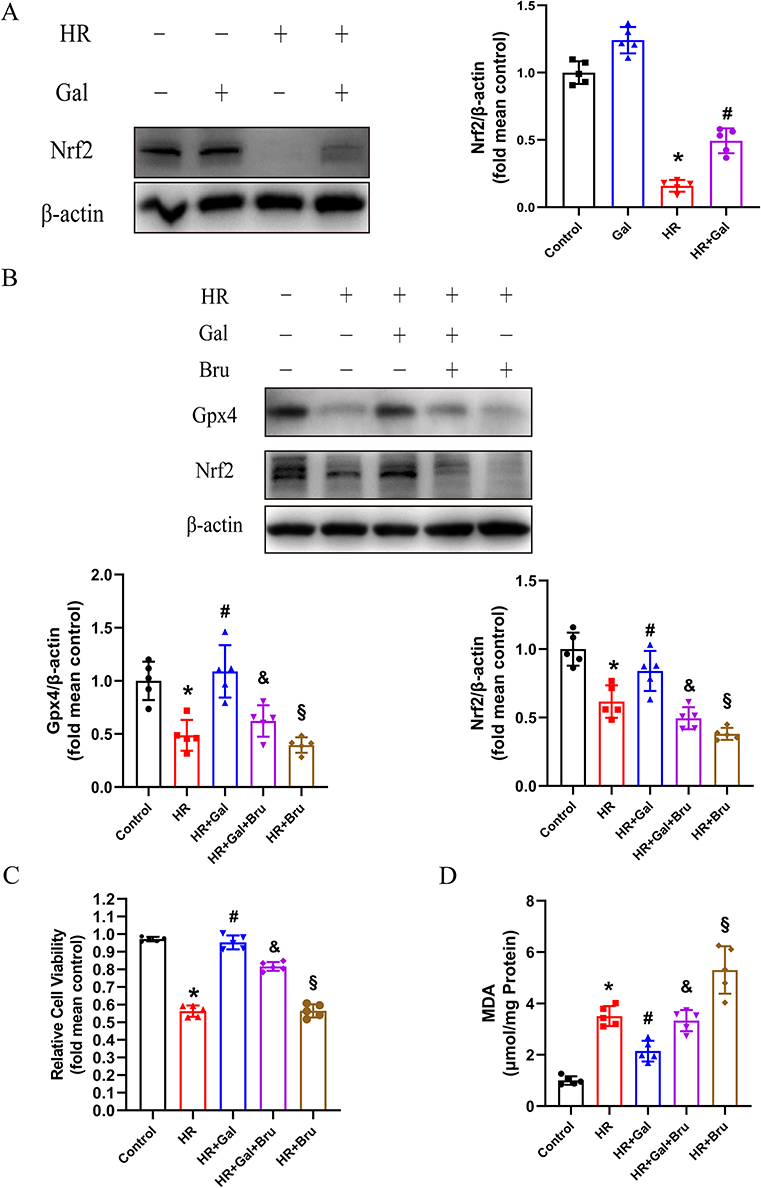 |
Figure 6 Galangin (Gal) inhibited ferroptosis via the Nrf2/Gpx4 pathway activation in vitro. (A) Representative images of Western Blot and quantitative analysis of Nrf2 in each group. Summary data are presented as means ± SD; n = 5. *P < 0.05 vs control group, #P < 0.05 vs HR group. (B) Representative pictures of Western Blot and quantitative analysis of Gpx4 and Nrf2 expression in different groups. Bru at 1 μM was added into NRCs at the onset of HR. (C) Cell vitality in different groups was determined with CCK-8 assay. (D) MDA levels in NRCs subjected to HR and treated with or without Gal or Bru (1 μM). Summary data are presented as means ± SD; n = 5. *P < 0.05 vs control group, #P < 0.05 vs HR group, &P < 0.05 vs HR + Gal group, §P < 0.05 vs HR + Gal + Bru group. Abbreviations: HR, hypoxia-reoxygenation; NRCs, neonatal rat cardiomyocytes; MDA, malondialdehyde; Bru, Brusatol; SD, standard deviation; CCK-8, Cell Counting Kit-8. |
Discussion
This study indicated that (1) ferroptosis was involved in MIRI development; (2) Gal alleviated myocardial injury in HR-treated NRCs and mice after IR injury; (3) Gal inhibited IR-induced myocardial injury through ferroptosis inhibition; (4) Gal suppressed ferroptosis through Nrf2/Gpx4 signaling pathway activation. Figure 7 shows these findings.
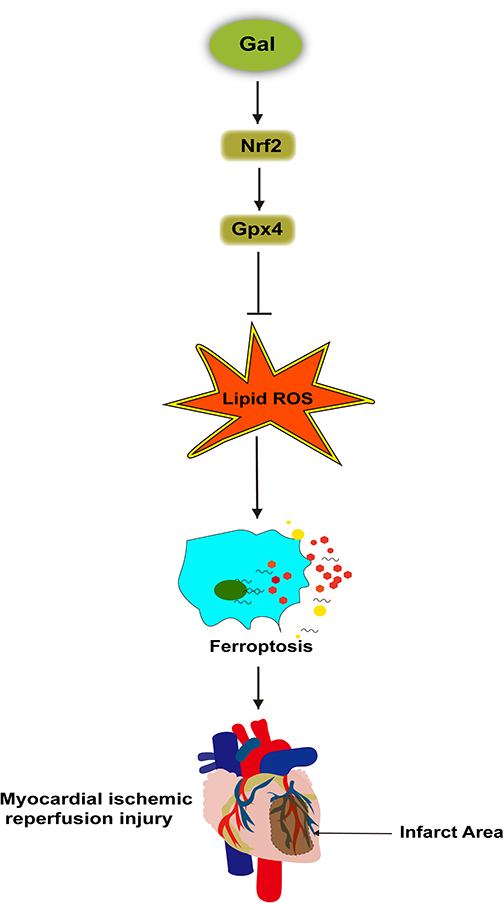 |
Figure 7 Galangin (Gal) stimulated the Nrf2/Gpx4 pathway to inhibit ferroptosis and alleviate myocardial injury after MIR. |
A previous study indicated that cardiomyocyte apoptosis begins during ischemia followed by an obvious increase during reperfusion.36 Nutrient deficiency or stress during MIR triggers autophagy. On one hand, aged or damaged proteins and organelles are degraded into amino acids and fatty acids for recycling and energy generation during baseline autophagy. On the other hand, excessive autophagy may harm the cells.8 Besides, SIRT regulation could inhibit NLRP3-mediated pyroptosis and reduce myocardial I/R injury.7 RIPK3 inhibition or knockdown abolished necroptosis and reduced infarct size after myocardial infarction or MIR.37 Ferroptosis was recently reported to be involved in MIRI progression, and ferroptosis inhibitors decreased infarct size and improved cardiac dysfunction.11,12 Thus, multiple regulated cell death types may be involved in MIRI pathophysiology. Whereas, the roles of different types of programmed cell deaths in MIRI were controversial. This study revealed that apoptosis, ferroptosis, necroptosis, and autophagy activated during MIR and ferroptosis seemed to play a more crucial role than other types of cell death. Consistently, similar findings reported that HR-treated cardiomyocytes demonstrated the highest viability and least cell injury after ferroptosis inhibitor administration compared to other programmed cell death inhibitors.38 Therefore, we aimed to determine effective drugs that inhibit ferroptosis and explore their acting targets to alleviate MIRI.
Alpinia officinarum Hance and Propolis are traditional Chinese herbal medicines widely used in recipes and health food. Recently, Gal, which is an active flavonoid extracted from these medicines, has attracted much attention for its potential extensive pharmacological effects,15,16,39–42 especially in cardio-protection.16,17,43 For example, Gal could prevent isoproterenol-induced myocardial fibrosis,44 reduce blood pressure and improve endothelium-dependent vasodilation impairment in hypertension.45 Gal ameliorates cardiac dysfunction and hypertrophy induced by renal artery stenosis in rats.46 The present study revealed that Gal decreased cell death in NRCs exposed to HR, alleviated cardiac myofibrils damage, reduced myocardial infarct size, and mitigated cardiac dysfunction in mice after IR injury. Moreover, several studies have demonstrated that mitochondria dysfunction is essential in MIRI process.47,48 One of the important characteristics of MIRI is the altered mitochondrial structure and function.49,50 Our study indicated that Gal attenuated mitochondrial damage and restored mitochondrial membrane potential, thereby protecting the cells from MIRI. Altogether, we provided solid evidence that Gal is beneficial against a series of cardiovascular diseases. The relationship between flavonoids and MIRI has been previously studied. In particular, vitexin could protect against MIRI via increasing MFN2 expression, thereby reducing mitochondrial damage and apoptosis.51 Naringenin administration attenuated MIRI by decreasing oxidative stress and endoplasmic reticulum stress via cGMP-PKGIα signaling.52 However, the mechanism by which Gal alleviates MIRI remains unclear and needs further investigation.
Gal was shown beneficial to cardiovascular diseases by mitigating oxidative stress in many reports.17,46 Additionally, Gal could mediate iron ions chelation and attenuate iron deposition in hepatocytes.18,53 Moreover, Gal may exert anti-ferroptosis effects via activating the PI3K/AKT/CREB signaling pathway.54 Thus, we hypothesized that Gal could protect against MIRI by inhibiting ferroptosis, which has not been previously discussed. Our study verified that Gal could suppress ferroptosis by lessening both lipid peroxides level and iron deposition in cardiomyocytes after MIR. Mitochondria are the main sites of ROS production,55 and the protection of mitochondria and clearance of mitochondrial ROS participate in defending against ferroptosis.56 Reportedly, Gal alleviates mitochondria dysfunction in the brain after acute ischemic stroke57 and reduces mitochondria ROS in hyperglycemia-induced rats.58 Consistently, as discussed above, we revealed that Gal restored mitochondrial dysfunction following MIRI. Thereby, Gal may suppress ferroptosis in cardiomyocytes partly by ameliorating mitochondria damage and decreasing mitochondria ROS. Erastin is the most commonly used ferroptosis stimulator. It mainly binds with SLC7A11, which is a key component of the cystine-glutamate antiporter, blocking cystine entering into cells and reducing lipid ROS generation.32 Our findings indicated that Gal attenuates erasin-induced ferroptosis, which further verifies the effects of Gal against ferroptosis in cardiomyocytes.
Nrf2/Gpx4 pathway has exerted protective effects against cardiovascular diseases by inhibiting oxidative stress59,60 and ferroptosis.61 The nuclear translocation of Nrf2 could promote Gpx4 transcription and decrease ROS production, thereby counteracting doxorubicin (DOX)-induced cardiotoxicity.60 Additionally, Nrf2/Gpx4 pathway activation could suppress ferroptosis in cardiomyopathy.62 Moreover, Nrf2/Gpx4 pathway was reported to be involved in MIRI protection. Nrf2 inhibition with ML385 blunted the activity of Gpx4 and enhanced ferroptosis, thereby exacerbating the myocardial injury during MIR.63 Consistently, we revealed that Gal blocked ferroptosis by the Nrf2/Gpx4 signaling pathway stimulation during HR exposure. As previously reported, Gal mitigated cardiac hypertrophy and oxidative stress induced by renal artery stenosis through the Nrf2 expression upregulation.46 Additionally, Gal attenuated cerebral ischemia-reperfusion injury by ferroptosis inhibition through the SLC7A11/GPX4 axis activation.64 The present study revealed that Gal may inhibit MIRI by the Nrf2/Gpx4 signaling pathway activation. Therefore, we speculate that Nrf2/Gpx4-mediated ferroptosis regulation plays a crucial role in cardiovascular disease protection. Nowadays, the association between Nrf2 and IR injury development is controversial. Some studies indicated that Nrf2 expression was downregulated in cardiomyocytes after HR exposure.65,66 However, another study proposed an elevated Nrf2 expression in cardiomyocytes in the HR injury.67 The paradox of Nrf2 expression might be determined by reperfusion duration.68 The nuclear translocation of Nrf2 was enhanced to remove the excess ROS radicals at the beginning of HR, while Nrf2 in cells might be depleted as the reperfusion exposure continues.
However, there are some limitations in this study: Firstly, as cell death types other than ferroptosis were also proved to be involved in the development of MIRI, we did not detect the effects on them with Gal. Secondly, more experiments are needed to prove that which programmed cell death types are most dominant in MIRI. Additionally, we only used Nrf2 inhibitor Brusatol in vitro study, but not in vivo study.
Conclusion
In conclusion, we indicated that Gal attenuates myocardial ischemic reperfusion-induced ferroptosis by targeting Nrf2/Gpx4 signaling pathway. Nutritional or pharmacological intervention with Gal might appear as a promising way to prevent or treat MIRI.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No.82000249 to Jing Yan, No. 81873460 to Aihua Chen.), the Natural Science Foundation of Changsha City (No. kq2202048 to Tao Yang), the Guangdong Basic and Applied Basic Research Foundation (Grant No. 2020A1515111028 to Jing Yan), the Scientific Research Funds of Health Commission of Hunan Province (No. B202303016247 to Tao Yang) and the Natural Science Foundation of Hunan Province (No. 2019JJ80053 to Luping Jiang). Wen Ou made great contributions for Data Curation, Visualization, Investigation, Formal Analysis during the revision.
Disclosure
The authors declare no conflict of interest.
References
1. Lee JM, Rhee TM, Hahn JY, et al. Multivessel percutaneous coronary intervention in patients with ST-segment elevation myocardial infarction with cardiogenic shock. J Am Coll Cardiol. 2018;71:844–856. doi:10.1016/j.jacc.2017.12.028
2. Ibanez B, James S, Agewall S, et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: the task force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J. 2018;39:119–177. doi:10.1093/eurheartj/ehx393
3. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. doi:10.1056/NEJMra071667
4. Heusch G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat Rev Cardiol. 2020;17:773–789. doi:10.1038/s41569-020-0403-y
5. Wang Z, Yang N, Hou Y, et al. L-Arginine-loaded gold nanocages ameliorate myocardial ischemia/reperfusion injury by promoting nitric oxide production and maintaining mitochondrial function. Adv Sci. 2023:e2302123. doi:10.1002/advs.202302123
6. Zhang H, Yin Y, Liu Y, et al. Necroptosis mediated by impaired autophagy flux contributes to adverse ventricular remodeling after myocardial infarction. Biochem Pharmacol. 2020;175:113915. doi:10.1016/j.bcp.2020.113915
7. Ding S, Liu D, Wang L, Wang G, Zhu Y. Inhibiting MicroRNA-29a protects myocardial ischemia-reperfusion injury by targeting SIRT1 and suppressing oxidative stress and NLRP3-mediated pyroptosis pathway. J Pharmacol Exp Ther. 2020;372:128–135. doi:10.1124/jpet.119.256982
8. Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–1010. doi:10.1038/nrm2529
9. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi:10.1016/j.cell.2012.03.042
10. Bai Q, Liu J, Wang G. Ferroptosis, a regulated neuronal cell death type after intracerebral hemorrhage. Front Cell Neurosci. 2020;14:591874. doi:10.3389/fncel.2020.591874
11. Fang X, Wang H, Han D, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116:2672–2680. doi:10.1073/pnas.1821022116
12. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59:298–308. doi:10.1016/j.molcel.2015.06.011
13. Tomar A, Vasisth S, Khan SI, et al. Galangin ameliorates cisplatin induced nephrotoxicity in vivo by modulation of oxidative stress, apoptosis and inflammation through interplay of MAPK signaling cascade. Phytomedicine. 2017;34:154–161. doi:10.1016/j.phymed.2017.05.007
14. Aladaileh SH, Abukhalil MH, Saghir SAM, et al. Galangin activates Nrf2 signaling and attenuates oxidative damage, inflammation, and apoptosis in a rat model of cyclophosphamide-induced hepatotoxicity. Biomolecules. 2019;9:346. doi:10.3390/biom9080346
15. Wang L, Liu H, He Q, et al. Galangin ameliorated pulmonary fibrosis in vivo and in vitro by regulating epithelial-mesenchymal transition. Bioorg Med Chem. 2020;28:115663. doi:10.1016/j.bmc.2020.115663
16. Wang HB, Huang SH, Xu M, et al. Galangin ameliorates cardiac remodeling via the MEK1/2-ERK1/2 and PI3K-AKT pathways. J Cell Physiol. 2019;234:15654–15667. doi:10.1002/jcp.28216
17. Abukhalil MH, Althunibat OY, Aladaileh SH, et al. Galangin attenuates diabetic cardiomyopathy through modulating oxidative stress, inflammation and apoptosis in rats. Biomed Pharmacother. 2021;138:111410. doi:10.1016/j.biopha.2021.111410
18. Salama SA, Elshafey MM. Galangin mitigates iron overload-triggered liver injury: up-regulation of PPARγ and Nrf2 signaling, and abrogation of the inflammatory responses. Life Sci. 2021;283:119856. doi:10.1016/j.lfs.2021.119856
19. Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem. 1999;274:26071–26078. doi:10.1074/jbc.274.37.26071
20. Harada N, Kanayama M, Maruyama A, et al. Nrf2 regulates ferroportin 1-mediated iron efflux and counteracts lipopolysaccharide-induced ferroportin 1 mRNA suppression in macrophages. Arch Biochem Biophys. 2011;508:101–109. doi:10.1016/j.abb.2011.02.001
21. Chorley BN, Campbell MR, Wang X, et al. Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res. 2012;40:7416–7429. doi:10.1093/nar/gks409
22. Fan R, Sui J, Dong X, Jing B, Gao Z. Wedelolactone alleviates acute pancreatitis and associated lung injury via GPX4 mediated suppression of pyroptosis and ferroptosis. Free Radic Biol Med. 2021;173:29–40. doi:10.1016/j.freeradbiomed.2021.07.009
23. Ge MH, Tian H, Mao L, et al. Zinc attenuates ferroptosis and promotes functional recovery in contusion spinal cord injury by activating Nrf2/GPX4 defense pathway. CNS Neurosci Ther. 2021;27:1023–1040. doi:10.1111/cns.13657
24. Ling Y, Chen G, Deng Y, et al. Polydatin post-treatment alleviates myocardial ischaemia/reperfusion injury by promoting autophagic flux. Clin Sci. 2016;130:1641–1653. doi:10.1042/CS20160082
25. Zha WJ, Qian Y, Shen Y, et al. Galangin abrogates ovalbumin-induced airway inflammation via negative regulation of NF-κB. eCAM. 2013;2013:767689. doi:10.1155/2013/767689
26. Shu YS, Tao W, Miao QB, Lu SC, Zhu YB. Galangin dampens mice lipopolysaccharide-induced acute lung injury. Inflammation. 2014;37:1661–1668. doi:10.1007/s10753-014-9894-1
27. Luo Y, Apaijai N, Liao S, et al. Therapeutic potentials of cell death inhibitors in rats with cardiac ischaemia/reperfusion injury. J Cell Mol Med. 2022;26:2462–2476. doi:10.1111/jcmm.17275
28. Zhang HR, Bai H, Yang E, et al. Effect of moxibustion preconditioning on autophagy-related proteins in rats with myocardial ischemia reperfusion injury. Ann Transl Med. 2019;7:559. doi:10.21037/atm.2019.09.66
29. Yan J, Yan JY, Wang YX, et al. Spermidine-enhanced autophagic flux improves cardiac dysfunction following myocardial infarction by targeting the AMPK/mTOR signalling pathway. Br J Pharmacol. 2019;176:3126–3142. doi:10.1111/bph.14706
30. Li N, Wang W, Zhou H, et al. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic Biol Med. 2020;160:303–318. doi:10.1016/j.freeradbiomed.2020.08.009
31. Li Y, Chen B, Yang X, et al. S100a8/a9 signaling causes mitochondrial dysfunction and cardiomyocyte death in response to ischemic/reperfusion injury. Circulation. 2019;140:751–764. doi:10.1161/CIRCULATIONAHA.118.039262
32. Dixon SJ, Patel DN, Welsch M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife. 2014;3:e02523. doi:10.7554/eLife.02523
33. Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi:10.1016/j.cell.2013.12.010
34. Fang X, Cai Z, Wang H, et al. Loss of cardiac ferritin h facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ Res. 2020;127:486–501. doi:10.1161/CIRCRESAHA.120.316509
35. Gai C, Liu C, Wu X, et al. MT1DP loaded by folate-modified liposomes sensitizes erastin-induced ferroptosis via regulating miR-365a-3p/NRF2 axis in non-small cell lung cancer cells. Cell Death Dis. 2020;11:751. doi:10.1038/s41419-020-02939-3
36. Lazou A, Iliodromitis EK, Cieslak D, et al. Ischemic but not mechanical preconditioning attenuates ischemia/reperfusion induced myocardial apoptosis in anaesthetized rabbits: the role of Bcl-2 family proteins and ERK1/2. Apoptosis. 2006;11:2195–2204. doi:10.1007/s10495-006-0292-5
37. Zhou H, Zhu P, Guo J, et al. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol. 2017;13:498–507. doi:10.1016/j.redox.2017.07.007
38. Ma X, Xu J, Gao N, Tian J, Song T. Dexmedetomidine attenuates myocardial ischemia-reperfusion injury via inhibiting ferroptosis by the cAMP/PKA/CREB pathway. Mol Cell Probes. 2023;68:101899. doi:10.1016/j.mcp.2023.101899
39. Huang YC, Tsai MS, Hsieh PC, et al. Galangin ameliorates cisplatin-induced nephrotoxicity by attenuating oxidative stress, inflammation and cell death in mice through inhibition of ERK and NF-kappaB signaling. Toxicol Appl Pharmacol. 2017;329:128–139. doi:10.1016/j.taap.2017.05.034
40. Liang X, Wang P, Yang C, et al. Galangin inhibits gastric cancer growth through enhancing STAT3 mediated ROS production. Front Pharmacol. 2021;12:646628. doi:10.3389/fphar.2021.646628
41. Xuan H, Ou A, Hao S, Shi J, Jin X. Galangin protects against symptoms of dextran sodium sulfate-induced acute colitis by activating autophagy and modulating the gut microbiota. Nutrients. 2020;12:347. doi:10.3390/nu12020347
42. Chen G, Liu J, Jiang L, et al. Galangin reduces the loss of dopaminergic neurons in an LPS-evoked model of parkinson’s disease in rats. Int J Mol Sci. 2017;19:12. doi:10.3390/ijms19010012
43. Prasatthong P, Meephat S, Rattanakanokchai S, et al. Galangin resolves cardiometabolic disorders through modulation of AdipoR1, COX-2, and NF-κB expression in rats fed a high-fat diet. Antioxidants. 2021;10:769. doi:10.3390/antiox10050769
44. Thangaiyan R, Arjunan S, Govindasamy K, Khan HA, Alhomida AS, Prasad NR. Galangin attenuates isoproterenol-induced inflammation and fibrosis in the cardiac tissue of albino wistar rats. Front Pharmacol. 2020;11:585163.
45. Chaihongsa N, Maneesai P, Sangartit W, Potue P, Bunbupha S, Pakdeechote P. Galangin alleviates vascular dysfunction and remodelling through modulation of the TNF-R1, p-NF-κB and VCAM-1 pathways in hypertensive rats. Life Sci. 2021;285:119965. doi:10.1016/j.lfs.2021.119965
46. Chaihongsa N, Maneesai P, Sangartit W, et al. Cardiorenal dysfunction and hypertrophy induced by renal artery occlusion are normalized by galangin treatment in rats. Biomed Pharmacother. 2022;152:113231. doi:10.1016/j.biopha.2022.113231
47. Gumpper-Fedus K, Park KH, Ma H, et al. MG53 preserves mitochondrial integrity of cardiomyocytes during ischemia reperfusion-induced oxidative stress. Redox Biol. 2022;54:102357. doi:10.1016/j.redox.2022.102357
48. Ale-Agha N, Jakobs P, Goy C, et al. Mitochondrial telomerase reverse transcriptase protects from myocardial ischemia/reperfusion injury by improving complex I composition and function. Circulation. 2021;144:1876–1890. doi:10.1161/CIRCULATIONAHA.120.051923
49. Miljkovic JL, Burger N, Gawel JM, et al. Rapid and selective generation of H(2)S within mitochondria protects against cardiac ischemia-reperfusion injury. Redox Biol. 2022;55:102429. doi:10.1016/j.redox.2022.102429
50. Sun S, Yu W, Xu H, et al. TBC1D15-Drp1 interaction-mediated mitochondrial homeostasis confers cardioprotection against myocardial ischemia/reperfusion injury. Metabolism. 2022;134:155239. doi:10.1016/j.metabol.2022.155239
51. Xue W, Wang X, Tang H, et al. Vitexin attenuates myocardial ischemia/reperfusion injury in rats by regulating mitochondrial dysfunction induced by mitochondrial dynamics imbalance. Biomed Pharmacother. 2020;124:109849. doi:10.1016/j.biopha.2020.109849
52. Yu LM, Dong X, Zhang J, et al. Naringenin attenuates myocardial ischemia-reperfusion injury via cGMP-PKGIα signaling and in vivo and in vitro studies. Oxid Med Cell Longev. 2019;2019:7670854. doi:10.1155/2019/7670854
53. Park SS, Bae I, Lee YJ. Flavonoids-induced accumulation of hypoxia-inducible factor (HIF)-1alpha/2alpha is mediated through chelation of iron. J Cell Biochem. 2008;103:1989–1998. doi:10.1002/jcb.21588
54. Chen K, Xue R, Geng Y, Zhang S. Galangin inhibited ferroptosis through activation of the PI3K/AKT pathway in vitro and in vivo. FASEB J. 2022;36:e22569. doi:10.1096/fj.202200935R
55. Zhang Y, Wang Y, Xu J, et al. Melatonin attenuates myocardial ischemia-reperfusion injury via improving mitochondrial fusion/mitophagy and activating the AMPK-OPA1 signaling pathways. J Pineal Res. 2019;66:e12542. doi:10.1111/jpi.12542
56. Li Q, Liao J, Chen W, et al. NAC alleviative ferroptosis in diabetic nephropathy via maintaining mitochondrial redox homeostasis through activating SIRT3-SOD2/Gpx4 pathway. Free Radic Biol Med. 2022;187:158–170. doi:10.1016/j.freeradbiomed.2022.05.024
57. Li S, Wu C, Zhu L, et al. By improving regional cortical blood flow, attenuating mitochondrial dysfunction and sequential apoptosis galangin acts as a potential neuroprotective agent after acute ischemic stroke. Molecules. 2012;17:13403–13423. doi:10.3390/molecules171113403
58. Aloud AA, Veeramani C, Govindasamy C, Alsaif MA, Al-Numair KS. Galangin, a natural flavonoid reduces mitochondrial oxidative damage in streptozotocin-induced diabetic rats. Redox Rep. 2018;23:29–34. doi:10.1080/13510002.2017.1365224
59. Zhang Y, Wu Q, Liu J, et al. Sulforaphane alleviates high fat diet-induced insulin resistance via AMPK/Nrf2/GPx4 axis. Biomed Pharmacother. 2022;152:113273. doi:10.1016/j.biopha.2022.113273
60. Lu Z, Liu Z, Fang B. Propofol protects cardiomyocytes from doxorubicin-induced toxic injury by activating the nuclear factor erythroid 2-related factor 2/glutathione peroxidase 4 signaling pathways. Bioengineered. 2022;13:9145–9155. doi:10.1080/21655979.2022.2036895
61. Wei Z, Shaohuan Q, Pinfang K, Chao S. Curcumin attenuates ferroptosis-induced myocardial injury in diabetic cardiomyopathy through the Nrf2 pathway. Cardiovasc Ther. 2022;2022:3159717. doi:10.1155/2022/3159717
62. Wang Y, Yan S, Liu X, et al. PRMT4 promotes ferroptosis to aggravate doxorubicin-induced cardiomyopathy via inhibition of the Nrf2/GPX4 pathway. Cell Death Differ. 2022;2022:1.
63. Wang Z, Yao M, Jiang L, et al. Dexmedetomidine attenuates myocardial ischemia/reperfusion-induced ferroptosis via AMPK/GSK-3β/Nrf2 axis. Biomed Pharmacother. 2022;154:113572. doi:10.1016/j.biopha.2022.113572
64. Guan X, Li Z, Zhu S, et al. Galangin attenuated cerebral ischemia-reperfusion injury by inhibition of ferroptosis through activating the SLC7A11/GPX4 axis in gerbils. Life Sci. 2021;264:118660. doi:10.1016/j.lfs.2020.118660
65. Zheng D, Liu Z, Zhou Y, et al. Urolithin B, a gut microbiota metabolite, protects against myocardial ischemia/reperfusion injury via p62/Keap1/Nrf2 signaling pathway. Pharmacol Res. 2020;153:104655. doi:10.1016/j.phrs.2020.104655
66. Li P, Lin N, Guo M, Huang H, Yu T, Zhang L. REDD1 knockdown protects H9c2 cells against myocardial ischemia/reperfusion injury through Akt/mTORC1/Nrf2 pathway-ameliorated oxidative stress: an in vitro study. Biochem Biophys Res Commun. 2019;519:179–185. doi:10.1016/j.bbrc.2019.08.095
67. Zhang L, Wang Y, Li C, et al. Dan hong injection protects against cardiomyocytes apoptosis by maintaining mitochondrial integrity through Keap1/nuclear factor erythroid 2-Related factor 2/JNK pathway. Front Pharmacol. 2020;11:591197. doi:10.3389/fphar.2020.591197
68. Zhang H, Liu Y, Cao X, et al. Nrf2 promotes inflammation in early myocardial ischemia-reperfusion via recruitment and activation of macrophages. Front Immunol. 2021;12:763760. doi:10.3389/fimmu.2021.763760
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.