")
Back to Journals » OncoTargets and Therapy » Volume 12
G-protein-signaling modulator 2 expression and role in a CD133+ pancreatic cancer stem cell subset
Authors Dang SC , Qian XB, Jin W, Cui L, Chen JX, Gu M
Received 17 September 2018
Accepted for publication 20 December 2018
Published 23 January 2019 Volume 2019:12 Pages 785—794
DOI https://doi.org/10.2147/OTT.S187670
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr XuYu Yang
Sheng-Chun Dang,1 Xiao-Bao Qian,1 Wei Jin,2 Lei Cui,1 Ji-Xiang Chen,1 Min Gu3
1Department of General Surgery, Affiliated Hospital of Jiangsu University, Zhenjiang, Jiangsu 212001, People’s Republic of China; 2Department of Obstetrics and Gynecology, ChangShu No. 2 People’s Hospital, Changshu, Jiangsu 215500, People’s Republic of China; 3Department of Oncology, Zhenjiang Hospital of Traditional Chinese and Western Medicine, Zhenjiang, Jiangsu 212001, People’s Republic of China
Background: To investigate the expression and role of G-protein-signaling modulator 2 (GPSM2) in a CD133+ pancreatic stem cell subset.
Materials and methods: Pancreatic cancer stem cells (PCSCs) from the cell line PANC-1 were sorted into CD133+ and CD133- subsets by flow cytometry. The tumorigenic potential of the subsets was assessed by subcutaneous tumor formation experiments in nude mice. Differential expression of GPSM2 was examined by real-time quantitative-PCR (qPCR) and Western blotting. To silence GPSM2 expression, a shRNA lentiviral vector targeting GPSM2 was constructed and stably transfected into CD133+ PCSCs. The inhibitory efficiency of the GPSM2 gene was verified by qPCR and Western blotting. The proliferation, colony formation, and migration abilities of the transfected CD133+ pancreatic cancer cells were assessed by MTT, soft agar colony formation, and Transwell assays.
Results: CD133+ and CD133- cell subsets were successfully isolated from PANC-1 cells. The CD133+ subset subcutaneously formed tumors in nude mice that were significantly bigger (343.05±57.59 mm3 vs 176.86±32.58 mm3, P<0.01) and denser (4.13±0.37 g vs 1.07±0.21 g, P<0.01) than those of the CD133- group. The GPSM2 mRNA and protein expression was significantly higher in CD133+ cells than in CD133- cells. Stable downregulation of GPSM2 expression reduced the proliferation, colony formation, and migration abilities of CD133+ PANC-1 cells (P<0.05).
Conclusion: The CD133+PANC-1 cells have obvious stem cell characteristics and increased GPSM2 expression. Downregulation of GPSM2 significantly reduces the proliferation and migration ability of the cells. Therefore, GPSM2 may provide an important target for regulating PCSCs.
Keywords: neoplastic stem cells, GPSM2 protein, human, lentivirus, flow cytometry, CD133 antigen, cell proliferation, migration
Introduction
Pancreatic cancer is a common malignancy of the digestive system, with a 5-year survival of <5%.1 Rapid invasion, high metastasis, and resistance to chemotherapeutic drugs are important causes of death in pancreatic cancer patients.2,3 Recent studies have shown that tumors contain a subset of cells called cancer stem cells (CSCs). This subset has the ability to self-renew and differentiate into heterogeneous tumors by asymmetric cell division (ACD),4 which is a process that is specific to stem cells.5–7 CSCs are highly resistant to conventional chemotherapeutic drugs and are associated with tumor recurrence and metastasis.8 Pancreatic cancer stem cells (PCSCs), which were first discovered in 2007,9 play an important role in pancreatic cancer metastasis and drug resistance.10–12 Although increasingly more studies have supported the existence of PCSCs, the mechanisms for regulating PCSCs remain to be elucidated.
G-protein-signaling modulator 2 (GPSM2) is an important factor that regulates the localization and mitotic spindle pole orientation of cells.13 Fukukawa et al have shown that GPSM2 is upregulated in breast cancer tissues and plays an essential role in cell division.14 In our previous study, we demonstrated that GPSM2 is overexpressed in pancreatic cancer tissues. Furthermore, GPSM2 overexpression has been correlated with the TNM staging, tumor differentiation, and prognosis of pancreatic cancer.15 In vitro experiments have confirmed that overexpression of GPSM2 enhances the migration ability of human pancreatic cancer cells,16 though its specific function in PCSCs has not been characterized. To further explore the contribution of GPSM2 to the regulation of pancreatic cancer, we assessed the differential expression of GPSM2 in PCSCs and evaluated its effect on the proliferation and migration abilities of PCSCs.
Materials and methods
Reagents and equipment
The following materials were used in this study: High DMEM (HyClone, Logan, UT, USA, catalog #: SH30022.01B), FBS (GIBCO, Rockville, MD, USA, catalog #: 16400–044), penicillin-streptomycin (Beijing Xiasi Biotechnology, Beijing, China, catalog #: SV30010), puromycin, trypsin (Worthington Biochemical Co., Lakewood, NJ, USA, catalog #: LS003703), Lipofectamine 2000 Transfection Reagent (Invitrogen, Grand Island, NY, USA, catalog #: 11668-027), fluorescein isothiocyanate-labeled mouse anti-human CD133 monoclonal antibodies (Thermo Fisher Scientific, Waltham, MA, USA, catalog #: 11-1339-42) and its corresponding isotype control mouse IgG2b antibodies (Thermo Fisher Scientific, catalog #: 11-4732-81), DNA Gel Recovery Kits (Tiangen Biotech Co., Ltd., Beijing, China, catalog #: DP209-03), AxyPrep plasmid preparation kits (Axygen Biosciences, Union City, CA, USA, catalog #: AP-MD-P-25), lentiviral vector LV-3 (pGLVH1/GFP+Puro), pHelper1.0, pHelper2.0 (GenePharma Co., Ltd., Shanghai, China), restriction endonucleases BamHI and EcoRI and T4 DNA ligase (New England Biolab, Ipswich, MA, USA), RT-qPCR reagent kits, TRIZOL, RNA lysis solution (Nanjing Biosky Co., Ltd., Nanjing, China), mouse anti-human GPSM2 antibody (Zhenjiang Hope Biotechnology Co., Ltd., Zhenjiang, China), anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (Zhenjiang Hope Biotechnology Co., Ltd., Zhenjiang, China), horseradish peroxidase-conjugated goat anti-mouse IgG antibody (Sunlidobio, Nanjing, China, catalog #: AbmaRT- M21001L), phenylmethylsulfonyl fluoride (Nanjing Wohong catalog #: 329-98-6), Pepstatin A (Shanghai Yuanye Bio-Technology Co., Ltd., Shanghai, China, catalog #: 26305-03-3, lot:10557), and diethylpyrocarbonate-treated water (Beijing Biotopped Life Science Co., Ltd., Amresco catalog #: E174). Primers used in this study were synthesized by Sangon Biotech (Shanghai) Co., Ltd. (Shanghai, China).
Experimental animals and cell lines
Six-week-old BALB/c mice weighing 18–20 g (n=16) were housed in a specific-pathogen-free grade animal facility with 24°C temperature and 40% humidity and fed sterile food and autoclaved water. Surgical protocols for all animals were approved by the Animal Experimental Committee of Jiangsu University (Jiangsu Province, China) and were performed in accordance with the Guide for the Care and Use of Laboratory Animals issued by the National Institute of Health.
Experimental cell lines used in this study included PANC-1 human pancreatic cancer cells (The Cell Bank of Type Culture Collection of Chinese Academy of Sciences, catalog #: TCHu98) and 293 T cells (The Cell Bank of Type Culture Collection of Chinese Academy of Sciences, catalog #: GNHu17). These two cell lines were incubated in DMEM containing 10% FBS at 37°C in 5% CO2.
Sorting of PCSCs by flow cytometry
PANC-1 human pancreatic cancer cells in logarithmic growth phase were harvested by washing twice in PBS, digestion with 0.25% trypsin, and low-speed centrifugation at 1,000 rpm for 5 minutes, followed by resuspension in 200 μL high-quality DMEM at 1×106 cells/mL. Twenty microliters of CD133-fluorescein isothiocyanate antibody or mouse IgG2b antibody were added to 1×106 cells in the isotype control group, followed by incubation at 4°C in the dark for 2 hours and centrifugation at 1,000 rpm for 5 minutes. After discarding the supernatant and washing twice in PBS, the cell pellets were resuspended in 500 μL PBS to prepare single cell suspensions and filtered through a nylon mesh filter (40-μm pore size) before cell sorting by flow cytometry. The cell sorting was repeated, and the twice-sorted cells were recovered. The purity of the CD133+ cells was verified to be >95% in three independent experiments. All of the aforementioned experimental procedures were performed under aseptic conditions.
Analysis of subcutaneous tumor formation
CD133−PANC-1 and CD133+PANC-1 cells were independently resuspended in a 1:1 mixture of PBS and BD Matrigel at 5×106 cells/mL. Six-week-old nude mice were randomly divided into two groups (n=8 mice per group): the CD133−PANC-1 and the CD133+PANC-1 group. A 1-mL syringe was used to inoculate 0.20 mL of cell suspension into the axillae of the mice (1×106 cells/mL per mouse). The sham group was injected with 0.2 mL of 1:1 PBS and BD Matrigel per mouse. The tumor volumes were calculated based on the long (L) and short diameters (I) (mm) measured by Vernier calipers according to the formula V = (L × I 2/2). Tumor growth curves were plotted using the mean ± SD of the tumor volumes. All nude mice were sacrificed by cervical dislocation 4 weeks after injection to harvest and measure the weight of tumors.
Assessment of GPSM2 protein expression by Western blotting
Cells were collected and washed twice in PBS, followed by the addition of 400 μL lysis buffer and sonication for 40 minutes on ice. After 12,000 rpm centrifugation at 4°C for 15 minutes, the supernatant was collected and the protein concentration and purity were evaluated. The extracted proteins were denatured at 100°C for 5 minutes, separated by 10% SDS-PAGE, and subsequently transferred to a nitrocellulose membrane. Membranes were blocked in tris-buffered saline with Tween-20 containing 5% skim milk for 2 hours and then incubated with 1:200 anti-GPSM2 antibody overnight at 4°C. After thorough washing, the membranes were incubated with 1:2,000 horseradish peroxidase-conjugated goat anti-mouse secondary antibody at room temperature for 1 hour. The membranes were then washed in PBS and developed in 3,3′-diaminobenzidine tetrahydrochloride solution. GAPDH (1:400 anti-GAPDH antibody) was used as an internal reference for Western blotting. Quantity One analysis software (Bio-Rad, Berkeley, CA, USA) was used for analyzing the gray value of protein bands.
Assessment of GPSM2 mRNA expression by RT-qPCR
RNA was extracted by the Trizol method, reverse transcribed at 42°C for 50 minutes and pre-denatured at 95°C for 5 minutes to obtain single-stranded cDNA template. Fluorescence-based RT-qPCR (25 μL final volume) was performed as follows: 95°C denaturation for 3 minutes; and 40 cycles of 95°C denaturation for 30 seconds, 55°C annealing for 20 seconds, and 72°C elongation for 20 seconds. The GPSM2 and GAPDH primer sequences for RT-qPCR (designed using Primer5 software) are shown in Table 1. The relative mRNA expression of GPSM2 was calculated using the formula RQ=2−ΔΔCT. The silencing efficiency of the GPSM2 shRNA was calculated by the following equation: Silencing efficiency (%) = (1 − relative expression of GPSM2 mRNA) × 100%.
 | Table 1 GPSM2 and GAPDH primers |
Construction of recombinant plasmids
GPSM2 shRNA for interference of GPSM2 RNA was designed as an siRNA sense-loop-antisense strand as follows: GTTCTCCGAACGTGTCACGTT-tcaagag-AACGTGACACGTTCGGAGAAC-tt. The target sequence (5′-TTCTCCGAACGTGTCACGT-3′) was selected using the Ambion online tool. To prepare the GPSM2 shRNA, primer sequences were designed with BamHI and EcoRI restriction sites at the 5′ end as follows: 5′-GATCCGTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAACTTTTTTG-3′ (forward primer) and 5′-AATTCAAAAAAGTTCTCCGAACGTGTCACGTTCTCTTGAAACGTGACACGTTCGGAGAACG-3′ (reverse primer). The control shNC was designed with 5′-TTCTCCGAACGTGTCACGT-3′ as the target sequence, which was not predicted to match any known genes according to NCBI BLAST. Similarly, primer sequences for the control shRNA were designed as 5′-GATCCGTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAACTTTTTTG-3′ (forward primer) and 5′-AATTCAAAAAAGTTCTCCGAACGTGTCACGTTCTCTTGAAACGTGACACGTTCGGAGAACG-3′ (reverse primer). All primers were synthesized commercially. To construct recombinant plasmids, the lentiviral core vector pGLV3/H1/GFP + Puro was digested with BamHI and EcoRI and then ligated with double-stranded shRNA and shNC.
Construction of stable shGPSM2 cells
The GPSM2 shRNA and shNC lentiviral vectors were co-transfected into 293 T cells with the packaging plasmids pHelper1.0 and pHelper2.0. Viral supernatants were collected and filtered at 24 and 48 hours after the transfection. To prepare stable knockdown cells, CD133+ PCSCs were inoculated into a 10-cm culture dish 24 hours before transfection and cultured in DMEM containing 10% FBS and penicillin–streptomycin. To ensure cell viability, the medium was replaced with 8 mL fresh DMEM containing 10% FBS without penicillin–streptomycin 2 hours before the transfection and incubated until 60%–70% confluency. The cells were then divided into three groups: Blank control, shNC, and shGPSM2 groups, which were co-incubated with PBS + puromycin plasmid, shNC virus solution + puromycin plasmid, and shGPSM2 virus solution + puromycin plasmid, respectively. The cells were incubated at 37°C in 5% CO2 for 4 hours. After co-transfection, the cells were incubated with fresh complete medium, which was replaced with fresh complete medium containing 3 μg/mL puromycin every 2 days for 45 days.
Assessment of cell viability by MTT assay
Stably transfected GPSM2 shRNA cells (the shGPSM2 group), stably transfected shNC control cells (the shNC group), and non-transfected CD133+ negative control stem cells (the Blank group) were seeded at a density of 5,000 cells/well in triplicate in 96-well plates. Wells without cells were used as an additional control. The cells were incubated for 24, 48, or 72 hours, and then 10 μL Thiazolyl blue (5 g/L MTT) working solution was added. The cells were continuously incubated at 37°C for 4 hours, followed by vortexing in dimethyl sulfoxide for 10 minutes. The absorbance (OD) at 595 nm wavelength was measured using a microplate reader. Cell growth inhibition rates were calculated as follows: Inhibition rate = ([OD of the control well − OD of the experimental well]/[OD of the control well − OD of the blank well]). MTT assays were repeated 3 times.
Soft agar colony formation assay
To prepare plates for colony formation assay, fully dissolved agar solution (2%) was diluted at 1:4 ratio in pre-warmed fresh complete culture medium at 37°C. The final mixture (2 mL of 0.5% agar medium) was added to culture dishes and was solidified at room temperature. Then, cells were digested in trypsin solution without EDTA, washed twice in PBS, and adjusted to a density of 1×103 cells/mL. Single-cell suspension 1 mL was combined with 1 mL of 0.5% agar medium to yield a 0.25% semi-solid agar medium (soft agar medium), which was immediately poured over the bottom layer and allowed to solidify at room temperature. The cells in the agar dish were incubated at 37°C in 5% CO2 for 2 weeks and then were stained with 0.4% crystal violet solution. The number of colonies (representing >10 cells) in four randomly selected fields (100× magnification) was counted in each group. Colony formation assays were repeated twice.
Assessment of migration ability by Transwell assay
Cells were trypsinized to prepare single-cell suspensions at 2×105 cells/mL. A total of 800 μL of medium containing 10% serum was added to the lower chambers of 24-well plates, and 150 μL cell suspension was added to the upper Transwell chambers. The cells were cultured at 37°C in 5% CO2 for 24 hours. Subsequently, the upper Transwell chambers were carefully removed with tweezers, and the liquid was aspirated. The upper chambers were then transferred to wells containing 800 μL 70% ethanol at room temperature for 10–30 minutes, washed twice in PBS, and transferred to a well containing 100% ethanol at room temperature for 20 minutes to fix the cells. Subsequently, the slides were transferred to a new well and stained with 800 μL 2% crystal violet solution at room temperature for 15–30 minutes. After washing the chamber with PBS, the cells on the bottom surface of the upper Transwell chamber were carefully wiped off with a cotton swab. The chamber membranes were completely air dried and then mounted on a glass slide. The cells were photographed and counted in four randomly selected fields using an inverted microscope (100×). These experiments were repeated twice.
Statistical analysis
Data are presented as mean ± SD (x– ± s). SPSS 23.0 (IBM SPSS Inc., Chicago, IL, USA) and GraphPad 5.0 software were used for statistical analysis. The independent sample t-test was used to assess the differences between paired independent samples. One-way ANOVA was used for comparison between groups. The least significant difference method was used for pairwise comparisons. *P<0.05 and **P<0.01 were considered to represent statistically significant differences.
Results
PANC-1 cells can be sorted into CD133− and CD133+ cell subsets that have different stem cell characteristics
To evaluate the potential role of GPSM2 in PCSCs, we first sorted PANC-1 cells into CD133+ and the CD133− subsets. Flow cytometry assays verified that the CD133+ stem cell subset had a much higher CD133+ ratio than the CD133− subset did (96.34%±1.25% vs 0.17±0.10; Figure 1A and B). To confirm that CD133 positivity is associated with the enhanced tumorigenicity of these cells, we performed a subcutaneous tumor formation experiment in nude mice. After 2 weeks of tumor cell inoculation, the tumors derived from the CD133+ subset were visibly larger than those derived from the CD133− subset (Figure 1C). The increased size of the CD133+ tumors could be observed from weeks 2–4, with the most obvious difference in volume after 4 weeks (343.05±57.59 mm3 vs 176.86±32.58 mm3, P<0.01; Figure 1D). At the end of the 4-week period, tumors were isolated from each group of mice (Figure 1E). Our results verified that the CD133+ tumors had a much greater mass than the CD133− tumors did (4.13±0.37 g vs 1.07±0.21 g, P<0.01; Figure 1F). These results suggest that the pancreatic cancer cells of the CD133+ subset have stem cell characteristics.
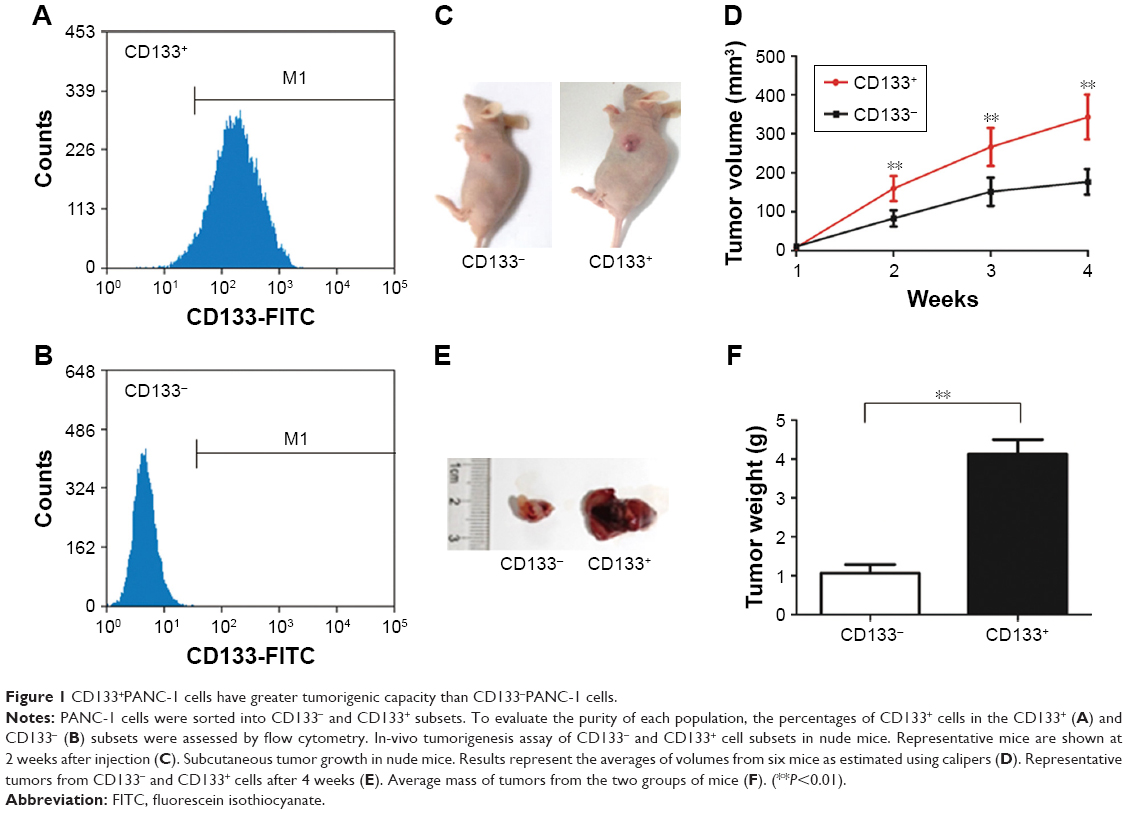 | Figure 1 CD133+PANC-1 cells have greater tumorigenic capacity than CD133−PANC-1 cells. |
CD133+PANC cells express elevated levels of GPSM2 mRNA and protein
To determine whether CD133 positivity in PANC-1 cells correlates with the expression of GPSM2, we assessed the GPSM2 mRNA and protein levels in the CD133− and CD133+ subsets. RT-qPCR results showed that the relative expression of GPSM2 mRNA was significantly higher in the CD133+ subset than in the CD133− subset (0.92±0.07 vs 0.09±0.04, P<0.01) or the unsorted PANC-1 cells (0.92±0.07 vs 0.72±0.17, P<0.05) (Figure 2A). Western blotting verified these findings for CD133+ compared with CD133− cells (0.90±0.08 vs 0.10±0.05, P<0.01) (Figure 2B). These results suggest that CD133+PANC cells express elevated levels of GPSM2.
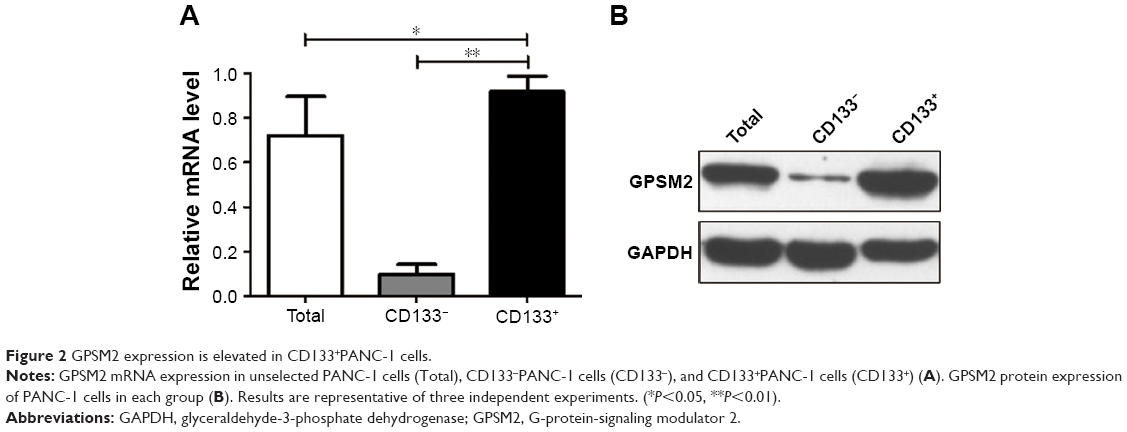 | Figure 2 GPSM2 expression is elevated in CD133+PANC-1 cells. |
GPSM2 downregulation inhibits the viability of CD133+ stem cells
To assess the function of shGPSM2 in CD133+PANC-1 cells, we prepared shGPSM2 and shNC viral particles and stably transfected them into CD133+PCSCs. Efficient knockdown was verified both by RT-qPCR (0.02±0.00 vs 0.86±0.07, P<0.01; 97.69%±0.00% silencing efficiency) (Figure 3A) and Western blotting (0.05±0.03 vs 0.89±0.13, P<0.01, Figure 3B). We performed MTT assays to compare the proliferation rate of the shGPSM2 cells to the proliferation rates of shNC and untransfected (Blank) cells. At 48 and 72 hours after the inoculation, the proliferation rate of the shGPSM2 cells was significantly weaker than that of the shNC cells or Blank cells (P<0.05, Figure 3C). Moreover, the cell growth inhibition rate of the shGPSM2 group was significantly higher than that of the shNC group (Table 2). These results suggest that GPSM2 downregulation inhibits PCSC viability.
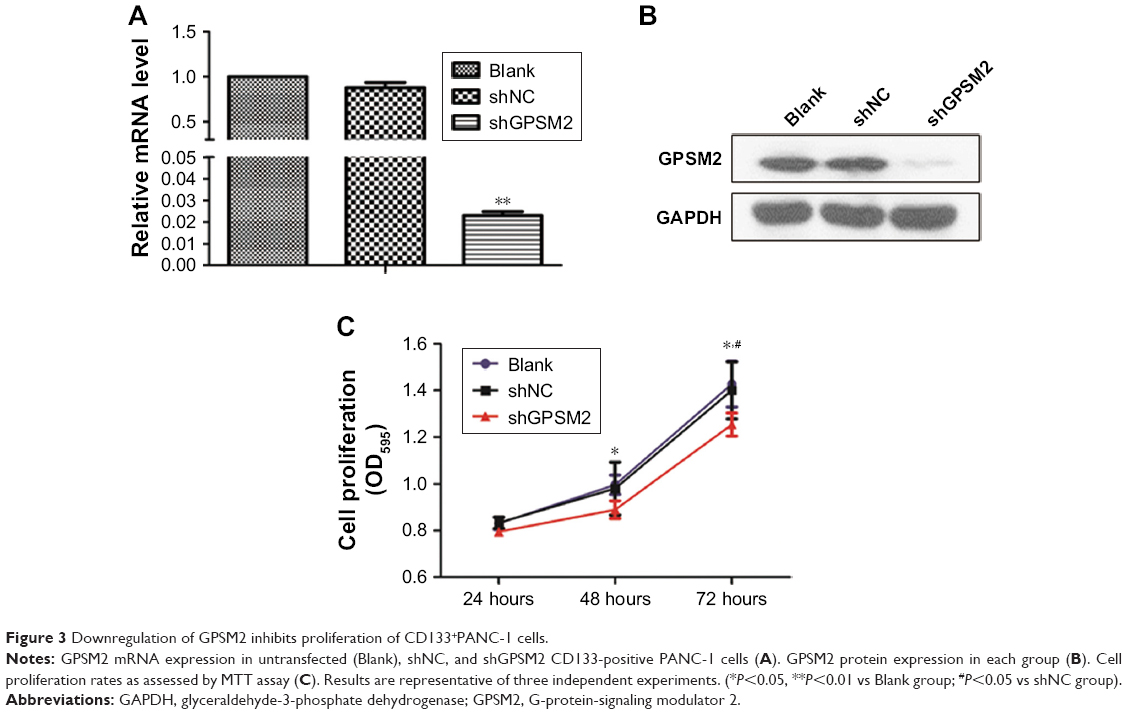 | Figure 3 Downregulation of GPSM2 inhibits proliferation of CD133+PANC-1 cells. |
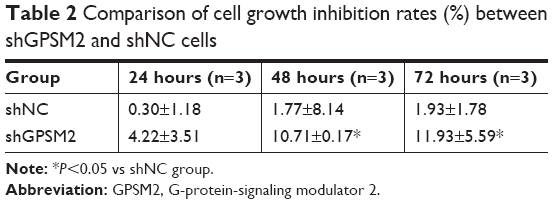 | Table 2 Comparison of cell growth inhibition rates (%) between shGPSM2 and shNC cells |
GPSM2 downregulation inhibits the colony formation ability of CD133+ stem cells
To determine whether GPSM2 may have a role in mediating the colony formation ability of PCSCs, we performed soft agar colony formation assays. The number of colonies formed by the shGPSM2 cells was significantly lower than that of the stably transfected shNC cells (17.33±2.52 vs 51.67±4.04, P<0.05) and the Blank control cells (17.33±2.52 vs 54.33±3.51, P<0.05) (Figure 4). There were no significant differences in the number of colonies formed in the shNC and Blank groups (P>0.05). These results suggest that GPSM2 downregulation inhibits the colony formation ability of PCSCs.
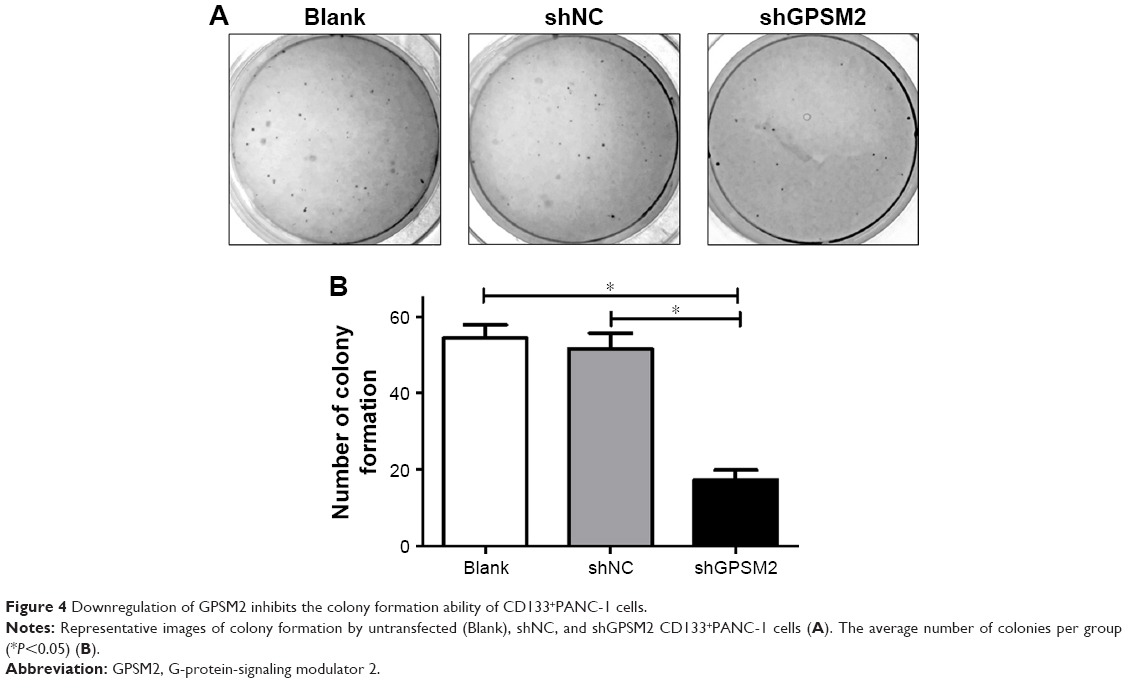 | Figure 4 Downregulation of GPSM2 inhibits the colony formation ability of CD133+PANC-1 cells. |
GPSM2 downregulation inhibits the migration ability of CD133+ stem cells
To determine whether GPSM2 also has a role in mediating the migration ability of PCSCs, we performed Transwell assays. The number of cells migrating to the lower chamber for the shGPSM2 group was significantly lower than for the shNC group (224.00±46.47 vs 403.3±54.29, P<0.05) and the Blank group (224.00±46.47 vs 389.00±77.16, P<0.05) (Figure 5). No significant differences were found between the shNC and the Blank groups (P>0.05). These results suggest that GPSM2 downregulation inhibits the migration ability of PCSCs.
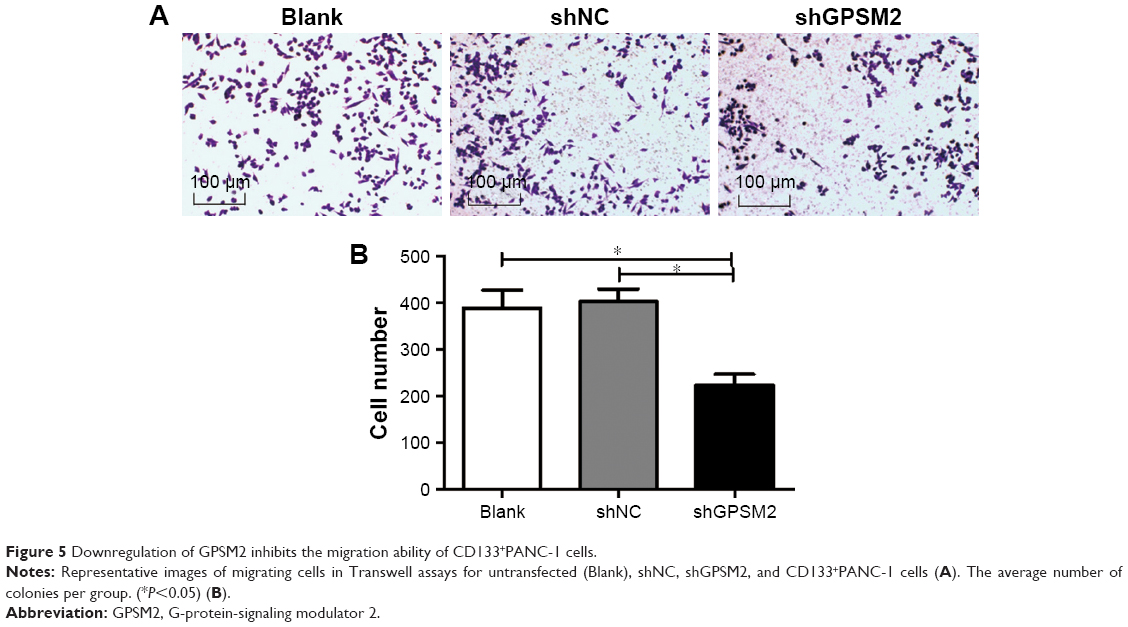 | Figure 5 Downregulation of GPSM2 inhibits the migration ability of CD133+PANC-1 cells. |
Discussion
Pancreatic cancer is highly malignant and is one of the most deadly cancers in the world. There will be 458,918 new pancreatic cancer cases and 432,242 cancer deaths worldwide in 2018 according to GLOBOCAN 2018.17 The incidence rate of pancreatic cancer has shown significant increase in China in recent years.18 Although the surgical treatment of pancreatic cancer has improved, the 5-year survival rate is still <5%.19 Furthermore, therapies that target different genes have shown limited efficacy.20 The high mortality rate of pancreatic cancer is due, in part, to the difficulty in obtaining early diagnoses,21 the resistance to radiotherapy and chemotherapy,22 and the prevalence of distant metastases,21 which are key problems in the treatment of pancreatic cancer.
Notably, the presence of CSCs is closely related to the mechanism of drug resistance and metastasis.22 As early as in 1973, Moore et al first proposed the theory of CSCs.23 According to current understanding, the cell subsets in tumor tissues that are involved in tumor formation, malignant transformation, self-repairing capacities, and other specialized properties, are known as CSCs or tumor-initiating cells.24–26 Bonnet and Dick discovered the first CSCs in leukemia in 1997. CSCs were subsequently discovered in breast cancer, which was the first solid tumor shown to have them.27 To date, CSCs have been found in a variety of tumor tissues, including glioblastoma,28 pancreatic cancer,9,29 melanoma,30 prostate cancer,31 liver cancer,32 gastric cancer,33 breast cancer,34 and colorectal cancer.35 Given the ability of self-renewal and multi-directional differentiation of CSCs, they have a key role in cancer invasion, drug resistance, and recurrence.36–39 Therefore, CSCs may provide attractive therapeutic targets.
For pancreatic cancer, numerous studies have shown that CSCs account for only 0.2%–0.8%,12 which makes their identification and isolation exceedingly challenging. Recognized tumor stem cell markers include CD44, CD24, CD133, epithelial specific antigen, aldehyde dehydrogenase, and c-Met.10,40 Among these, CD133 has been confirmed to be an important marker of PCSCs. Hermann et al isolated CD133+ cells from human pancreatic cancer cells by flow cytometry, and CD133+ cells could be serially passaged in nude mice, indicating that CD133+ pancreatic cancer cells have high and sustained carcinogenic capacity.41 Therefore, we selected CD133 as a surface marker for screening PCSCs in this study and used flow cytometry to sort CD133+ and CD133− cell subsets in human PANC-1 cells. These cell subsets were assessed by subcutaneous tumor formation experiments in nude mice, which verified that the tumor volume and mass of the CD133+ group were significantly larger than that of the CD133− group, which confirms that the CD133+ cell subsets had stem cell characteristics and is consistent with the results of Hermann et al.
Research has shown that when CDCs undergo ACD, they also undergo symmetric cell division. ACD plays an important role in maintaining CSC subsets and is associated with tumor resistance, invasion, and metastasis.42,43 GPSM2, as a G-protein-signaling modulator, is involved in cell migration and plays an important role in mitotic spindle orientation.44 In addition, GPSM2 mutations cause autosomal recessive Chudley–McCullough syndrome, leading to congenital sensorineural deafness,13,45–48 which is due to the role of GPSM2 in establishing planar polarity and spindle orientation during ACD. In addition, studies of Drosophila neuroblasts have shown that GPSM2 binds to the Drosophila NuMA homolog, Mud, providing an attachment point for tubulin to regulate spindle orientation to promote ACD.5,6 Moreover, GPSM2 directly interacts with NuMA in mouse mammary stem cells, resulting in ACD of mammary stem cells.49 In this study, Western blotting and RT-qPCR showed that GPSM2 protein and mRNA expression in the CD133+ cell subsets was significantly higher than in the CD133− cell subsets, suggesting that GPSM2 is overexpressed in PCSCs and might be involved in the regulation of PCSC growth and differentiation.
To further evaluate the effect of GPSM2 on PCSCs, we prepared stable GPSM2 shRNA knockdown CD133+ cells. MTT and colony formation assays showed that silencing of GPSM2 expression reduced the proliferation and colony formation ability of CD133+PANC-1 cells. The shGPSM2 cells also had reduced migration ability in Transwell assays. These results suggest that downregulation of GPSM2 expression effectively inhibits the proliferation and migration abilities of PCSCs. Moreover, in vitro studies suggested that GPSM2 accelerates cell growth, cell cycle, migration, and invasion, and inhibits apoptosis by activating the PI3K/AKT pathway in hepatocellular carcinoma.50 Further experimentation may identify the specific regulatory mechanisms by which GPSM mediates these processes.
Because PCSC growth relies on the regulation of the tumor microenvironment, the effects of GPSM2 in the body and tumor tissues might differ from the in vitro effects observed in this study. However, our results provide evidence for the differential expression of GPSM2 in PCSCs, as well as its effects on the proliferation and migration abilities of PCSCs. This study provides a theoretic basis for further investigation of the underlying mechanism of GPSM2 in regulating PCSCs, which could suggest a therapeutic direction for the development of novel treatments for pancreatic cancer.
Acknowledgments
The Six Talent Peaks Project in Jiangsu Province (No 2016-WSN-007), the Jiangsu 333 Project Foundation (No BRA2017560), the Zhenjiang Science and Technology Committee (No SH 2018061), Changshu Science and Technology Committee (No 20170016) and the Changshu Health Commisson Science and Technology Project (No csws201801) supported this study.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. | ||
Thota R, Pauff JM, Berlin JD. Treatment of metastatic pancreatic adenocarcinoma: a review. Oncology. 2014;28(1):70–74. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
O’Connor ML, Xiang D, Shigdar S, et al. Cancer stem cells: a contentious hypothesis now moving forward. Cancer Lett. 2014;344(2):180–187. | ||
Siller KH, Cabernard C, Doe CQ. The NuMA-related Mud protein binds Pins and regulates spindle orientation in Drosophila neuroblasts. Nat Cell Biol. 2006;8(6):594–600. | ||
Yu F, Kuo CT, Jan YN. Drosophila neuroblast asymmetric cell division: recent advances and implications for stem cell biology. Neuron. 2006;51(1):13–20. | ||
Fuja TJ, Schwartz PH, Darcy D, Bryant PJ. Asymmetric localization of LGN but not AGS3, two homologs of Drosophila pins, in dividing human neural progenitor cells. J Neurosci Res. 2004;75(6):782–793. | ||
Subramaniam D, Kaushik G, Dandawate P, Anant S. Targeting cancer stem cells for chemoprevention of pancreatic cancer. Curr Med Chem. 2018;25(22):2585–2594. | ||
Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67(3):1030–1037. | ||
Rasheed ZA, Matsui W. Biological and clinical relevance of stem cells in pancreatic adenocarcinoma. J Gastroenterol Hepatol. 2012;27(Suppl 2):15–18. | ||
Xia J, Chen C, Chen Z, Miele L, Sarkar FH, Wang Z. Targeting pancreatic cancer stem cells for cancer therapy. Biochim Biophys Acta. 2012;1826(2):385–399. | ||
Li Y, Kong D, Ahmad A, Bao B, Sarkar FH. Pancreatic cancer stem cells: emerging target for designing novel therapy. Cancer Lett. 2013;338(1):94–100. | ||
Almomani R, Sun Y, Aten E, et al. GPSM2 and Chudley-McCullough syndrome: a Dutch founder variant brought to North America. Am J Med Genet A. 2013;161A(5):973–976. | ||
Fukukawa C, Ueda K, Nishidate T, Katagiri T, Nakamura Y. Critical roles of LGN/GPSM2 phosphorylation by PBK/TOPK in cell division of breast cancer cells. Genes Chromosomes Cancer. 2010;49(10):861–872. | ||
Peng Y, Cui L, Shi JQ. Expression of G-protein signaling modulator 2 in pancreatic cancer tissues. J Jiangsu Univ. 2016;26(3):231–234. | ||
Wei B, Zhang JX, Cui L, Jg Q, Qian XB, Dang SC. Effects of GPSM2 over-expression on migration ability of human pancreatic cancer cells. Chin J Gen Surg. 2017;26(3):311–316. | ||
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. | ||
Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. | ||
Mohammed S, van Buren G, Fisher WE. Pancreatic cancer: advances in treatment. World J Gastroenterol. 2014;20(28):9354–9360. | ||
Chiorean EG, Coveler AL. Pancreatic cancer: optimizing treatment options, new, and emerging targeted therapies. Drug Des Devel Ther. 2015;9:3529–3545. | ||
He XY, Yuan YZ. Advances in pancreatic cancer research: moving towards early detection. World J Gastroenterol. 2014;20(32):11241–11248. | ||
Long J, Zhang Y, Yu X, et al. Overcoming drug resistance in pancreatic cancer. Expert Opin Ther Targets. 2011;15(7):817–828. | ||
Moore MA, Williams N, Metcalf D. In vitro colony formation by normal and leukemic human hematopoietic cells: characterization of the colony-forming cells. J Natl Cancer Inst. 1973;50(3):603–623. | ||
Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58(1):267–284. | ||
Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138(5):822–829. | ||
Dick JE. Breast cancer stem cells revealed. Proc Natl Acad Sci USA. 2003;100(7):3547–3549. | ||
Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730–737. | ||
Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63(18):5821–5828. | ||
Li C, Lee CJ, Simeone DM. Identification of human pancreatic cancer stem cells. Methods Mol Biol. 2009;568:161–173. | ||
Frank NY, Margaryan A, Huang Y, et al. ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res. 2005;65(10):4320–4333. | ||
Miki J, Furusato B, Li H, et al. Identification of putative stem cell markers, CD133 and CXCR4, in hTERT-immortalized primary nonmalignant and malignant tumor-derived human prostate epithelial cell lines and in prostate cancer specimens. Cancer Res. 2007;67(7):3153–3161. | ||
Lundin A, Driscoll B. Lung cancer stem cells: progress and prospects. Cancer Lett. 2013;338(1):89–93. | ||
Zhao Y, Feng F, Zhou YN. Stem cells in gastric cancer. World J Gastroenterol. 2015;21(1):112–123. | ||
Geng SQ, Alexandrou AT, Li JJ. Breast cancer stem cells: multiple capacities in tumor metastasis. Cancer Lett. 2014;349(1):1–7. | ||
O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445(7123):106–110. | ||
Zhang PY, Yang YJ, Xue Y, et al. Cancer stem cells: targeting tumors at the source. Eur Rev Med Pharmacol Sci. 2015;19(10):1821–1828. | ||
Colak S, Medema JP. Cancer stem cells – important players in tumor therapy resistance. FEBS J. 2014;281(21):4779–4791. | ||
Vermeulen L, de Sousa e Melo F, Richel DJ, Medema JP. The developing cancer stem-cell model: clinical challenges and opportunities. Lancet Oncol. 2012;13(2):e83–e89. | ||
Francipane MG, Chandler J, Lagasse E. Cancer stem cells: a moving target. Curr Pathobiol Rep. 2013;1(2):111–118. | ||
Li C, Wu JJ, Hynes M, et al. c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology. 2011;141(6):2218–2227. | ||
Hermann PC, Huber SL, Herrler T, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1(3):313–323. | ||
Caussinus E, Gonzalez C. Induction of tumor growth by altered stem-cell asymmetric division in Drosophila melanogaster. Nat Genet. 2005;37(10):1125–1129. | ||
Bu P, Chen KY, Lipkin SM, Shen X. Asymmetric division: a marker for cancer stem cells in early stage tumors? Oncotarget. 2013;4(7):950–951. | ||
Mapelli M, Gonzalez C. On the inscrutable role of inscuteable: structural basis and functional implications for the competitive binding of NuMA and inscuteable to LGN. Open Biol. 2012;2(8):120102. | ||
Diaz-Horta O, Sirmaci A, Doherty D, et al. GPSM2 mutations in Chudley-McCullough syndrome. Am J Med Genet A. 2012;158A(11):2972–2973. | ||
Doherty D, Chudley AE, Coghlan G, et al. GPSM2 mutations cause the brain malformations and hearing loss in Chudley-McCullough syndrome. Am J Hum Genet. 2012;90(6):1088–1093. | ||
Hamzeh AR, Nair P, Mohamed M, et al. A novel nonsense GPSM2 mutation in a Yemeni family underlying Chudley-McCullough syndrome. Eur J Med Genet. 2016;59(6–7):337–341. | ||
Koenigstein K, Gramsch C, Kolodziej M, et al. Chudley-McCullough syndrome: variable clinical picture in twins with a novel GPSM2 mutation. Neuropediatrics. 2016;47(3):197–201. | ||
Culurgioni S, Mari S, Bonetti P, et al. Insc:LGN tetramers promote asymmetric divisions of mammary stem cells. Nat Commun. 2018;9(1):1025. | ||
He XQ, Zhang YF, Yu JJ, et al. High expression of G-protein signaling modulator 2 in hepatocellular carcinoma facilitates tumor growth and metastasis by activating the PI3K/AKT signaling pathway. Tumour Biol. 2017;39(3):1010428317695971. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.