Back to Journals » International Journal of Nanomedicine » Volume 17
Functionalized Mesoporous Silica Nanoparticles for Drug-Delivery to Multidrug-Resistant Cancer Cells
Authors Igaz N ![]() , Bélteky P
, Bélteky P ![]() , Kovács D, Papp C
, Kovács D, Papp C ![]() , Rónavári A
, Rónavári A ![]() , Szabó D, Gácser A, Kónya Z
, Szabó D, Gácser A, Kónya Z ![]() , Kiricsi M
, Kiricsi M ![]()
Received 3 March 2022
Accepted for publication 6 July 2022
Published 14 July 2022 Volume 2022:17 Pages 3079—3096
DOI https://doi.org/10.2147/IJN.S363952
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Nóra Igaz,1 Péter Bélteky,2 Dávid Kovács,1,3 Csaba Papp,4 Andrea Rónavári,2 Diána Szabó,5 Attila Gácser,4 Zoltán Kónya,2,6 Mónika Kiricsi1
1Department of Biochemistry and Molecular Biology, University of Szeged, Szeged, Hungary; 2Department of Applied and Environmental Chemistry, University of Szeged, Szeged, Hungary; 3Institut de Pharmacologie Moléculaire et Cellulaire, Université Côte d’Azur, Inserm, CNRS, Valbonne, France; 4HCEMM-USZ Fungal Pathogens Research Group, Department of Microbiology, University of Szeged, Szeged, Hungary; 5Department of Oto-Rhino-Laryngology and Head & Neck Surgery, Szeged, Hungary; 6Eötvös Loránd Research Network, Reaction Kinetics and Surface Chemistry Research Group, Szeged, Hungary
Correspondence: Mónika Kiricsi, Department of Biochemistry and Molecular Biology, University of Szeged, Közép fasor 52, Szeged, H-6726, Hungary, Tel +36 (62) 544-887, Email [email protected] Zoltán Kónya, Department of Applied and Environmental Chemistry, University of Szeged, Rerrich square 1, Szeged, H-6720, Hungary, Tel +36 (62) 544620, Email [email protected]
Background: Multidrug resistance is a common reason behind the failure of chemotherapy. Even if the therapy is effective, serious adverse effects might develop due to the low specificity and selectivity of antineoplastic agents. Mesoporous silica nanoparticles (MSNs) are promising materials for tumor-targeting and drug-delivery due to their small size, relatively inert nature, and extremely large specific surfaces that can be functionalized by therapeutic and targeting entities. We aimed to create a fluorescently labeled MSN-based drug-delivery system and investigate their internalization and drug-releasing capability in drug-sensitive MCF-7 and P-glycoprotein-overexpressing multidrug-resistant MCF-7 KCR cancer cells.
Methods and Results: To track the uptake and subcellular distribution of MSNs, particles with covalently coupled red fluorescent Rhodamine B (RhoB) were produced (RhoB@MSNs). Both MCF-7 and MCF-7 KCR cells accumulated a significant amount of RhoB@MSNs. The intracellular RhoB@MSN concentrations did not differ between sensitive and multidrug-resistant cells and were kept at the same level even after cessation of RhoB@MSN exposure. Although most RhoB@MSNs resided in the cytoplasm, significantly more RhoB@MSNs co-localized with lysosomes in multidrug-resistant cells compared to sensitive counterparts. To examine the drug-delivery capability of these particles, RhoB@Rho123@MSNs were established, where RhoB-functionalized nanoparticles carried green fluorescent Rhodamine 123 (Rho123) - a P-glycoprotein substrate – as cargo within mesopores. Significantly higher Rho123 fluorescence intensity was detected in RhoB@Rho123@MSN-treated multidrug-resistant cells than in free Rho123-exposed counterparts. The exceptional drug-delivery potential of MSNs was further verified using Mitomycin C (MMC)-loaded RhoB@MSNs (RhoB@MMC@MSNs). Exposures to RhoB@MMC@MSNs significantly decreased the viability not only of drug-sensitive but of multidrug-resistant cells and the elimination of MDR cells was significantly more robust than upon free MMC treatments.
Conclusion: The efficient delivery of Rho123 and MMC to multidrug-resistant cells via MSNs, the amplified and presumably prolonged intracellular drug concentration, and the consequently enhanced cytotoxic effects envision the enormous potential of MSNs to defeat multidrug-resistant cancer.
Keywords: mesoporous silica nanoparticles, drug-delivery, fluorescently labeled MSNs, functionalized MSNs, multidrug resistance
Graphical Abstract:

Introduction
The development of multidrug resistance (MDR) is one of the main reasons behind the low efficiency or the failure of chemotherapy. This represents a major hurdle, especially in breast cancer treatment, where neoadjuvant chemotherapy is routinely applied to manage early-stage operable or advanced breast cancer.1 The emerging MDR phenotype can be the result of various molecular events. Such is the overexpression and/or increased efflux activity of membrane-bound ABC transporters like P-glycoprotein (Pgp, MDR1, ABCB1),2 multidrug resistance-associated protein 1 (MRP1, ABCC1)3 or breast cancer resistance protein (BCRP, ABCG2, MXR),4 that have broad substrate specificity and are capable of eliminating chemotherapeutic agents from cancer cells, resulting in a significantly attenuated cytotoxic efficiency and concomitantly, a higher survival rate of cancer cells upon first-line traditional chemotherapy.5 Nevertheless, apart from ABC transporters, other mechanisms might also contribute to the development of chemoresistance in cancer cells, including drug inactivation reactions, alteration of DNA damage response, apoptotic dysregulation, epithelial-mesenchymal transition, and various epigenetic events.6,7
Another critical issue of chemotherapy is the specificity of the employed anticancer agents which forms the basis of the intensive ongoing research to find the ultimate compounds to specifically manipulate cancerous, especially MDR cells without exerting unpredictable toxicities on healthy tissues.8 The low specificity of antitumor drugs administered clinically in the maximum tolerated doses (MTD) often leads to a broad range of undesired side effects such as nausea, fatigue, diarrhea, myalgia, hand-foot syndrome, or in some cases to nephro- and neurotoxicity.9,10 In order to increase the efficiency and specificity of anticancer therapy, and to combat MDR cancer, the performance of various nanomaterials has recently been tested in relevant oncotherapeutic settings.11–14 Due to the unique characteristics, such as small size, controllable drug release, and specific passive and active targeting capability, carefully designed smart nanosystems might become the ultimate solution for overcoming drug resistance and suppressing unwanted side effects. Passive targeting exploits the unique features of the tumor vasculature, where the fenestrated endothelium of blood vessels allows the extensive passage and accumulation of nanomaterials, and the defective lymphatic drainage retains the nanoparticles within the tumor to a higher extent compared to normal tissues and organs.15 Additionally, the functionalization of nanocarriers with various tumor-specific ligands further enhances the direct, active targeting of tumors and improves the internalization of the particles by cancer cells.16
Silica nanoparticles (SNs) hold a huge potential in the biomedical field. Owing to a wide range of available synthesis methods, reproducible and controlled preparation of SNs can be achieved to assure the appropriate nanoparticle size and shape, the formation of uniform and tunable pores, and to offer the possibility of modification of both the internal and external surfaces of the nanoparticles with different moieties for further functionalization.17 Mesoporous silica nanoparticles (MSNs) consisting of a honeycomb-like porous structure – with hundreds of cylindrical pores in the 2–50 nm range as independent reservoirs that can encapsulate enormous amounts of the desired drug molecules – are considered the most suitable for drug-delivery and controlled release purposes.18–20 Studies have already revealed that MSNs are biocompatible, as they do not affect the viability and the morphology of human cells (at concentrations below 100 μg/mL).21 The large external and internal surfaces of MSNs can be modified selectively, nucleic acids, proteins, or other types of targeting ligands, fluorescent dyes can be coupled to the exterior surface of MSNs, and state-of-the-art antitumor drugs, pro-drugs, contrast agents, or fluorescent molecules can be encapsulated inside the mesopores.22 Further modification of the MSN structure might be needed to avoid leakage of encapsulated anticancer agents or other bioactive molecules from MSN-based delivery systems by various pH-, redox-, enzyme-, magnetic-responsive gatekeepers or to achieve drug release from MSNs by pH- or stimuli-sensitive manner.23–26
These results suggest that there is a rapidly growing scientific interest in delineating the potential of utilizing MSNs for chemotherapeutic drug-delivery, in particular into multi-chemoresistant cancer cells, and defining the factors that determine cancer selectivity and controlled drug release of MSNs upon cancer therapy. In fact, in this study, our aim was to design and test a fluorescently labeled drug-delivery system based on MSNs, where the intracellular localization of the nanoparticles as well as the intracellular presence and the cytotoxic effect of the mesopore-released cargo can be monitored. For this, amino-functionalized MSNs were labeled on their surface covalently with Rhodamine B fluorescent dye, and then first Rhodamine 123 was loaded into the internal pores of the particles. The drug-delivery and the cytotoxicity of these functionalized MSNs were examined and compared in drug-sensitive MCF-7 and P-glycoprotein-overexpressing multidrug-resistant MCF-7 KCR cancer settings to reveal their potential in defeating MDR. Finally, as a proof-of-concept, the drug-delivery potential and anticancer properties of the MSN carrier were investigated on MCF-7 and MDR MCF-7 KCR breast cancer cells using RhoB-labeled MSN particles encapsulating the anti-proliferative agent Mitomycin C.
Materials and Methods
Cell Culture
Drug-sensitive MCF-7 and multidrug-resistant MCF-7 KCR human breast adenocarcinoma cells were provided by Prof. Imre Boros. The MCF-7 cell line was originally obtained from ATCC. The drug-resistant MCF-7 KCR cell line was developed from MCF-7 under selection pressure using doxorubicin from 10 nM to 1 μM concentration.27,28 To maintain the MDR phenotype MCF-7 KCR cells were kept in cell culture medium complemented with 1 µM doxorubicin (Sigma-Aldrich, Saint Louis, MO, USA) for a week followed by a week of drug-free period. All cell lines were maintained in RPMI-1640 (Lonza, Basel, Switzerland) medium complemented with 10% FBS (EuroClone, Pero, Italy), 2 mM L-glutamine (Biowest, Nuaille, France), 0.01% streptomycin, and 0.005% penicillin (Biowest, Nuaille, France). Cells were cultured under standard conditions in a 37 °C incubator containing 5% CO2 in 95% humidity.
Preparation of Mesoporous Silica Nanoparticles
All chemicals were purchased from Sigma-Aldrich (Saint Louis, MO, USA). In the initial steps of the synthesis, amino-functionalized mesoporous silica nanoparticles (MSN) were produced through a sol-gel method by Finlay et al, with slight modifications.29 Firstly, 250 mg of cetyl trimethyl ammonium bromide (CTAB) was dissolved in 120 mL deionized water in which 1 mL 1 M NaOH was added, then the solution was heated to 80 °C. Once the temperature has been reached, 1080 μL tetraethyl orthosilicate (TEOS) and 120 μL aminopropyl triethoxysilane (APTES), corresponding to the molar ratio of 10:1 TEOS:APTES were added to the mixture, and the resulting gel was stirred for 2 h at 80 °C.
Once the procedure was finished, the particles were washed repeatedly to remove the surfactant using ultracentrifugation on 5900 g for 25 min, with a few drops of 1 M NaOH added before each centrifugation, to electrostatically facilitate the precipitation of MSN. This was repeated until the supernatant was clear and not foaming. The final step of MSN synthesis was the drying of the particles using lyophilization.
Selection of the Suitable Dye for MSN Labeling
To develop a fluorescently labeled MSN drug-delivery system, first, the appropriate fluorescent dyes had to be selected. We opted for testing the suitability of the red fluorescent Rhodamine B and the green fluorescent Rhodamine 123. To determine whether a given dye meets all the requirements, a series of flow cytometry tests were carried out on free dye-exposed cancer cells. We examined how much dye is taken up by drug-sensitive and multidrug-resistant cells and determined whether the dye is exported from the MDR cancer cells by efflux pump activity or not. For this, MCF-7, and MCF-7 KCR cells were seeded into 6-well plates and left to grow. After 24 hours, cells were trypsinized and collected by centrifugation (400 g, 5 min). Cell pellets were resuspended in a serum-free culture medium containing either 5.2 µM Rhodamine B or 5.2 µM Rhodamine 123 and were incubated with the dyes for 20 min at 37°C. In separate MCF-7 and MCF-7 KCR samples, the P-glycoprotein inhibitor verapamil was used in 20 µM concentration for 20 minutes concomitantly with the fluorescent dyes Rhodamine B or Rhodamine 123 (in 5.2 µM concentration). After incubation, cells were washed twice with pre-warmed PBS and the fluorescence intensities of Rhodamine B or Rhodamine 123 were determined by a FACScalibur flow cytometer (BD Biosciences, San Jose, California, USA). Raw data were analyzed using FlowJo V10 software.
Preparation of Functionalized Mesoporous Silica Nanoparticles
Silica nanoparticles covalently marked by Rhodamine B (RhoB@MSNs) were prepared through amide formation. In a typical synthesis, 0.5 mg Rhodamine B and 0.047 g 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) were dissolved in 50 mL deionized water and stirred for 30 min at room temperature during which activated ester groups could form at the carboxyl functions of the dye. Subsequently, 0.63 g MSN was added into the solution and the coupling reaction was performed at room temperature with overnight stirring. Any unbound Rhodamine dye was removed with ultracentrifugation, which was repeated until the supernatant was colorless. At this point, the sample was either considered finished, or 1 mg Rhodamine 123 was added to 0.63 g RhoB@MSNs to produce RhoB@Rho123@MSNs or 10 mg Mitomycin C was added to 20 mg RhoB@MSNs to generate RhoB@MMC@MSNs, respectively. The samples were stirred overnight at 40 °C to facilitate adsorption. Finally, all samples were once again freeze-dried and the resulting pink (RhoB@MSNs), orange (RhoB@Rho123@MSNs), and violet (RhoB@MMC@MSNs) powders were kept at 4 °C until further use.
Characterization of Mesoporous Silica Nanoparticles
The synthesized MSNs were characterized by transmission electron microscopy (TEM), dynamic light scattering (DLS), zeta potential measurement, N2 adsorption porosimetry, and Fourier-transform infrared spectroscopy (FT-IR). The size and morphology of MSNs were assessed through TEM, using an FEI Tecnai G2 20 × Twin instrument (Tecnai G2 Philips CM10, Amsterdam, The Netherlands) with 200 kV accelerating voltage. The colloidal properties of the particles were measured by dynamic light scattering and zeta potential using a Malvern Zetasizer Nano ZS, the specific surface area of the mesoporous particles was investigated by a 3H-2000 BET-A surface area analyzer, finally, identification of the functional groups of the chemical components and verification of the synthesis of amino-functionalized and Rhodamine B-labeled MSNs was performed by FT-IR using a Bruker Vertex 70 spectrophotometer. The FT-IR spectra were collected at a spatial resolution of 4 cm−1 in the transmission mode, between 4000 and 450 cm −1.
The drug loading efficiency of Rhodamine 123 and Mitomycin C in RhoB@MSNs was assessed based on the paper by Lei et al.30 For this, we used UV-Vis spectroscopy relying on the characteristic absorbance of the dye and the drug. The absorbance spectra of different MSN-structures were recorded on an Ocean Optics 355 DH-2000-BAL UV–Vis spectrophotometer (Halma PLC, Largo, FL, USA), within the range from 200 to 600 nm using a 10-mm path length quartz cuvette. First, to describe the calibration curve, a dilution series was prepared from the given compound to cover the 0–1 absorption range. After determining the calibration curve and the equation, a solution of the given compound and silica particles were made at the proper concentrations (described in the Materials and Methods section), then mixed in 100 mL distilled water, stirred overnight and the resulting samples were collected by centrifugation. Thereafter, the absorbance of the supernatant was examined and based on the calibration curve the concentration of the supernatant and the loaded silica product was calculated. The amount of Rhodamine 123 and Mitomycin C were determined by the characteristic absorbance maximum and the drug-loading efficiency (LE(%)) was calculated by the following equation:

in which ma was the added mass of Rhodamine 123 or Mitomycin C, mb was the mass of Rhodamine 123 or Mitomycin C in supernatant, and mc was the total mass of RhoB@Rho123@MSN or RhoB@MMC@MSN input of the reaction system.
The drug release behavior of the RhoB@MMC@MSNs and RhoB@Rho123@MSNs was also evaluated.31,32 To measure drug release from the MSNs, first we performed calibration experiments using Rhodamine 123 and Mitomycin C via measuring the absorbance of Mitomycin C at 365 nm and the Rhodamine 123 at 503 nm. Rhodamine 123 and Mitomycin C release from the substrates was assessed as follows. The release profile was obtained by soaking the loaded MSNs in 50 mL of stirred distilled water; 200.57 mg of RhoB@Rho123@MSN and 38.56 mg of RhoB@MMC@MSN were placed in a seamless semipermeable cellulose dialysis membrane (length: 8 cm, (cut-off: 12–14 kDa) Merck Ltd., Budapest, Hungary). The solution was kept at 310 K and the pH was regulated (pH:7.3–7.4). In order to avoid limitation of the delivering rate by external diffusion constraints, continuous stirring at 500 rpm was maintained during the assays. The drug concentration in the liquid phase was evaluated by UV spectrometry at 503 (Rhodamine 123) or 365 nm (Mitomycin C), respectively. One milliliter of sample was taken at predetermined time intervals (at 5, 10, 15, 30, 60, 75, 90, 120, 180, 240, 300 min and 24h) and absorbance of the solvated drug was determined. These absorbance values were convertible to concentration dimension when the relationship between these parameters is determined by calibration process (the error of the calculated concentrations was less than ±2%). Based on the previously published work, for the mathematical modelling of the registered dissolution profiles (relative concentration (ct/c0) of released drug as a function of time) the first-/second-order-, Higuchi-, Weibull-, Korsmeyer–Peppas-, and Hopfenberg-models were applied to fit the experimental data in nonlinear parameter estimation way, and the goodness the fits were measured by the value of R2.32
Assessing the Intracellular Accumulation of Functionalized MSNs
To examine the intracellular accumulation of RhoB@MSNs and RhoB@Rho123@MSNs in MCF-7 and MCF-7 KCR cells, Rhodamine B and Rhodamine 123 fluorescence intensities were measured. Cells in 1×104 cells/well density were seeded into 96-well plates and were left to grow. On the following day, cells were exposed to RhoB@MSNs or RhoB@Rho123@MSNs in various concentrations (0; 0.2; 0.4; 0.6; 0.8; 1; 1.2; 1.4 mM of MSN). After 24-hour incubation samples were washed 3 times with PBS, and the Rhodamine B or the Rhodamine 123 fluorescence intensity was determined using Synergy HTX plate reader (BioTek, Winooski, Vermont, USA). The applied parameters were Ex: 485/20; Em: 528/20 in case of the Rhodamine 123 fluorescence measurements and Ex: 530/25; Em: 620/40 filters were used for the Rhodamine B fluorescence intensity determination. The measured values were normalized to the fluorescence intensity of the cell-free wells containing RhoB@MSNs or RhoB@Rho123@MSNs, according to the previously described protocol. GraphPad Prism 6 software was used for statistical analysis, two-way ANOVA Sidak’s multiple comparisons test was applied on experimental data.
Toxicity of Functionalized MSNs
To investigate the viability of MCF-7 and MCF-7 KCR cells upon treatments with the different MSNs, 104 cells/well were seeded into 96-well plates and were exposed to RhoB@MSNs, or to RhoB@Rho123@MSNs in various concentrations (0; 0.2; 0.4; 0.6; 0.8; 1; 1.2; 1.4 mM of MSNs). After 24-hour treatments, we removed the MSN-containing media, washed the cells, and incubated them with 0.5 mg/mL MTT (Sigma-Aldrich, Saint Louis, MO, USA) in culture medium for 1 hour at 37°C. Formazan crystals were solubilized in DMSO (Molar Chemicals, Halásztelek, Hungary), and the absorbance at 570 nm (Background: 630 nm) was determined using Synergy HTX plate reader (BioTek, Winooski, Vermont, USA). Statistical analysis was performed in GraphPad Prism 6 software using two-way ANOVA Sidak’s multiple comparisons test.
Uptake of Functionalized MSNs by Cancer Cells
Flow cytometry was utilized when the uptake of MSN nanoparticles by cancer cells was examined. In these cases, 3×105 MCF-7 or MCF-7 KCR cells were seeded into 6-well plates in complete culture media (RPMI medium complemented with 10% FBS, 2 mM L-glutamine, 0.01% streptomycin and 0.005% penicillium). On the following day, cells were treated with 0.1 mM RhoB@MSNs in the above described complete culture media for 24 hours. Then, the RhoB@MSN-containing medium was removed, samples were washed with PBS, and cells were incubated in serum-free media for 24 or 48 hours. As a reference, in independent samples, cells were treated only for 24 hours in full culture media containing 0.1 mM RhoB@MSNs. Before flow cytometry analysis, all samples were washed with PBS, trypsinized, centrifuged (400 g, 5 min), resuspended in 200 µL PBS and measured by FlowSight Imaging Flow Cytometer (Amnis, EMD Millipore, Burlington, MA, USA). Data were analyzed by Ideas Software Version 6.2 (Amnis, EMD Millipore, Burlington, MA, USA). Statistical analysis was performed in GraphPad Prism 6 software using two-way ANOVA Sidak’s multiple comparisons test.
Intracellular Localization of Functionalized MSNs
To distinguish between intracellular and membrane-associated MSNs, the cell membranes of MCF-7 and MCF-7 KCR cells were labeled with PKH67 green fluorescent dye (Sigma-Aldrich, Saint Louis, MO, USA) according to the manufacturer’s instructions. After membrane staining with PKH67, 3×105 cells were seeded into 6-well plates and left to grow. On the following day, cells were treated with 0.1 mM RhoB@MSNs for 24 hours. Then, the samples were prepared for flow cytometry according to the previously described protocol. The obtained fluorescence data were analyzed with FlowSight Imaging Flow Cytometer using Ideas Software Version 6.2 (Amnis, EMD Millipore, Burlington, MA, USA).
The intracellular localization, more specifically, the cytosolic, the potential mitochondrial or lysosomal localization of RhoB@MSNs was investigated also by fluorescent microscopy. For this, MCF-7, and MCF-7 KCR cells were seeded into 24-well plates containing glass coverslips in each well (VWR International, Radnor, PA, USA). On the following day, cells were exposed to 0.1 mM RhoB@MSNs and after 24-hour treatments, cells were washed twice in PBS. The localization of RhoB@MSNs was detected owing to its red fluorescence; lysosomes were visualized by green fluorescent LysoTracker Green DND-26 (Cell Signaling Technology, Danvers, MA, USA) applied in 50 nM concentration for 5 minutes; in parallel samples, mitochondria were stained with green fluorescent MitoTracker Green FM (Cell Signaling Technology, Danvers, MA, USA) applied on cells in 100 nM concentration for 15 minutes. In case of MCF-7 KCR cells, verapamil was applied on cells 1 hour before Lyso-, or MitoTracker staining. All samples were visualized by Zeiss Axio Observer 7.0 fluorescence microscope (Zeiss Microscopy, Jena, Germany) and photos were taken by Zeiss Axiocam 503 mono camera (Zeiss Microscopy, Jena, Germany). To test co-localization of lysosomes and RhoB@MSNs, CellProfiler 3.1.8. was used with a custom-made pipeline. Statistical analysis was performed in GraphPad Prism 6 software using unpaired t-test.
Anticancer Efficiency of Free and of MSN-Delivered Mitomycin C
To determine if Mitomycin C (MMC) is a substrate of Pgp or not, 1×104 of either MCF-7 or MCF-7 KCR cells were seeded into 96-well plates, and on the following day, cells were exposed to MMC (in 5.78; 11.56; 23.13; 46.25; 92.5; 185 µM concentrations). A set of MCF-7 and MCF-7 KCR samples were treated with 20 µM of the Pgp inhibitor verapamil 1 hour prior to MMC treatments. As a read-out, MTT assays were performed, as described above.
Next, to assess the efficiency of drug-delivery to MCF-7 and MCF-7 KCR cells, we compared the anti-cancer effect of the free MMC and the MSN-delivered MMC (RhoB@MMC@MSNs). In these cases, 1×104 cells were seeded into 96-well plates and left to grow overnight. On the next day, free MMC and MMC-loaded MSNs were added to the media of cells in various concentrations (5.78; 11.56; 23.13; 46.25; 92.5; 185 µM MMC). After 24-hour treatments, cells were washed with PBS and then incubated with 0.5 mg/mL MTT reagent for 1 hour at 37°C. Finally, the absorbance of the dissolved formazan (absorbance at 570 nm, background: 630 nm) was measured in Synergy HTX plate reader (BioTek, Winooski, VT, USA). Statistical analysis was performed in GraphPad Prism 6 software using two-way ANOVA Sidak’s multiple comparisons test.
Results
Synthesis and Functionalization of MSNs
For this study, first amino-functionalized mesoporous silica nanoparticles (MSNs) were produced through a sol-gel method. Based on TEM images and DLS measurements, the utilized sol-gel process was successful in synthesizing mesoporous spherical nanoparticles of approximately 100 nm average diameter as illustrated in Figure 1A–C. The FT-IR analysis on the samples revealed characteristic Si-O-Si (1078 cm−1) and Si-OH (950 cm−1) vibrations, the NH2 scissoring vibration (1600 cm−1), and the band at 3400 cm−1 attributed to the primary amine N-H stretching are in good agreement with the literature data proving that the method provided amino-functionalized MSNs (Figure 1D). Regarding the nitrogen adsorption isotherm of the sample, its specific surface area was 1423.5 m2/g, which is in the typical range of similar MCM 41 type materials and drug-delivery systems.33–35
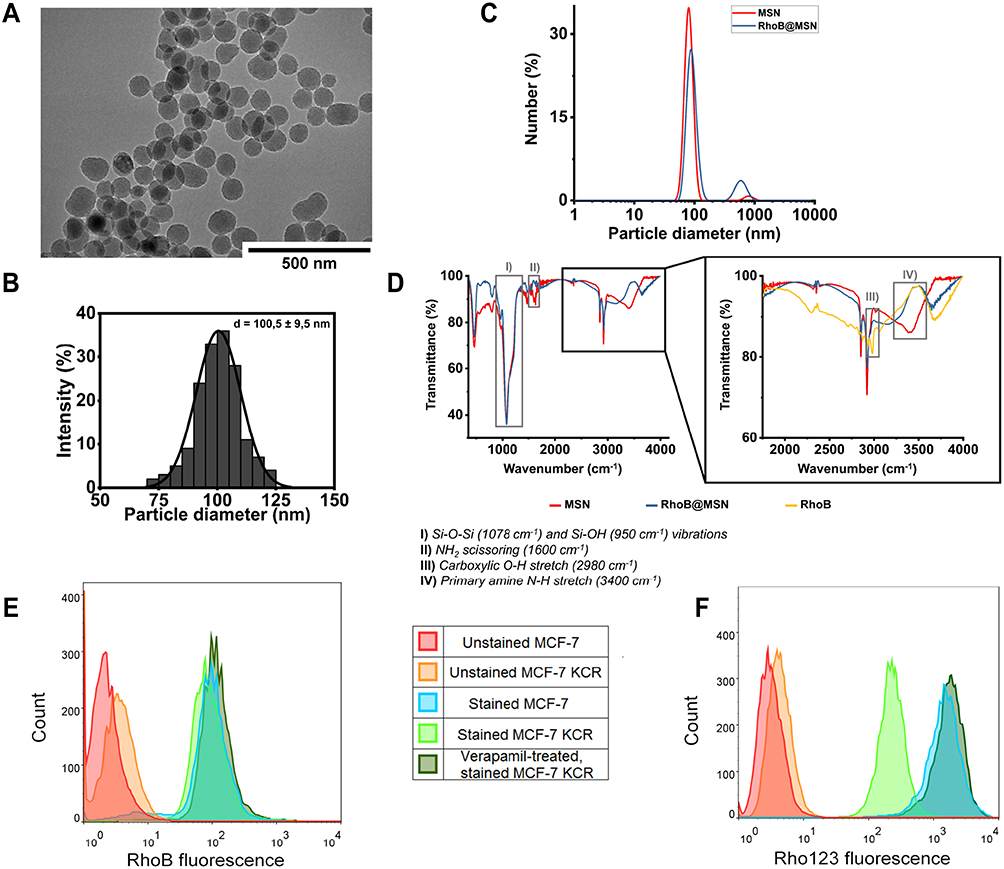 |
Figure 1 Characterization of the obtained MSNs and Rhodamine B-functionalized MSNs. (A) Representative transmission electron microscopic image of amino-functionalized MSNs. Scale bar: 500 µm (B). Size distribution of the amino-functionalized MSNs was determined based on TEM image analysis. The particles on TEM images were measured and their diameter was illustrated on this graph. (C) Using dynamic light scattering the average size of amino-functionalized (MSN) and Rhodamine B-functionalized MSNs (RhoB@MSN) was measured via the determination of hydrodynamic diameter of the particles (D). FT-IR spectra of amino-functionalized MSNs (MSN), Rhodamine B (RhoB) and of Rhodamine B-functionalized MSNs (RhoB@MSN) (E). Histograms representing the Rhodamine B fluorescence intensity upon Rhodamine B treatment of MCF-7 and MCF-7 KCR cells and of Verapamil-treated MCF-7 KCR cells (F). Histograms representing the Rhodamine 123 fluorescence intensity of Rhodamine 123-exposed MCF-7 and MCF-7 KCR cells and of Verapamil-treated MCF-7 KCR cells. |
Then, using the obtained MSN-samples, three differently labeled and loaded functionalized MSN-delivery systems were synthesized. To be able to track the localization of the particles within cells following treatments, red fluorescent Rhodamine B dye was coupled to the surface of the MSNs yielding RhoB@MSNs. To visualize the cargo-releasing capability of the particles, Rhodamine 123 green fluorescent dye was loaded into the mesopores of the synthesized RhoB@MSNs, resulting in RhoB@Rho123@MSNs. Finally, to verify the advantageous drug-delivery properties of the fluorescently-labeled MSN carrier – especially in MDR settings – with a clinically applied anti-proliferative agent Mitomycin C (MMC), RhoB@MMC@MSNs was prepared, where the mesopores encapsulated MMC instead of Rho123. Prior to MSN functionalization, the two fluorescent dyes were tested to reveal if they were substrates of P-glycoprotein. Flow cytometry results revealed that both dyes could be internalized by the studied breast cancer cells indicated by high fluorescence intensities for RhoB and for Rho123 in both MCF-7 as well as MCF-7 KCR cells (Figure 1E and F). Importantly, these experiments affirmed that Rho123 is a substrate of Pgp, however RhoB is not. Histograms obtained for RhoB-exposed MCF-7 and MCF-7 KCR, as well as for verapamil-treated MCF-7 KCR cells were all overlapping, indicating that when Pgp is inhibited by verapamil, no change occurs in the amount of the intracellularly retained fluorescent dye (Figure 1E). However, when Pgp-overexpressing multidrug-resistant MCF-7 KCR cells were treated with verapamil to block Pgp-driven dye efflux, the Rho123 fluorescence intensity increased, and MCF-7 KCR cells retained approximately the same amount of Rho123 dye as MCF-7 cells (Figure 1F). Therefore, we decided to couple RhoB to the surface of MSNs and use Rho123 as a model cargo within mesopores. The functionalized MSNs were then prepared accordingly. The free amino functional groups on the surface of MSNs were used for the covalent conjugation of the nanoparticles with the fluorescent dye Rhodamine B through a coupling reaction between MSN amino and RhoB carboxyl groups mediated by EDC. The success of the functionalization was demonstrated by the subsequent DLS (Figure 1C), zeta potential and FT-IR (Figure 1D) analysis of the particles. The particle number-based size distributions provided by DLS revealed, that while both MSNs and RhoB@MSNs demonstrated slight aggregation leading to secondary peaks at pH 7, the primary diameter of the silica nanoparticles remained unaffected by the coupling reaction. The zeta potential of MSNs was −25.7 mV, while +19.3 mV was measured for RhoB@MSNs. The sign change in the zeta potential of the particles was due to the fact, that on neutral pH, the surface of silica has a negative charge due to the acid/base equilibrium of silanol groups, while Rhodamine B possesses a quaternary nitrogen, leading to the positive potential measured on the functionalized particles.36 Ultimately, FT-IR spectroscopy was used to determine the exact nature of the surface functionalization by comparing the spectra of MSNs, RhoB@MSNs and RhoB (Figure 1D). At lower wavenumbers, the IR spectrum of the dye is excluded, since its numerous peaks would make the comparison of the silica samples difficult.37 Based on the results, the chemical composition of the mesoporous silica nanoparticles can be verified, as strong Si-O-Si (1078 cm−1) and Si-OH (950 cm−1) vibrations were detected in both samples. The formation of amide bonds was assessed based on the presence/absence of related vibrations in specific samples. Namely, NH2 scissoring, and primary amine NH stretching vibrations were detected around 1600 and 3600 cm−1 in MSNs, respectively, which were absent in RhoB@MSNs, furthermore, the carboxylic OH stretching vibration (2980 cm−1) of the fluorescent dye was absent in either silica samples, leading to the conclusion, that the conjugation reaction was successful.
Following the chemical characterization of the dye-conjugated particles, a portion was loaded with either Rhodamine 123 or Mitomycin C. Then, the loading efficiency of RhoB@MSNs with the two cargo molecules was evaluated. Based on the measurements, in case of Rhodamine 123, loading efficiency (LE) was calculated to be 0.15% by measuring the absorbance of the supernatant at 503 nm. In case of Mitomycin C, LE(%) was calculated to be 30.33% by measuring the absorbance of the supernatant at 365 nm. These values are in good correlation with the dye/drug input amounts. In fact, we have utilized significantly less amount of Rhodamine 123 to be incorporated into MSN mesopores than Mitomycin C, since this dye shows strong fluorescence at very low concentrations, and we estimated that a smaller amount of the dye would be more suitable for the fluorescence measurements of the treated cancer cells.
Finally, the drug release profile of RhoB@Rho123@MSNs and RhoB@MMC@MSNs was assessed. The release kinetics of the drugs were studied as a function of time and the results are shown in Supplementary File. As it is visible, the materials exhibit somewhat different drug release profiles. The diffusion model that we have applied to study the delivery from this system considers the leaching of the medicament by water, which is able to enter the drug-matrix phase through pores. The drug is presumed to dissolve slowly into the fluid phase and diffuses from the system along the solvent filled capillary channels. While Mitomycin C release occurred faster in the first hour, and the curve saturated at 70%, in case of Rhodamine 123 the release was 75%, showing a slower, stable and controlled pattern. The latter feature increases the bioavailability and allows the reduction of the dosage. The observed profiles were similar in trend to the results of previously published studies showing controlled drug release from MSN.32,38 Based on the previously published work, for the mathematical modelling of the registered dissolution profiles (relative concentration (ct/c0) of released drug as a function of time) different models were applied to fit the experimental data in nonlinear parameter estimation way.32
Accumulation of RhoB@MSNs in Drug-Sensitive and Multidrug-Resistant Cancer Cells
Next, we characterized the effects induced by the Rhodamine B-labeled MSNs that did not yet contain any dye or drug to carry within mesopores, on drug-sensitive MCF-7 and on multidrug-resistant MCF-7 KCR cells. Thus, the potential toxic effects exerted by RhoB@MSNs as well as the uptake efficiency of RhoB@MSNs by these breast cancer cells were investigated. In accordance with other publications, RhoB@MSN treatments did not cause significant viability loss of either cell line in the tested concentration range compared to the untreated cells (Figure 2A), which supports the proposed biocompatibility of MSNs. Although RhoB@MSNs exerted no or low toxicity, a concentration-dependent increase in the Rhodamine B fluorescence intensity could be measured in MCF-7 cells following exposure to RhoB@MSNs, and more importantly, a similar degree of nanoparticle accumulation was observed in drug-resistant MCF-7 KCR cells as well (Figure 2B). These observations indicate that both MCF-7 and MCF-7 KCR cells can internalize RhoB@MSNs with a comparable degree.
 |
Figure 2 Accumulation of RhoB@MSNs in drug-sensitive and multidrug-resistant cancer cells (A). Viability of MCF-7 and MCF-7 KCR cells after RhoB@MSN treatments compared to the untreated control samples (B). Rhodamine B fluorescence intensity of MCF-7 and MCF-7 KCR cells following 24-hour RhoB@MSN exposure. Two-way ANOVA Sidak’s multiple comparisons test, *P<0.05; **P<0.01; #P<0.0001 (C). Representative histograms obtained by flow cytometry analysis of PKH67-labeled untreated and RhoB@MSN-treated MCF-7 cells (D). Histograms representing the internalized Rho@MSNs in RhoB@MSN-treated and control MCF-7 KCR cells (E). Relative amount of internalized RhoB@MSNs within RhoB-positive cells. Unpaired t-test. |
To verify that the detected fluorescent signal is the result of internalized particles, the cell membranes were labeled with green fluorescent PKH67 membrane dye then cells were treated with 0.1 mM RhoB@MSNs for 24 hours and finally the samples were prepared for flow cytometry. The software analysis differentiated the intracellular, or membrane-associated particles according to the overlap of red RhoB fluorescent signal and the green fluorescent PKH67 membrane dye (Figure 2C and D). The analysis revealed that 72.7 ± 8.1% of RhoB-positive MCF-7 cells internalized the RhoB@MSNs and 79.9 ± 7.9% of RhoB-positive MCF-7 KCR cells carried the RhoB@MSNs in their cytosol (Figure 2E).
Intracellular Localization of RhoB@MSNs
The subcellular localization of the internalized RhoB@MSNs was investigated by fluorescent microscopy. Fluorescent microscopic images confirmed that both MCF-7 (Figure 3A) and MCF-7 KCR (Figure 3B) cells can internalize RhoB@MSNs. In fact, no significant difference was observed in the percentage of RhoB-positive MCF-7 (36.1 ± 4.7%) and MCF-7 KCR (29.4 ± 2.8%) cells (Figure 3C). To detect the intracellular localization of MSNs, lysosomes and mitochondria were counterstained with Lyso- and MitoTracker green fluorescent dyes (Figure 3A and B). The red fluorescent RhoB@MSNs accumulated in the cytoplasm, and partly in the lysosomes of drug-sensitive MCF-7 cells (Figure 3A). Strong co-localization was observed between RhoB@MSNs and the green fluorescent LysoTracker in multidrug-resistant MCF-7 KCR cells (Figure 3B). Significant differences were observed between MCF-7 cells and MCF-7 KCR cells in the intracellular localization of RhoB@MSNs, namely significantly more RhoB@MSNs were found in lysosomes of MCF-7 KCR cells than in MCF-7 cells (Figure 3D). We could not detect co-localization between RhoB@MSNs and mitochondria in any of the samples.
 |
Figure 3 Subcellular localization of the internalized RhoB@MSNs (A). Representative images of RhoB@MSN-treated MCF-7 cells after MitoTracker as well as LysoTracker staining, respectively. Scale bar: 20 µm (B). Representative pictures of RhoB@MSN-exposed MCF-7 KCR cells after Mito-, and LysoTracker staining. Scale bar: 20 µm (C). Quantification of RhoB-positive MCF-7 and MCF-7 KCR cells based on microscopic images. Unpaired t-test (D). The percentage of RhoB@MSNs that co-localized with lysosomes, according to the fluorescent microscopic images. Unpaired t-test, Mann–Whitney test, ****P<0.0001. |
Time-Dependent Dynamics of the Intracellular Rhodamine B Fluorescence Intensity in RhoB@MSN-Treated MCF-7 and MCF-7 KCR Cells
Next, we wanted to test whether the internalized RhoB@MSNs stay permanently within drug-sensitive and multidrug-resistant cancer cells or get quickly eliminated. To investigate this, the time-dependent changes in the intracellular fluorescence intensity of RhoB were measured by flow cytometry 0, 24 and 48 hours after the MSN-containing medium was removed and replaced by FBS-free medium to avoid cell division.
No differences were observed in the percentage of RhoB@MSN-positive drug-sensitive MCF-7 cells at any time point. After the 24-hour RhoB@MSN treatment 37.4 ± 4.2% of MCF-7 cells were RhoB-positive, and 24 and 48 hours after the removal of the particle-containing media 37.4 ± 9.7% and 37.1 ± 8.9% of the MCF-7 cells were RhoB-positive, respectively (Figure 4A and C). Despite no changes in the percentage of RhoB-positive cells, a slight decrease in fluorescence intensity can be observed on the histograms within RhoB-positive MCF-7 cells (Figure 4A).
 |
Figure 4 Time-dependent dynamics of the intracellular fluorescence intensity of Rhodamine B in RhoB@MSN-treated MCF-7 and MCF-7 KCR cells (A). Representative histograms obtained by flow cytometric analysis of MCF-7 cells after 24-hour RhoB@MSN treatment and 24 or 48 hours after the removal of RhoB@MSN-containing media (B). Representative histograms of untreated, 24 h RhoB@MSN-treated MCF-7 KCR cells and of MCF-7 KCR cells 24 or 48 hours after the removal the RhoB@MSN treatments (C). Graph and Table representing the percentage of RhoB-positive MCF-7 and MCF-7 KCR cells after 0, 24 and 48 hours from the removal of RhoB@MSNs. Two-way ANOVA Sidak’s multiple comparisons test, ****P<0.0001. |
In case of the multidrug-resistant MCF-7 KCR cells, when the RhoB@MSN treatment was removed, 30.9 ± 4.8% of the cells was RhoB-positive (Figure 4B and C). Twenty-four hours after the removal of RhoB@MSN-containing media, no changes were detected in the relative amount of RhoB-positive MCF-7 KCR cells (33.9 ± 5.5%). However, 48 hours after switching for MSN-free media, the percentage of RhoB-positive cells was slightly, but not significantly reduced (27.9 ± 12.6%) (Figure 4B and C). On the representative histograms of MCF-7 KCR cells, a dynamic decrease in the fluorescence intensity is clearly visible (Figure 4B).
The fluorescent microscopic results were in accordance with those of flow cytometry, since there was only a slight, but not significant difference in the percentage of RhoB-positive cells between MCF-7 (36.1 ± 4.7%) and MCF-7 KCR cells (29.4 ± 2.8%) (Figure 3C). These results suggest that RhoB@MSNs are permanently trapped in sensitive MCF-7, and also in multidrug-resistant MCF-7 KCR cells.
The Efficiency of Drug-Delivery via MSNs
To investigate the efficiency of drug-delivery by MSNs, the green fluorescent dye Rhodamine 123, then the antineoplastic agent Mitomycin C were utilized as cargo, thus were loaded into the mesopores of RhoB@MSNs in separate synthesis events.
At the beginning, we used the green fluorescent Rhodamine 123 as a model compound and followed its delivery into drug-resistant and sensitive cancer cells both in free format and as the mesoporous payload of MSNs. Previously, by selecting the appropriate dyes for the synthesis of functionalized MSNs, we performed flow cytometry experiments on Rhodamine 123-treated MCF-7 and MCF-7 KCR cells, the latter ones with and without Verapamil treatment. We found that the Rhodamine 123 dye is the substrate of P-glycoprotein (Figure 1F). Further experiments were carried out by applying free Rhodamine 123 in increasing concentrations on MCF-7 and MCF-7 KCR cells, then the green fluorescence intensities of the cells were assessed. These data revealed Rho123 concentration-dependent increase in the fluorescence of MCF-7 cells; however, no fluorescence could be achieved for MCF-7 KCR cells following free Rhodamine 123 exposures (Figure 5A). This confirms that Rho123 dye is effectively eliminated by efflux pump activity of MDR cancer cells.
 |
Figure 5 The efficiency of MSN-based drug-delivery of the fluorescent cargo Rhodamine 123 (A). Intracellular Rho123 fluorescence intensity of MCF-7 and MCF-7 KCR cells treated with the free form of Rho123. Two-way ANOVA Sidak’s multiple comparisons test, **P<0.01; ****P<0.0001 (B). Viability of MCF-7 and MCF-7 KCR cells after RhoB@Rho123@MSN exposures compared to the untreated cells (C). Rhodamine B fluorescence intensity of MCF-7 and MCF-7 KCR cells after RhoB@Rho123@MSN treatments (D). The measured Rhodamine 123 fluorescence intensity after RhoB@Rho123@MSN exposure of MCF-7 and MCF7 KCR cells. Two-way ANOVA Sidak’s multiple comparisons test, ****P<0.0001. |
Next, MCF-7 and MCF-7 KCR cells were treated with the RhoB@Rho123@MSN system. Viability assays confirmed that similarly to RhoB@MSNs, these particles do not exert any toxic effect on either cell line (Figure 5B). Furthermore, both the intracellular Rhodamine B (Figure 5C) and Rhodamine 123 (Figure 5D) fluorescence intensities of the RhoB@Rho123@MSN-exposed cells could be measured. These results revealed that RhoB@Rho123@MSNs were able to efficiently deliver intracellularly the Pgp substrate Rhodamine 123 to both sensitive and drug-resistant MCF-7 KCR cells (Figure 5D). Although due to P-glycoprotein activity, the Rhodamine 123 fluorescence intensities are somewhat weaker in MCF-7 KCR cells after RhoB@Rho123@MSN treatments than those in their drug-sensitive counterparts, it is of utmost importance that a rather high intracellular concentration of Rhodamine 123 could be achieved in drug-resistant cells with the help of this MSN-delivery platform. This is an extremely relevant finding, since it suggests that when cytotoxic drugs are administered and transported via alternative drug-delivery approaches, multidrug-resistant cancer cells can be targeted and defeated efficiently.
Finally, as a proof-of-concept, we examined the impact of a commonly used anti-proliferative and anticancer agent Mitomycin C (MMC),39 after it reached cancer cells in free form and in our MSN-based system. For this aim, we generated RhoB@MMC@MSNs, where MMC was loaded into the porous channels of RhoB@MSNs. Before starting the experiments with RhoB@MMC@MSNs, we had to delineate whether MMC is recognized by Pgp as a substrate or not. A similar approach was used as in case of Rho123, since MCF-7 and multidrug-resistant MCF-7 KCR cells were treated with free MMC with or without Verapamil, however, this time the viability of the treated cancer cells was used as a read-out. According to the results, cell viability was significantly decreased for Pgp-overexpressing MDR MCF-7 KCR cells after they were treated with Verapamil + MMC, compared to MMC monotreatments (IC50 of Verapamil + MMC treatment = 100.2 µM; IC50 of MMC treatment ˃185 µM) (Figure 6A and B). Nevertheless, no difference was observed in MCF-7 cell viability following MMC and MMC + Verapamil exposures (IC50 of MMC treatment = 69.7 µM; IC50 of Verapamil + MMC treatment = 69.8 µM) (Figure 6A and B). These findings suggest that MMC is the substrate of efflux transport in drug-resistant cancer cells.
 |
Figure 6 The efficiency of MSN-based delivery of the anticancer agent Mitomycin C as a cargo (A). Viability of MCF-7 and MCF-7 KCR cells after Mitomycin C or Mitomycin C + Verapamil treatments. Two-way ANOVA Sidak’s multiple comparisons test, **P<0.005; ****P<0.0001 (B). IC50 values calculated from the viability results of Mitomycin C-, or Mitomycin C + Verapamil-treated MCF-7 and MCF-7 KCR cells (C). Viability of MCF-7 and MCF-7 KCR cells upon MMC or RhoB@MMC@MSN treatments. Two-way ANOVA Sidak’s multiple comparisons test, *P<0.05; **P<0.005; ****P<0.0001 (D). IC50 values determined from the viability results of MCF-7 and MCF-7 KCR cells after MMC or RhoB@MMC@MSN treatment. |
Then, the drug-delivery efficiency of MSNs was investigated using free MMC and RhoB@MMC@MSNs and the anti-proliferative and toxic effects of MMC were detected on the two cell lines. As expected, after 24-hour incubation, the free MMC treatment resulted in a concentration-dependent decrease in the viability of drug-sensitive MCF-7 cells (IC50 = 63.3 µM); therefore, MMC could exert its toxic activity on these cancer cells that do not overexpress Pgp (Figure 6C). However, the free MMC treatment did not affect the cell viability of multidrug-resistant MCF-7 KCR cells, showing only a slight toxic effect in the highest applied concentration. Indeed, the impact of free MMC on multidrug-resistant MCF-7 KCR cells is so weak, that the estimated IC50 value is out of the tested concentration range (IC50 ˃185 µM). Importantly, it is already evident at a glance that significantly lower cell viability of both MCF-7 and MCF-7 KCR cells was obtained following RhoB@MMC@MSN treatments compared to the cell viability recorded after free MMC exposures. Albeit the MDR MCF-7 KCR cells were not exactly as sensitive to the RhoB@MMC@MSN treatments than MCF-7 cells (IC50 on MCF-7 = 13.6 µM, IC50 on MCF-7 KCR = 35.9 µM) (Figure 6C and D), clearly, it is possible to efficiently eliminate multidrug-resistant cells with RhoB@MMC@MSNs and lower MMC concentrations are necessary to achieve this goal than when free MMC is used on MDR cancer cells. Therefore, we can conclude that both Rhodamine 123 accumulation and MMC anticancer efficiency on MCF-7 KCR cells were significantly increased when the agents were delivered via MSNs, thus by exploiting the excellent delivery propensity of MSNs higher intracellular concentration of anticancer agents can be achieved even in highly challenging multidrug-resistant cancer cells.
Discussion
The therapy of malignant diseases often leads to the development of multidrug resistance, which would ultimately cause the failure of traditional chemotherapy. By exploiting the passive tumor-targeting potentials of nanometer-sized materials increased efficiency and higher tumor-specificity could be achieved via nanoparticles. Moreover, functionalization of nanoparticles with tumor-specific ligands can further enhance the delivery and the accumulation of nanosystems in the tumor tissue and augment their uptake by cancer cells. Mesoporous silica nanoparticles (MSNs) are promising materials for tumor-targeting and drug-delivery purposes due to their two distinct surfaces. The large outer surface of the MSNs can be functionalized to enhance the targeting efficiency and cancer-specificity, while different anticancer agents can be loaded into the mesopores of the nanosystem to increase the concentration of antitumor drugs in the tumor tissue. It was previously shown that MSNs have an extremely high drug-loading capacity, thus are ideal carriers of various types of antineoplastic agents, their combinations and also for multimodal cancer therapy.40–42
One of the principal reasons behind the multidrug-resistant phenotype is the increased expression and activity of plasma membrane residing efflux transporters. In previous studies, it was verified that the ABC transporter Pgp is overexpressed in MCF-7 KCR cells,43,44 therefore this cell line represents an ideal in vitro model to investigate the drug-delivery capability of our MSN-based system on multidrug-resistant cells. In this study, our aim was to create a fluorescently labeled MSN-based drug-delivery system to investigate the delivery and the cytotoxicity of the cargo drugs for the purpose to eliminate multidrug-resistant MCF-7 KCR breast cancer cells. Furthermore, we wanted to examine and compare the impact and the faith of the carrier system on drug-sensitive and drug-resistant cancer cells. For this, first amino-functionalized MSNs were synthesized with an average size between 50 and 100 nm. This size range is suitable for cancer targeting, since the surface area and the loading capacity of such particles are very high and previously it was shown that the 50 nm sized particles are taken up by cancer cells with the highest degree.45 In our system, MSNs were functionalized with red fluorescent Rhodamine B dye, which was linked via amide bonds to the NH2 moieties on the outer surface of MSNs (RhoB@MSNs). Rhodamine B was selected as a surface marker of MSNs, because this dye is not the substrate of Pgp according to our flow cytometry results. To follow the drug-releasing capability and the efficiency of cargo-delivery by MSNs, the green fluorescent Rhodamine 123 dye or the anticancer agent Mitomycin C was loaded into the mesopores of the particles, respectively, yielding two different functionalized nanosystems (RhoB@Rho123@MSNs and RhoB@MMC@MSNs).
According to our results, MSNs (both RhoB@MSNs and RhoB@Rho123@MSNs) are biocompatible, since no toxic effect was detected upon exposing either MCF-7 or MCF-7 KCR cells to these materials in the tested concentration range, which indicates the feasibility of this system for drug-delivery purposes. Moreover, we verified by flow cytometry and fluorescent microscopy that drug-sensitive and MDR cancer cells are both able to take up and accumulate RhoB@MSNs to a similar degree. It is known that MSNs are internalized by cancer cells mainly via endocytosis, but it is also accepted that the particles can escape from endolysosomal vesicles, thus the cargo can be protected from degradation.46,47 This raised a question about the exact localization of MSNs following cellular uptake, especially in the case of MDR cancer cells. We could detect nanoparticles in the cytoplasm, partly in the lysosomes of MCF-7 cells. Significant difference was observed between MCF-7 and MCF-7 KCR cells in the number of lysosomally localized RhoB@MSNs, since more red fluorescing RhoB@MSNs were detected in the lysosomes of MCF-7 KCR cells. We concluded that these nanoparticles can be internalized by drug-sensitive, and more importantly by multidrug-resistant cancer cells as well, but in MCF-7 KCR cells more particles might be remaining within lysosomes.
Thus, the next question was whether this lysosomal presence of MSNs affects the efficiency of the delivered anticancer agents against MDR cells or not. For the subsequent experiments, we utilized cargo-carrying RhoB-labeled MSNs, where Rhodamine 123, a Pgp substrate green fluorescent dye was selected as a payload. We verified that drug loading was in fact efficient and that the cargo fluorescent dye can be effectively and dynamically released from the mesoporous structure of the RhoB@Rho123@MSNs. After treating MCF-7 and MCF-7 KCR cells with free Rho123 or RhoB@Rho123@MSN the green fluorescence intensities of the cells were compared. Our flow cytometry results confirmed that after free Rhodamine 123 treatments in contrary to MCF-7 cells, extremely low green fluorescence signal of MCF-7 KCR cells could be detected, suggesting that the efflux activity of Pgp in these drug-resistant cells can completely eliminate Rho123. However, the inhibition of Pgp with Verapamil resulted in increased Rhodamine 123 fluorescence of multidrug-resistant cancer cells. Importantly, high intracellular concentration of Rhodamine 123 could be achieved both in MCF-7 and MCF-7 KCR cells, when RhoB@Rho123@MSNs were applied instead of free Rho123, implying that dye-delivery by MSNs can circumvent the immediate elimination of Rho123 from MDR cancer cells. Finally, to validate the excellent drug-delivery capability of MSNs to MDR cancer cells with an anti-proliferative agent, MMC was loaded into the mesopores of MSNs (RhoB@MMC@MSNs). Again, efficient drug loading was verified and drug release tests revealed that the cargo antineoplastic drug can be effectively released from the mesoporous structure of the RhoB@MMC@MSNs. MMC is also the substrate of Pgp, and when it enters the drug-resistant cells in free form, without the inhibition of the efflux pumps, this anticancer agent would be readily eliminated from MDR cells to prevent cell death. However, MMC delivered as RhoB@MMC@MSN decreased more the viability of drug-sensitive as well as of multidrug-resistant cancer cells, than free MMC. As expected, a slightly higher IC50 value was obtained after RhoB@MMC@MSN treatments on MCF-7 KCR cells than on drug-sensitive MCF-7 cells; nevertheless, these IC50 values were much lower than those calculated based on the viability of free MMC-treated samples.
Conclusion
In summary, this study provides compelling data on the suitability of MSNs for carrying the anticancer agent Mitomycin C as cargo molecule into various cancer cells. Our results suggest that MSNs are appropriate drug delivery platforms, and apart from MMC, other highly efficient antineoplastic agents could also be supplied to the tumor tissue via this system, but prove also that with the help of the outstanding activity of these carriers even multidrug-resistant cancer cells can be defeated. The application of MSNs in tumor therapy could enhance the performance and the specificity of oncotherapy especially when multi-resistant and highly challenging cancer cells have to be destroyed. The numerous advantages of these materials could be exploited in the future also in multimodal treatment approaches.
Abbreviations
ATCC, American Type Culture Collection; Ex, excitation; Em, emission; FBS, fetal bovine serum; IC, inhibitory concentration; MDR, multidrug resistant; MMC, mitomycin C; MSNs, mesoporous silica nanoparticles; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide; RhoB, rhodamine B; Rho123, rhodamine 123; RPMI-1640, Roswell Park Memorial Institute 1640; PBS, phosphate-buffered saline; Pgp, P-glycoprotein.
Data Sharing Statement
All original data of this manuscript are available on request.
Ethics Approval and Consent to Participate
This article does not contain any studies with human participants or animals performed by any of the authors.
Acknowledgments
The graphical abstract was created with BioRender.com.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agreed to be accountable for all aspects of the work.
Funding
This research was funded by the Hungarian Government and the European Union within the frames of the Széchenyi 2020 Program through grants GINOP-2.3.2-15-2016-00035 and GINOP-2.3.2-15-2016-00038. Furthermore, this work was supported by the ÚNKP-21-5-SZTE-592 (M.K.), ÚNKP-21-5-SZTE-576 (A.R) and NTP-NFTÖ-21-B-0091 (N.I.) grants of the New National Excellence Program of the Ministry for Innovation and Technology and by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (BO/00878/19/8 for M.K., BO/00384/21/7 for A.R.). A.G. has received funding from the EU’s Horizon 2020 research and innovation program under grant agreement No. 739593.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Connolly RM, Stearns V. Current approaches for neoadjuvant chemotherapy in breast cancer. Eur J Pharmacol. 2013;717(1–3):58–66. doi:10.1016/j.ejphar.2013.02.057
2. Li Y, Yuan H, Yang K, Xu W, Tang W, Li X. The structure and functions of P-glycoprotein. Curr Med Chem. 2010;17:786–800. doi:10.2174/092986710790514507
3. Naito S, Hasegawa S, Yokomizo A, et al. Non‐P‐glycoprotein‐mediated atypical multidrug resistance in a human bladder cancer cell line. Jap J Cancer Res. 1995;86:1112–1118. doi:10.1111/j.1349-7006.1995.tb03028.x
4. Staud F, Pavek P. Breast cancer resistance protein (BCRP/ABCG2). Int J Biochem Cell Biol. 2005;37:720–725. doi:10.1016/j.biocel.2004.11.004
5. Nooter K, Stoter G. Molecular mechanisms of multidrug resistance in cancer chemotherapy. Pathol Res Pract. 1996;192:768–780. doi:10.1016/S0344-0338(96)80099-9
6. Tendulkar S, Dodamani S. Chemoresistance in ovarian cancer: prospects for new drugs. Anticancer Agents Med Chem. 2020. doi:10.2174/1871520620666200908104835
7. Ruan T, Liu W, Tao K, Wu C. A review of research progress in multidrug-resistance mechanisms in gastric cancer. Onco Targets Ther. 2020;Volume 13:1797–1807. doi:10.2147/OTT.S239336
8. Baji Á, Kiss T, Wölfling J, et al. Multicomponent access to androstano-arylpyrimidines under microwave conditions and evaluation of their anti-cancer activity in vitro. J Steroid Biochem Mol Biol. 2017;172:79–88. doi:10.1016/j.jsbmb.2017.06.001
9. Beusterien K, Grinspan J, Kuchuk I, et al. Use of conjoint analysis to assess breast cancer patient preferences for chemotherapy side effects. Oncologist. 2014;19(2):127–134. doi:10.1634/theoncologist.2013-0359
10. Magge RS, DeAngelis LM. The double-edged sword: neurotoxicity of chemotherapy. Blood Rev. 2015;29(2):93–100. doi:10.1016/j.blre.2014.09.012
11. Kovács D, Szoke K, Igaz N, et al. Silver nanoparticles modulate ABC transporter activity and enhance chemotherapy in multidrug resistant cancer. Nanomedicine. 2016;12:601–610. doi:10.1016/j.nano.2015.10.015
12. Kovács D, Igaz N, Marton A, et al. Core-shell nanoparticles suppress metastasis and modify the tumour-supportive activity of cancer-associated fibroblasts. J Nanobiotechnology. 2020;18. doi:10.1186/s12951-020-0576-x
13. Igaz N, Kovács D, Rázga Z, Kónya Z, Boros IM, Kiricsi M. Modulating chromatin structure and DNA accessibility by deacetylase inhibition enhances the anti-cancer activity of silver nanoparticles. Colloids Surf B Biointerfaces. 2016;146:670–677. doi:10.1016/j.colsurfb.2016.07.004
14. Igaz N, Szőke K, Kovács D, et al. Synergistic radiosensitization by gold nanoparticles and the histone deacetylase inhibitor SAHA in 2D and 3D cancer cell cultures. Nanomaterials. 2020;10(1):158. doi:10.3390/nano10010158
15. Shi Y, van der Meel R, Chen X, Lammers T. The EPR effect and beyond: strategies to improve tumor targeting and cancer nanomedicine treatment efficacy. Theranostics. 2020;10(17):7921–7924. doi:10.7150/thno.49577
16. Das RP, Gandhi VV, Singh BG, Kunwar A. Passive and active drug targeting: role of nanocarriers in rational design of anticancer formulations. Curr Pharm Des. 2019;25(28):3034–3056. doi:10.2174/1381612825666190830155319
17. Tang F, Li L, Chen D. Mesoporous silica nanoparticles: synthesis, biocompatibility and drug delivery. Adv Mater. 2012;24(12):1504–1534. doi:10.1002/adma.201104763
18. Wang Y, Zhao Q, Han N, et al. Mesoporous silica nanoparticles in drug delivery and biomedical applications. Nanomedicine: Nanotechnology, Biology and Medicine. 2015;11(2):313–327. doi:10.1016/j.nano.2014.09.014
19. Giri S, Trewyn BG, Lin VSY. Mesoporous silica nanomaterial-based biotechnological and biomedical delivery systems. Nanomedicine. 2007;2(1):99–111. doi:10.2217/17435889.2.1.99
20. Slowing II, Vivero-Escoto JL, Wu CW, Lin VSY. Mesoporous silica nanoparticles as controlled release drug delivery and gene transfection carriers. Adv Drug Deliv Rev. 2008;60:1278–1288. doi:10.1016/j.addr.2008.03.012
21. Lai CY, Trewyn BG, Jeftinija DM, et al. A mesoporous silica nanosphere-based carrier system with chemically removable CdS nanoparticle caps for stimuli-responsive controlled release of neurotransmitters and drug molecules. J Am Chem Soc. 2003;125. doi:10.1021/ja028650l
22. Bharti C, Gulati N, Nagaich U, Pal A. Mesoporous silica nanoparticles in target drug delivery system: a review. Int J Pharm Investig. 2015;5(3):124. doi:10.4103/2230-973x.160844
23. Barui S, Cauda V. Multimodal decorations of mesoporous silica nanoparticles for improved cancer therapy. Pharmaceutics. 2020;12(6):527. doi:10.3390/pharmaceutics12060527
24. Cai L, Zhu P, Huan F, et al. Toxicity-attenuated mesoporous silica Schiff-base bonded anticancer drug complexes for chemotherapy of drug resistant cancer. Colloids Surf B Biointerfaces. 2021. doi:10.1016/j.colsurfb.2021.111839
25. Zhu P, Xu Z, Cai L, Chen J. Porphyrin iron-grafted mesoporous silica composites for drug delivery, dye degradation and colorimetric detection of hydrogen peroxide. Nanoscale Res Lett. 2021;16. doi:10.1186/s11671-021-03501-6
26. Iturrioz-Rodríguez N, Correa-Duarte MA, Fanarraga ML. Controlled drug delivery systems for cancer based on mesoporous silica nanoparticles. Int J Nanomedicine. 2019;Volume 14:3389–3401. doi:10.2147/IJN.S198848
27. Kars MD, Işeri ÖD, Gündüz U, Ural AU, Arpaci F, Molnár J. Development of rational in vitro models for drug resistance in breast cancer and modulation of MDR by selected compounds. Anticancer Res. 2006;26:4559–4568.
28. Gopisetty MK, Adamecz DI, Nagy FI, et al. Androstano-arylpyrimidines: novel small molecule inhibitors of MDR1 for sensitizing multidrug-resistant breast cancer cells. Eur J Pharm Sci. 2021;156:105587. doi:10.1016/j.ejps.2020.105587
29. Finlay J, Roberts CM, Dong J, Zink JI, Tamanoi F, Glackin CA. Mesoporous silica nanoparticle delivery of chemically modified siRNA against TWIST1 leads to reduced tumor burden. Nanomedicine. 2015;11:1657–1666. doi:10.1016/j.nano.2015.05.011
30. Lei W, Sun C, Jiang T, et al. Polydopamine-coated mesoporous silica nanoparticles for multi-responsive drug delivery and combined chemo-photothermal therapy. Mater Sci Eng C. 2019;105:110103. doi:10.1016/j.msec.2019.110103
31. Varga N, Hornok V, Janovák L, Dékány I, Csapó E. The effect of synthesis conditions and tunable hydrophilicity on the drug encapsulation capability of PLA and PLGA nanoparticles. Colloids Surf B Biointerfaces. 2019;176:212–218. doi:10.1016/j.colsurfb.2019.01.012
32. Juhász Á, Ungor D, Berta K, Seres L, Csapó E. Spreadsheet-based nonlinear analysis of in vitro release properties of a model drug from colloidal carriers. J Mol Liq. 2021. doi:10.1016/j.molliq.2021.115405
33. Chen H, Fu S, Fu L, Yang H, Chen D. Simple synthesis and characterization of hexagonal and ordered al–mcm–41 from natural perlite. Minerals. 2019;9(5):264. doi:10.3390/min9050264
34. Huang X, Young NP, Townley HE. Characterization and comparison of mesoporous silica particles for optimized drug delivery. Nanomater Nanotechnol. 2014;4:2. doi:10.5772/58290
35. Kruk M, Jaroniec M, Sayari A. Adsorption study of surface and structural properties of MCM-41 materials of different pore sizes. J Phy Chem B. 1997;101:583–589. doi:10.1021/jp962000k
36. Xu P, Wang H, Tong R, Du Q, Zhong W. Preparation and morphology of SiO2/PMMA nanohybrids by microemulsion polymerization. Colloid Polym Sci. 2006;284(7):755–762. doi:10.1007/s00396-005-1428-9
37. Pandurangappa M, Kumar KS. Micellar mediated trace level mercury quantification through the rhodamine B hydrazide spirolactam ring opening process. Anal Methods. 2011;3(3):715. doi:10.1039/c0ay00693a
38. Kiss T, Katona G, Mérai L, et al. Development of a hydrophobicity-controlled delivery system containing levodopa methyl ester hydrochloride loaded into a mesoporous silica. Pharmaceutics. 2021;13(7):1039. doi:10.3390/pharmaceutics13071039
39. Volpe A, Racioppi M, D’Agostino D, Cappa E, Filianoti A, Bassi PF. Mitomycin C for the treatment of bladder cancer. Minerva Urologica e Nefrologica. 2010;62:133–144.
40. He Q, Shi J, Chen F, Zhu M, Zhang L. An anticancer drug delivery system based on surfactant-templated mesoporous silica nanoparticles. Biomaterials. 2010. doi:10.1016/j.biomaterials.2010.01.015
41. Wang K, Lu J, Li J, et al. Current trends in smart mesoporous silica-based nanovehicles for photoactivated cancer therapy. J Control Release. 2021;339:445–472. doi:10.1016/j.jconrel.2021.10.005
42. Feng S, Lu J, Wang K, et al. Advances in smart mesoporous carbon nanoplatforms for photothermal–enhanced synergistic cancer therapy. Chem Eng J. 2022. doi:10.1016/j.cej.2022.134886
43. Toth M, Boros IM, Balint E. Elevated level of lysine 9-acetylated histone H3 at the MDR1 promoter in multidrug-resistant cells. Cancer Sci. 2012;103(4):659–669. doi:10.1111/j.1349-7006.2012.02215.x
44. Gopisetty MK, Kovács D, Igaz N, et al. Endoplasmic reticulum stress: major player in size-dependent inhibition of P-glycoprotein by silver nanoparticles in multidrug-resistant breast cancer cells. J Nanobiotechnology. 2019;17. doi:10.1186/s12951-019-0448-4
45. Lu F, Wu SH, Hung Y, Mou CY. Size effect on cell uptake in well-suspended, uniform mesoporous silica nanoparticles. Small. 2009;5:1408–1413. doi:10.1002/smll.200900005
46. Lu J, Liong M, Sherman S, et al. Mesoporous silica nanoparticles for cancer therapy: energy-dependent cellular uptake and delivery of paclitaxel to cancer cells. Nanobiotechnology. 2007;3(2):89–95. doi:10.1007/s12030-008-9003-3
47. He Q, Shi J. MSN anti-cancer nanomedicines: chemotherapy enhancement, overcoming of drug resistance, and metastasis inhibition. Adv Mater. 2014;26(3):391–411. doi:10.1002/adma.201303123
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.