Back to Journals » International Journal of Nanomedicine » Volume 16
Functionalized DMP-039 Hybrid Nanoparticle as a Novel mRNA Vector for Efficient Cancer Suicide Gene Therapy
Authors Gao Y ![]() , Men K
, Men K ![]() , Pan C, Li J
, Pan C, Li J ![]() , Wu J, Chen X
, Wu J, Chen X ![]() , Lei S
, Lei S ![]() , Gao X, Duan X
, Gao X, Duan X
Received 7 May 2021
Accepted for publication 7 July 2021
Published 30 July 2021 Volume 2021:16 Pages 5211—5232
DOI https://doi.org/10.2147/IJN.S319092
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Yan Gao,1,* Ke Men,1,* Congbin Pan,1 Jingmei Li,1 Jieping Wu,1 Xiaohua Chen,2 Sibei Lei,1 Xiang Gao,1 Xingmei Duan2
1State Key Laboratory of Biotherapy and Cancer Center, West China Hospital of Sichuan University, Chengdu, Sichuan Province, People’s Republic of China; 2Department of Pharmacy, Sichuan Academy of Medical Sciences & Sichuan Provincial People’s Hospital, Personalized Drug Therapy Key Laboratory of Sichuan Province, School of Medicine, University of Electronic Science and Technology of China, Chengdu, 610072, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ke Men
State Key Laboratory of Biotherapy and Cancer Center, West China Hospital of Sichuan University, Chengdu, 610041, People’s Republic of China
Email [email protected]
Xingmei Duan
Department of Pharmacy, Sichuan Academy of Medical Sciences & Sichuan Provincial People’s Hospital, Personalized Drug Therapy Key Laboratory of Sichuan Province, School of Medicine, University of Electronic Science and Technology of China, Chengdu, 610072, People’s Republic of China
Email [email protected]
Background: Gene therapy has emerged as a new strategy for cancer therapy. As an alternative nucleic acid material, messenger ribonucleic acid (mRNA) is being increasingly utilized in cancer gene therapy. However, unfulfilled requirements and a lack of ideal mRNA delivery vectors persist.
Methods: We developed an advanced mRNA delivery system, DMP-039, by fusing a cell-penetrating peptide, cRGD-R9, and a cationic nano-sized DMP backbone together. The DMP gene vector backbone was synthesized by the self-assembly of DOTAP lipid and mPEG-PCL polymer. Introduction of the cRGD-R9 peptide onto the DMP backbone was performed to elevate the mRNA delivery capacity, which resulted in a peptide-functionalized hybrid delivery system.
Results: The average size of the synthesized DMP-039 was 268.9 ± 12.4 nm (PDI = 0.382), with a potential of 17.4 ± 0.5 mV. The synthesized DMP-039 hybrid nanoparticles exhibited high mRNA delivery efficiency through multiple mechanisms during transmembrane transportation. By loading the encoding mRNA from the suicide gene Bim, a locally administered mBim/DMP-039 complex strongly inhibited growth in two colon cancer models. Moreover, intravenous administration of the mBim/DMP-039 complex efficiently suppressed C26 pulmonary metastatic tumor progression with high safety. The in vivo distribution, degradation, and excretion were also investigated in detail.
Conclusion: Our results suggest that the DMP-039 peptide-functionalized hybrid nanoparticle is an advanced candidate for mRNA-based suicide gene therapy.
Keywords: mRNA, Bim, cell-penetrating peptide, gene therapy
Introduction
Because the mortality rate for cancer continues to increase, it remains a major public health problem. Colon cancer is the third most frequently diagnosed cancer with the fourth highest mortality rate across all age groups worldwide.1–3 At present, the traditional methods for treating colon cancer include surgery, chemotherapy, targeted therapy, and immunotherapy.4,5 Gene therapy has emerged as a new treatment method for colon cancer, with potential therapeutic strategies including apoptosis induction, immune stimulation, and direct cytotoxicity.6–8 Several preclinical- and clinical-phase anti-colon cancer gene therapy studies are now underway.9,10
As an important therapeutic strategy, a suicide gene is capable of inducing cell apoptosis and thus has a suppressive effect.11 Apoptosis is the process of physiological cell suicide controlled by genes to maintain the stability of the human body.12 Introducing additional suicide genes, either endogenous or exogenous in nature, has been proven to be effective in cancer gene therapy.12 As a crucial member of the Bcl-2 family, Bim (also known as BCL2L11) is known for inhibiting Bcl-2 activity and activating BAX-BAK1 proteins, thus triggering mitochondrial apoptosis.13
In recent years, Bim has been applied in the treatment of various diseases, including chronic infection, ischemia, diabetes, Alzheimer’s disease, and infectious disease.14–18 Thus, there is great interest in the development of Bim for use in cancer therapy. In tumorigenesis, to achieve abnormal proliferation, cancer cells have developed their own mechanisms to prevent Bim from working. In tumor metastasis, cancer cells leave their primary site and colonize in a new environment by attaching to other normal cells. However, apoptosis can reduce cell attachment and block tumor metastasis. Thus, introducing additional Bim has been suggested so that it can act as a potential suicide gene target in cancer suicide gene therapy.
Messenger ribonucleic acid (mRNA) is a single-stranded RNA molecule that plays an important role in transferring the genetic code from the genome to ribosomes for intracellular protein production. It also plays a role as an alternative therapeutic nucleic acid material that can be used instead of conventional plasmid DNA. Relative to its counterpart, mRNA possesses several advantages in delivering target gene information.19 For example, mRNA is free of redundant elements and has simpler sequences than plasmid DNA, and thus, the delivery capacity of an mRNA molecule is larger than that of DNA of similar length. After the mRNA is delivered into cells, mRNA can translate the target protein in the cytoplasm without entering the nucleus.
With the maturity of synthesis technology, in vitro transcript mRNA (IVT mRNA) with proper capping and tailing modifications can be easily prepared on a large scale, which would prevent bacterial host contamination caused by plasmid fermentation. For these reasons, several mRNA-based drugs or therapeutic strategies have been developed and tested for different diseases. In 2020, two mRNA-based vaccines, including BNT162b1 from Pfizer (New York, NY, USA) and BioNTech (Mainz, Germany), and mRNA-1273 from Moderna (Cambridge, MA, USA), were approved by the United States Food and Drug Administration for the prevention of coronavirus disease 2019.20,21 These nanoparticle-based mRNA drugs have exhibited strong potential worldwide.22 Several mRNA-based therapeutic candidates are also undergoing clinical or pre-clinical evaluations for diseases such as isolated methylmalonic acidemia, influenza, varicella-zoster virus, and a wide variety of cancers.23–26 However, there are still limitations to the development of mRNA-based therapeutics, one of which is delivery efficiency. The linear conformation of mRNA molecules is usually irregular, which results in difficulty in condensing the molecules for delivery. The varied lengths of mRNAs encoding different genes thus require a strong capability for gene binding so that they can be used in a potential delivery system. In addition, it is also crucial to ensure the stability of the mRNA cargo upon delivery. Therefore, it is necessary to develop advanced mRNA delivery systems with high levels of efficiency and safety.
In recent years, there have been many efforts to develop ideal vectors for mRNA delivery with satisfactory delivery efficiency. Systems such as multi-component nanoparticles, modified polyethyleneimine (PEI)-derivatives, and peptide–protein conjugates have been created and applied in mRNA-based gene therapy studies.27–29 Despite these efforts, the toxicity and complexity of developed vectors increased along with the enhancement of mRNA delivery capacity, which created an obstacle impeding further application. These complex structures and components increased the difficulty of manufacturing and quality control. A mass of introduced cationic groups and organic solvents used in the process of chemical modification elevated the toxicity of the vectors themselves. This further limited their in vivo application regardless of local or systemic administration. Therefore, there is a great demand for an advanced and efficient mRNA delivery vector with a simple composition.
In our previous work, the self-assembly of a cationic lipid N-[1-(2,3-dioleoyloxy) propyl]-N,N,N-trimethylammonium methyl-sulfate (DOTAP) with a biodegradable copolymer methoxypoly(ethylene glycol)poly(ε-caprolactone) (mPEG-PCL) into nano-sized micelles resulted in the formation of a DMP nano-backbone.30,31 There was high efficiency in the delivery of small interfering ribonucleic acid (siRNA) and plasmid DNA by this hybrid DMP nanoparticle. The simple and biodegradable structure of the DMP nanoparticle renders it an ideal non-viral gene vector with high biocompatibility and safety.
DMP nanoparticles have been successfully applied in multiple cancer gene therapy studies. In particular, their viability in systemic administration during gene therapy has been evaluated and verified. These properties suggest that DMP nanoparticles are excellent candidates to act as the backbone for mRNA delivery vectors. Furthermore, DMP nanoparticles are also open to functional modification. Because of their self-assembly ability, a wide variety of functional groups such as proteins, peptides, chemical groups, and fluorescent moieties can be easily introduced to the DMP backbone, thus providing further opportunities to upgrade the structure and function with limited involvement of organic solvents.
As a promising class of peptides, cell-penetrating peptides (CPPs) can promote membrane penetration and cellular internalization of conjugated cargos.32 CPPs have been widely applied in the delivery of siRNA, protein, plasmid DNA, and chemical compounds.33–36 Moreover, CPPs have the ability to specifically recognize cell surface biomarkers, which further contributes to their application. However, due to our limited understanding of their action mechanisms, in many cases, using multiple CPPs to modify delivered cargo is rare. In addition, it is difficult to control the modification process involving two or more CPPs.
In this study, we fused two CPPs of R9 with cRGD. The latter, which contains a cyclic arginine-glycine-aspartate, is reported to have extra abilities to promote cell uptake. This fused CPP was then introduced to the DMP nano-backbone, producing a CPP functionalized cationic hybrid nanoparticle, labeled as DMP-R9-cRGD (DMP-039). We hypothesized that the modification of the R9-cRGD peptide on the DMP backbone could result in a double-promotional effect on cellular uptake.
In this work, the properties and mRNA delivery capacity of the DMP-039 nanoparticles were characterized in detail. By binding with encoding mRNA from the suicide gene Bim, the therapeutic potential of the Bim mRNA (mBim)/DMP-039 complex was evaluated in multiple colon cancer animal models, and its in vivo distribution, degradation, and excretion were also investigated in detail.
Methods
DOTAP-mPEG-PCL (DMP) micelles were synthesized according to the protocol used in our previous study. cRGD-R9 (039) was synthesized by Shanghai APeptide Co., Ltd. (Shanghai, China). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and polyethyleneimine (Mw=25,000, PEI25K) were purchased from Sigma-Aldrich. C26 (a Mus musculus colon carcinoma cell line, American Type Culture Collection (ATCC)) and 293T (human embryonic kidney cell line, ATCC) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS, Cell-Box, Aoke Biotechnology, Chengdu) at 37°C under a humidified atmosphere of 5% CO2. Experiments with plasmids were propagated in Escherichia coli and purified using a Plasmid DNA Mini Kit (Omega Bio-tek). BALB/c mice were obtained from Beijing HFK Biotechnology Co., Ltd. (Beijing, China) and maintained under specific pathogen-free conditions. All mouse experiments were conducted under the guidance of the Animal Ethics Committee of the General Administration of Health Research of Sichuan University and The Guidelines and Standard Operating Procedure (SOP) for Laboratory Animals of Sichuan University.
In vitro Transcription of mRNA
We used a T7 polymerase-based in vitro transcription method to prepare IVT mRNA. First, we used PVAX1-Bim plasmid DNA (pBim) as a PCR template to obtain a linear IVT Bim template. The sequence of the forward primer for pBim is 5ʹ-TAATACGACTCACTATAGGGAGAATGGCCAAGCAACCTTCT-3ʹ, and the sequence of the reverse primer is 5ʹ-ATGCCTTCTCCATACCAGACG-3ʹ. The murine Bim mRNA (mBim) was synthesized using the mMESSAGE-mMACHINETM T7 Transcription Kit (Thermo) with 0.5 μg template, and then, the mRNA was purified with the MEGAclearTM Transcription Clean-Up Kit (Thermo) according to the manufacturer’s instructions. The final yield of purified mBim was approximately 40 μg, and it was electrophoresed on an agarose gel to confirm that it was a full-length segment.
Preparation and Characterization of Nanoparticles
DMP-039 was synthesized by conjugating DMP with CPPs. We first prepared DMP micelles by DOTAP self-assembly with mPEG-PCL. Briefly, N-[1-(2,3-dioleoyloxy) propyl]-N,N,N-trimethylammonium methyl-sulfate (cationic lipid DOTAP, Avanti) and mPEG-PCL polymer (Ruixibio, China) (1:9, w/w) were co-dissolved in methylene dichloride. Then, the methylene dichloride was removed by rotary evaporation for 45 min, with the subsequent formation of a lipid film. Distilled water at 55°C was added for re-hydration and self-assembly into micelles. Before conjugating with cRGD-R9, the linker maleimide-PEG-PCL was added to DMP, which formed maleimide-activated DMP (DMP-Mal). Then, cRGD-R9 was dissolved in 2-[4-(2-hydroxyethyl)-1-piperazinyl]ethanesulfonic acid (HEPES, 50 mM) buffer, DMP-Mal (1:30, w/w) was added, and the solution was reacted at 4°C overnight. After that, DMP-R9-cRGD (DMP-039) was dialyzed in distilled water with a Slide-A-Lyzer® Dialysis Cassette 2000 MWCO (Thermo) to obtain a final concentration of 7.75 mg/mL. The prepared DMP-039 was stored at 4°C for further use.
The nanoparticles were formed by mixing DMP-039 with mRNA at a ratio of 25:1 with distilled water. The mixed nanoparticles were incubated at room temperature for 15 min. The particle size and zeta potential of DMP-039 were determined by dynamic light scattering (Nano ZS, Malvern). The morphology of the DMP-039 nanoparticles was examined by transmission electron microscopy (TEM) (Tecnai G2 F20 S-TWIN, FEI, USA). All results were the mean value of three test runs.
mRNA Gel Retarding Assay
We used different molecular ratios of DMP-039 nanoparticles to evaluate the binding ability of mRNA. When DMP-039 binds with mRNA, the molecular weight increases, the migration in the gel will be blocked, and the bands that appear will be shortened. mBim (0.5 μg) mixed with DMP-039 at different ratios was electrophoresed on a 1% (w/v) agarose gel for 15 min at 120 V. The gel was then stained with GoldView II Nuclear Staining Dye (Solarbio), and the bands were detected with an E-gel imager (Bio-Rad, ChemiDox XRS, USA).
Quantitative Real-Time PCR
We extracted total RNA from C26 cells transfected with mBim/DMP-039 (1:25, w/w) and pBim/CLP (DNA:Protamine:CLP=1:2:2, w/w/w) to measure the level of Bim mRNA. TRIzol™ reagent (Vazyme) was added to the lysed sample, which was incubated and then centrifuged to precipitate the mRNA. The extracted total mRNA was dissolved with RNase-free water. After verification by gel electrophoresis, the mRNA was used as a cDNA template by SuperScript II (Vazyme). cDNA analysis was performed with a SYBR Green ER quantitative PCR SuperMix Universal kit (Vazyme). Mouse β-actin was chosen as a reference to quantitate the mBim expression level. The PCR primers for mBim (forward: 5ʹ-TTTGACACAGACAGGAGCCC, reverse: 3ʹ-CAGCTCCTGTGCAATCCGTA) and β-actin (forward: 5ʹ-CCCAGGCATTGCTGACAGG, reverse: 5ʹ-TGGAAGGTGGACAGTGAGGC) were synthesized and purified.
RNase Protection Assay
To investigate the ability of DMP-039 to protect mRNA against RNase A (Solarbio, Beijing), naked mBim or DMP-039/mBim was incubated with RNase A at a final concentration of 0.25 mg/mL. After incubation at 37°C for different amounts of time, sodium dodecyl sulfate (SDS, 0.24 mg/mL) was added for denaturation at 70°C for 10 min. mRNA was extracted from DMP-039 on ice for 5 min with the assistance of heparin (1 mg/mL). The samples were examined by electrophoresis on a 1% (w/v) agarose gel for 15 min at 120 V.
In vitro Transfection of DMP-039
The transfection efficiency of the DMP-039/mRNA complex was measured with a flow cytometer. Briefly, 293T and C26 cells were inoculated in 24-well plates (3×105 cells per well) overnight at 37°C under a humidified atmosphere of 5% CO2. The cells in each well were transfected with the DMP-039/mRNA (1 μg) complex, which encoded the enhanced green fluorescent protein (EGFP) gene, and the medium was replaced with DMEM for expression. After 4 hours, the medium was replaced with full medium. PEI25K/EGFP (1:1, w/w) and DMP/EGFP (25:1, w/w) were used as transfection controls. After 48 hours, the cells were photographed with a fluorescence microscope, and flow cytometry (NovoCyte Flow Cytometer, ACEA Biosciences, USA) was used to determine the transfection efficiency of the DMP-039/mRNA complex.
Cytotoxicity Assay
The cytotoxicity of DMP-039 nanoparticles in vitro was detected by the MTT assay. Briefly, 293T cells were plated in 96-well plates (5×103 cells per well) overnight at 37°C under a humidified atmosphere of 5% CO2. The plates were treated with different concentrations of DMP-039 nanoparticles, using PEI25K as a standard control. After transfecting for 48 hours in the incubator, 20 μL of MTT solution was added to each well to stop the reaction. After incubating in a 37°C incubator for 4 hours, the medium in the plate was replaced with 150 μL DMSO, and then, the plates were incubated at room temperature for 10 min with shaking. After that, the absorbance at 570 nm was recorded (Spectramax Absorbance Reader, Molecular Devices, USA).
Anti-Proliferation Assay
To assess the growth inhibition ability of the mBim/DMP-039 complex, C26 cells were plated in a 96-well plate (1×103 cells per well) overnight at 37°C under a humidified atmosphere of 5% CO2. The C26 cells were transfected with DMP-039 and mBim (0.5 μg)/DMP-039 or pBim (0.5 μg)/CLP according to well-established protocols. After 96 hours, the anti-proliferation was measured using the MTT assay.
Clonogenic Assay
This assay is an important technique to detect the cell proliferation capacity of DMP-039. mBim (2 μg)/DMP-039 or pBim (2 μg)/CLP was added to a 6-well plate that contained 1×103 C26 cell dilutions. The plates were incubated, and cells continued to culture and formed colonies in 1–2 weeks. When colonies were clearly visible to the naked eye, the supernatant was discarded, and the cells were carefully rinsed twice with PBS. The colonies were fixed with paraformaldehyde for 15 min, and then stained with crystal violet (0.1%) for at least 30 min. The 6-well plate was inverted on a transparent film, and the colonies were counted to determine the inhibition rate.
In vitro Apoptosis Assay
To verify the apoptosis mechanism after delivery of the Bim gene, DMP-039, mBim (1 μg)/DMP-039, or pBim (1 μg)/CLP were transfected into C26 cells (2×105 cells per well in 24-wells). After 24 hours, cell samples from each group were harvested, centrifuged, and re-suspended in Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide staining solution (Genechem, Shanghai). Flow cytometry was used to measure the cell apoptosis rate.
Western Blot Analysis
The proteins of mitochondria are important components of the apoptotic pathway. C26 cells (1×105 cells per well in 6-well plates) were transfected with the mBim/DMP-039 complex. After 48 hours, cells were collected, and protein samples were extracted with RIPA/IP (Beyotime). In order to ensure that the loading volume for each protein sample was consistent, it was necessary to determine the concentration of total proteins with Coomassie brilliant blue G-250 (Beyotime). Next, the proteins were separated by polyacrylamide gel electrophoresis (Beyotime) and transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore). The PVDF membrane was blocked with 5% non-fat milk (Servicebio Technology, Wuhan). Then we probed the target protein with the primary antibody overnight at 4°C as follows: Bim (1:1000, Abcam), Bcl-2 (1:2000, Abcam), cytochrome c (1:5000, ab133504), caspase-9/cleaved caspase-9 (1:2000, Abcam), and β-actin (1:1000, Abcam). The PVDF membrane was washed and incubated with goat-rabbit (1:10,000, Cell Signaling Technology) secondary antibody. The target proteins were detected using a chemiluminescence system (ChemiScope Touch, CLINX, Shanghai).
Cellular Uptake Mechanism of the DMP-039/mRNA Complex
C26 cells were plated in 24-well plates (3×104 cells) to determine the cellular uptake mechanism of the DMP-039/mRNA complex. C26 cells were first treated with different inhibitors for 30 min while being blocked with different concentrations of cRGD-R9 (0.04, 0.08, 0.12, 0.16, and 0.2 mg/mL), chlorpromazine hydrochloride (CPZ, 1 μg/mL), methyl-β-cyclodextrin (M-β-CD, 8.3 mM), amiloride (2 mM), or genistein (0.3 mM). These concentrations of the different inhibitors did not induce cytotoxicity. After treatment with the different inhibitors, cells were transfected with EGFP/DMP-039 (1:25, w/w) or EGFP/DMP (1:25, w/w) for 4 hours, and then, the medium in each well was replaced with DMEM containing 10% FBS. After transfection for 48 hours, flow cytometry was used to determine the transfection efficiency rate. As for fluorescence microscopy, 3×104 cells were seeded in chamber slides (Millicell, 4 well-glass) and cultured overnight. Then, the C26 cells were treated with different inhibitors according to well-established protocols. After transfecting for 48 hours, nuclei were stained with Hoechst (1 mg/mL, Solarbio), and plasma membranes were stained with Dil (10 mg/mL, Beyotime). A fluorescence microscope (ZEN880, Bamboo Living) was used to capture and analyze the fluorescence emission of different inhibitors.
In vivo Fluorescence Imaging
To evaluate the distribution of DMP-039 in vivo, we used an in vivo imaging system (Caliper Life Sciences) to record the experimental results. Before imaging, mice were intravenously injected with normal saline or 300 μL of DMP conjugated with Cy7 dye (ex:756 nm, em: 779 nm). After injection at 0, 2, 5, 10, 15, and 30 min, mice were euthanized, and their hearts, livers, spleens, lungs, and kidneys were immediately harvested. Then, the organs were placed into the imaging chamber, and the images of each mouse were recorded by PerkinElmer with exposure times of 4 s. Simultaneously, fluorescent images with a region of intensity (ROI) for each organ were analyzed by Living Image software 3.2.
Biodegradation Study in vivo
To evaluate the biocompatibility and biodegradation of DMP-039 in vivo, male Sprague-Dawley rats (200–220 g) were intravenously injected with DMP-039 (3 mg). After 24 hours, metabolism cages were used to collect the urine and feces of the DMP-039 group and the control group, which were stored at −80°C. For extraction of feces, fecal samples (2 g) were crushed with a food processor, dissolved in 10 mL acetonitrile, mixed well by vortexing, and centrifuged at 8000 rpm for 15 min. Then, the supernatant was removed and stored at 4°C for later use. For extraction of urine, 800 μL of urine was added to 1.2 mL acetonitrile, mixed well by vortexing, and centrifuged at 100,000 x g for 15 min. The supernatant was removed and stored at 4°C for later use. Furthermore, for the feces and urine samples, the solvent was removed by nitrogen gas at 37°C, and then, the sample was re-dissolved in 200 μL of acetonitrile. The supernatant was collected and stored at 4°C for biodegradation analysis using an AB SCIEX Qtrap 5500 (UFLC Lc-30AD, Shimadzu).
In vivo Tumor Inhibition Assay
To assess the therapeutic effect of the mBim/DMP-039 complex in an abdominal cavity metastatic model, 6–8 week-old BALB/c female mice were intraperitoneally injected with C26 tumor cells (2 × 105 cells in 100 μL). On day 2, the mice were randomly divided into four groups and were intraperitoneally injected every day for 17 treatments with normal saline (NS), DMP-039 (250 μg), mBim (10 μg)/DMP-039 complex, or pBim (5 μg)/CLP complex. On day 18, all mice were weighed before euthanasia. Then, their tumors and five internal organs (heart, liver, spleen, lungs, and kidneys) were harvested. After the tumors were weighed, all the tissues and organs were stored in 4% paraformaldehyde for further use.
We then assessed the therapeutic effect of the mBim/DMP-039 complex in the subcutaneous tumor model. Mice were subcutaneously injected with C26 tumor cells (1.5 × 106 cells in 100 μL), and on day 5, the mice were randomly divided into four groups. Every day, the mice received 19 treatments of NS, DMP-039 (250 μg), mBim (10 μg)/DMP-039 complex or pBim (5 μg)/CLP complex through intratumoral administration. The changes in tumor volume were monitored every second day after injection. On day 24, when the tumor size reached 1000 mm3 (calculated as 0.5 × length × width2), all mice were euthanized, and their tumors and the five internal organs were immediately harvested. After the tumors were weighed, all the tissues and organs were stored in 4% paraformaldehyde for further use.
We established a metastatic lung tumor model to assess the therapeutic effect of the mBim/DMP-039 complex. Mice were intravenously injected with C26 tumor cells (2 × 105 cells in 100 μL). The next day, the mice were randomly divided into four groups and intravenously injected every day for 13 treatments with NS, DMP-039 (300 μg), mBim (12 μg)/DMP-039, or pBim (5 μg)/CLP. On day 15, the mice were euthanized, and the five internal organs were immediately harvested. After the lungs were weighed, all the tissues and organs were stored in 4% paraformaldehyde for further use.
Immunohistochemistry Analysis
To examine the protein expression in a tumor through a series of immunological reactions of specific antibodies, tumor tissues were fixed with 4% paraformaldehyde to maintain the original morphological structure. After dehydration and transparency treatment, the appropriate tissue parts were chosen for embedding in paraffin wax, and then, 3–4-μm-thick slices of tissue were created. The hydrated sections were stained with hematoxylin-eosin (H&E). After H&E staining of lung sections, ImageJ software was used to measure the area of the entire lung and the relative area of tumors in the lung.
To detect apoptosis, we used a terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) kit (Promega) to stain paraffin sections. After blocking with 3% hydrogen peroxide, the sections were stained with Bim (1:50, Abcam), caspase-9 (1:300, Abcam), and caspase-3 (1;1000, Abcam) primary antibodies overnight at 4°C to identify the apoptosis mechanisms of Bim in tumors. Paraffin sections were incubated with anti-CD31 (1:50, Abcam) to evaluate the expression level of vascular endothelial growth factor in tumor vessels. The tissue sections were also stained with CD8 (1:250, Abcam), IFN-γ (1:500, Abcam), CD206 (1:1000, Abcam), TNF-α (1:500, Abcam), and Ly-6C (1:400, Biolegend) to analyze the immune cells that infiltrated in the metastatic lung tumor model. After incubating overnight at 4°C, the tissue sections were treated with a horseradish peroxidase-conjugated secondary antibody, and the sections were subsequently observed with a fluorescence microscope (Olympus, Japan).
Blood Tests
Blood tests are a standard method for evaluating the safety of biomaterials in vivo. Female BALB/c mice were divided into two groups and then intravenously injected with NS or mBim/DMP-039 complex (10:250, μg:μg). After 24 hours, the blood was collected and analyzed for mean corpuscular volume (MCV), mean platelet volume (MPV), hemoglobin (HGB), mean corpuscular hemoglobin concentration (MCHC), red blood cells (RBCs), platelet distribution width (PDW), mean corpuscular hemoglobin (MCH), and lymphocytes (Lymph).
Statistical Analysis
All the data are defined as the mean ± standard deviation (SD), and P < 0.05 was considered statistically significant. The experimental data were analyzed by GraphPad Prism 8.0 software with one-way and two-way ANOVA.
Results
Preparation and Characterization of DMP-039 Nanoparticles
In this work, a DMP nano-backbone was first formed by self-assembly of the cationic lipid DOTAP and mPEG-PCL block polymer into nano-sized micelles. In order to fuse with CPPs, the DMP nano-backbone was rendered active with maleimide-PEG-PCL (Mal-PEG-PCL), forming DMP-maleimide nanoparticles. Then, cRGD fused with R9 peptide was conjugated to DMP-maleimide by maleimide-thiol Michael addition, resulting in the DMP-039 nanoparticles (Scheme 1). As shown in Figure 1A and B, the average diameter of the DMP-039 nanoparticles was 268.9 ± 12.4 nm (polymer dispersity index (PDI) = 0.382), with an average zeta potential of 17.4 ± 0.5 mV.
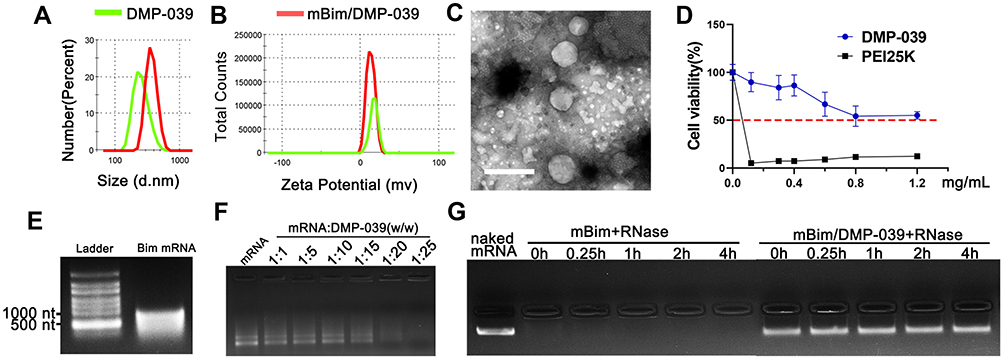 |
Figure 1 Characterization of the DMP-039/mRNA complex. (A) Size distribution of DMP-039 and mBim/DMP-039. (B) Zeta potential of DMP-039 and mBim/DMP-039. (C) Transmission electron microscopy (TEM) photomicrographs of DMP-039 stained by phosphotungstic acid solution (scale bar: 400 nm). (D) Cell viability assay of DMP-039 and PEI25K on 293T cells. (E) In vitro transcribed murine Bim mRNA (mBim) resolved by electrophoresis. (F) Gel retarding assay of the DMP-039/mRNA complex. (G) RNase protection assay of the mBim/DMP-039 complex. |
The morphology of the DMP-039 nanoparticles was then observed under TEM. As measured by TEM (Figure 1C), the morphology of the prepared DMP-039 nanoparticles was monodispersed with a diameter of nearly 250 nm, consistent with that of the size assessment results above. The cytotoxicity of the DMP-039 nanoparticles on 293T cells was assessed by MTT assay. According to our results, the IC50 of the DMP-039 nanoparticles was greater than 1.2 mg/mL, while that of PEI25K was merely 3 μg/mL (Figure 1D), suggesting a high degree of safety.
To prepare the mBim/DMP-039 complex, mRNA molecules were first synthesized using an in vitro transcription method followed by 5ʹ end-capping and 3ʹ end-poly(A) reaction. The purified murine mBim was then electrophoresed for verification. As shown in Figure 1E, a band of approximately 591 nt was illuminated on the gel, which was the expected gene length. The mBim/DMP-039 complex was then formed by mixing mBim with DMP-039. Relative to the DMP-039 nanoparticle, the particle size of the mBim/DMP-039 complex was increased to 381.6 ± 21.5 nm (PDI = 0.213), while a slight decrease in zeta potential was observed from 17.4 to 13.1 mV (Figure 1A and B). These results suggested that the mBim/DMP-039 complex was formed through electronic interaction and retained a nano-sized structure.
We then investigated the ability of DMP-039 to bind to mRNA, and ultimately, no obvious mRNA band was observed with a molar ratio of 25:1 (w/w) for DMP-039:mBim (Figure 1F). These results suggest that DMP-039 can effectively condense mRNA. We then further studied the ability of DMP-039 to protect against mRNA in vitro. As shown in Figure 1G, naked mBim was rapidly degraded in the presence of RNase (at final a concentration of 0.25 mg/mL), while no obvious degradation was observed in the mBim/DMP-039 complex groups. Moreover, no obvious change in the brightness of the mRNA band was observed at different time points for up to 4 hours, suggesting a strong ability of mRNA to protect DMP-039 against mBim. Therefore, our results suggest that the prepared DMP-039 nanoparticles can efficiently bind and protect mRNA molecules.
DMP-039 Efficiently Delivers mRNA in vitro
We then evaluated the efficiency of the DMP-039 nanoparticles to deliver mRNA in vitro. According to our results, DMP-039 delivered EGFP-encoded mRNA into C26 cells with high efficiency (Figure 2A). The transfection rate by the DMP-039/mRNA complex reached 41.29%, which was much higher than that of unmodified DMP (24.18%; P < 0.05) and that of the gold standard transfection agent, PEI25K (22.73%; P < 0.05) (Figure 2B). This strong mRNA delivery ability of DMP-039 was also verified with 293T cells. In this case, the transfection efficiency of DMP-039 reached 98.06%, while those values for DMP and PEI25K were 85.36% (P < 0.05) and 87.34% (P < 0.05) (Figure 2C), respectively.
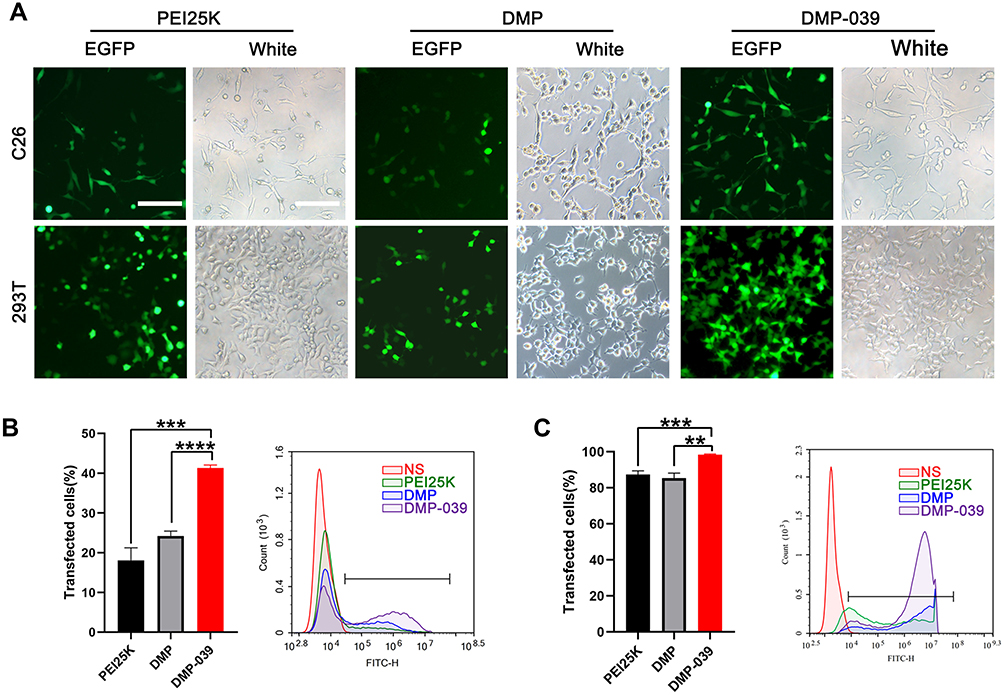 |
Figure 2 In vitro gene transfection by the DMP-039/mRNA complex. (A) Fluorescence microscopy images of C26 and 293T cells (scale bar: 100 μm). (B) The efficiency of transfection of C26 cells (***P < 0.001, ****P < 0.0001) and (C) 293T cells (**P < 0.01, ***P < 0.001) analyzed by flow cytometry. |
Relative to the DMP-039 group, treatment with PEI25K resulted in strong toxicity with obvious cell shrinkage, deformation, and damage. Therefore, our results suggested that the modification of the cRGD-R9 peptide on a DMP nano-backbone greatly contributed to the high mRNA delivery ability of the DMP-039 vector. As compared with both DMP and PEI25K, DMP-039 showed high efficiency for mRNA-based transfection into C26 cancer cells.
Cellular Uptake Mechanism Study of the DMP-039/mRNA Complex
We then studied the cellular uptake mechanism of the DMP-039 complex. In general, four endocytosis pathways, including clathrin-mediated, caveolin-mediated, lipid raft-mediated endocytosis, and pinocytosis, are considered the major pathways in the uptake process of nano-sized cargo. Therefore, we pretreated C26 cells before transfection with diverse inhibitors of the pathways mentioned above, including lipid raft-mediated endocytosis (methyl-β-cyclodextrin, M-β-CD), clathrin-mediated endocytosis (chlorpromazine hydrochloride, CPZ), caveolin-mediated endocytosis (genistein), and pinocytosis (amiloride).
As the results show in Figure 3A, relative to the untreated DMP-039/mRNA complex group, a lower intensity of EGFP fluorescence was observed in the M-β-CD–treated, CPZ-treated, and genistein-treated groups (white arrows). Moreover, as calculated by flow cytometry, relative to the untreated group, uptake rate reductions of 85%, 83%, and 77% (P < 0.05) were detected in these three groups. A smaller impact of nearly 30% was detected in the amiloride-treated group when compared to the untreated group (P < 0.05).
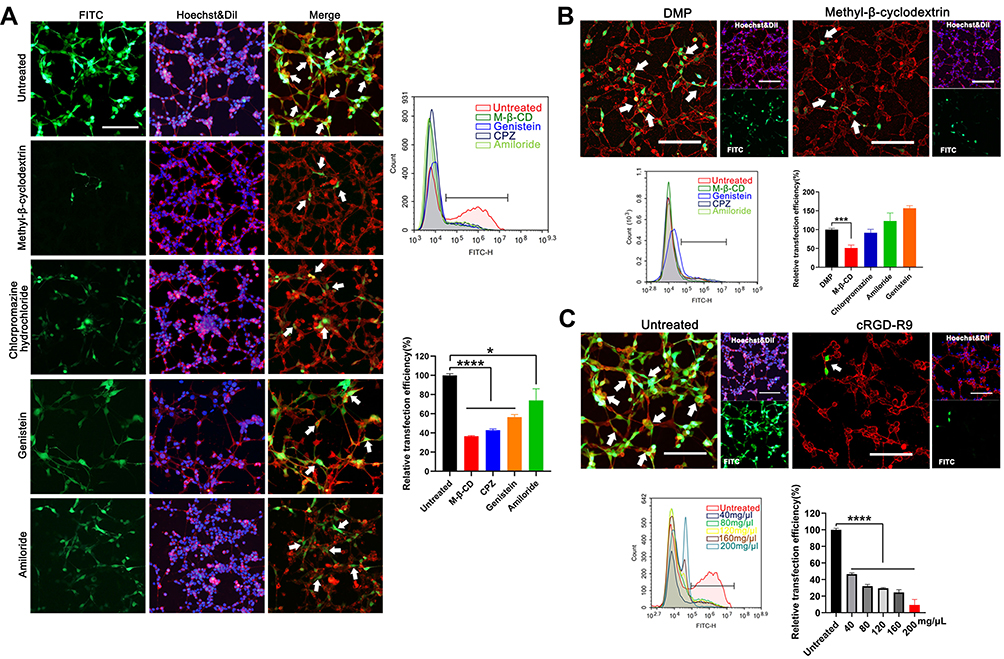 |
Figure 3 Study on the cellular uptake mechanism of the DMP-039/mRNA complex. (A) Fluorescent images of internalization of the DMP-039/mRNA complex after being treated with various inhibitors; the uptake rates were calculated by flow cytometry (scale bar: 100 μm) (*P < 0.05, ****P < 0.0001). Representative EGFP mRNA signals are marked by white arrows. (B) The internalization of the DMP/mRNA complex was found to be suppressed only by methyl-β-cyclodextrin, as determined by fluorescent microscopy and flow cytometry (scale bar: 100 μm) (***P < 0.001). Representative EGFP mRNA signals are marked by white arrows. (C) The internalization of the DMP-039/mRNA complex was obviously suppressed by cRGD-R9 (200 mg/μL), as determined by fluorescent microscopy and flow cytometry (scale bar: 100 μm) (****P < 0.0001). Representative EGFP mRNA signals are marked by white arrows. All cell nuclei were stained with Hoechst (blue), and plasma membranes were stained with Dil (red) (scale bar: 100 μm). |
The cellular uptake mechanism of the DMP/mRNA complex without CPP functionalization was also studied. As shown in Figure 3B, the cellular uptake of DMP-delivered mRNA was found to be only suppressed by M-β-CD, with an inhibition rate of 49% (P < 0.05). This suggested that a single main internalization pathway was involved in the delivery process of the DMP/mRNA complex. To further verify this, C26 cells were pretreated with free cRGD-R9 peptide for 30 min before transfection with the DMP-039/mRNA complex. As shown in Figure 3C, obvious attenuated fluorescence was observed, with an average inhibition rate up to 90.64% (P < 0.0001). Additionally, when the cRGD-R9 peptide was increased from 40 to 200 mg/μL, increased uptake suppression effects were detected in C26 cells, and the inhibition rate increased from 46% to 9% (P < 0.0001). These results further indicate a contributing role of the cRGD-R9 peptide in promoting mRNA delivery by DMP-039. In total, our results suggest that multiple uptake pathways were involved in the cellular uptake process of the DMP-039/mRNA complex. The functionalization of the DMP backbone with CPPs obviously promoted mRNA internalization by triggering more uptake pathways.
The mBim/DMP-039 Complex Efficiently Inhibits C26 Cancer Cell Growth in vitro
We then studied the anti-cancer mechanism of the mBim/DMP-039 complex with respect to C26 cells in vitro. Because DMP-039 is potent in mRNA delivery, we first determined whether treatment with the mBim/DMP-039 complex resulted in high levels of mBim and protein. As tested by qRT-PCR, a high level of mBim was detected in the mBim/DMP-039 complex group, with a 2×104-fold increase relative to DMP-039-treated cells (P < 0.05) (Figure 4A). Additionally, the expression level of Bim protein significantly increased in the mBim/DMP-039 complex group (Figure 4B).
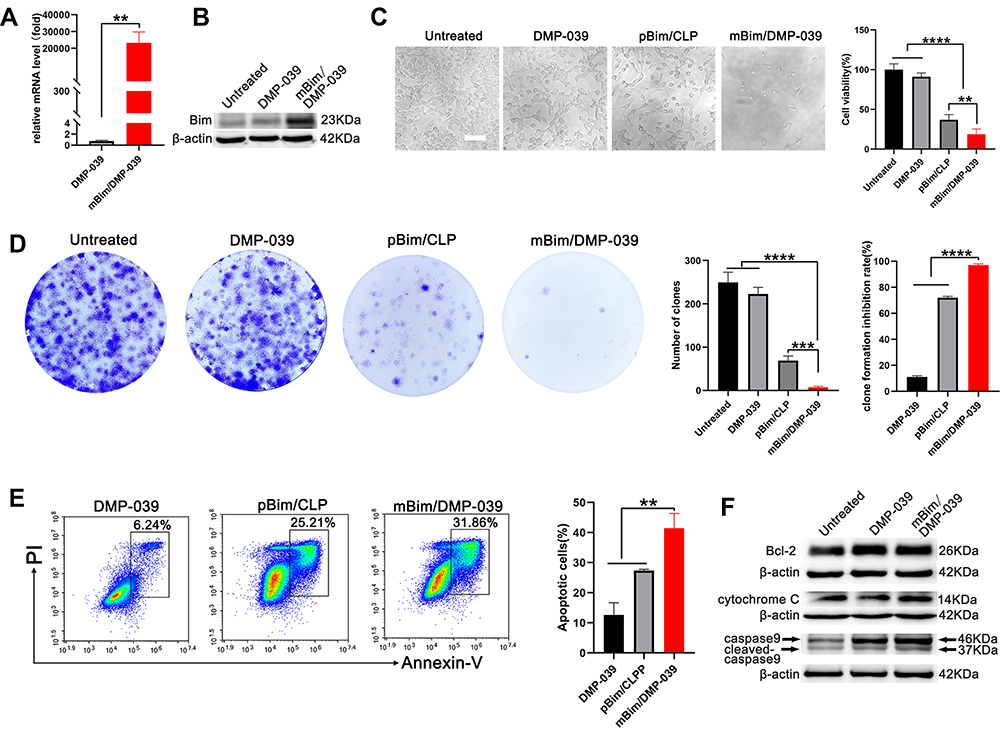 |
Figure 4 In vitro anti-cancer study of the mBim/DMP-039 complex. (A) The level of Bim mRNA in C26 cells after transfection (**P < 0.01). (B) The level of Bim protein in each treatment group. (C) Image of C26 cells after treatment with the mBim/DMP-039 complex (scale bar: 100 μm) (**P < 0.01, ****P < 0.0001). Cell viabilities were measured by the MTT assay. (D) Detection of the anti-proliferation effect of the mBim/DMP-039 complex by clonogenic assay. Inhibition rates were calculated based on clone numbers (***P < 0.001, ****P < 0.0001). (E) The mBim/DMP-039 complex efficiently induced apoptosis in C26 cells as determined by flow cytometry (**P < 0.01). (F) Western blot analysis of apoptosis-related proteins. |
These results suggest that DMP-039 can deliver mBim into C26 cells with high efficiency. The anti-proliferation effect of the mBim/DMP-039 complex was then studied by MTT assay. According to our results, treatment with the mBim/DMP-039 complex obviously decreased C26 cell viability (Figure 4C). Relative to the untreated group, only 18.67% of cells survived in the mBim/DMP-039 complex group, suggesting a lesser degree of cell viability than that in the DMP-039 group (91.07%; P < 0.05) (Figure 4C). The anti-proliferation ability of the mBim/DMP-039 complex group was found to be superior to that of the pBim/CLP group (81.33% vs 63.04%; P < 0.05).
The ability of the mBim/DMP-039 complex to inhibit cell growth was also evaluated by clonogenic assay. As the results show in Figure 4D, there were fewer C26 cell clones in the mBim/DMP-039 complex group, whose calculated average number of clones was 7 ± 2, and the average inhibition rate was 97% (P < 0.0001) (Figure 4D). On the contrary, those of the untreated and DMP-039 groups were 249 ± 19 (P < 0.0001) and 223 ± 12 (P < 0.0001) (Figure 4D), respectively. Relative to the mBim/DMP-039 complex group, treatment with the pBim/CLP complex resulted in additional C26 cell clones (69 ± 9; P < 0.05), consistent with the MTT results. These findings suggest that the mBim/DMP-039 complex can highly express mBim in cells and has a strong ability to inhibit growth in vitro.
As an important protein in the Bcl-2 family, it has been reported that Bim induces cell apoptosis. Hence, we subsequently studied whether the inhibition effect of the mBim/DMP-039 complex was a result of apoptosis induction. According to our results, relative to the DMP-039 groups, the mBim/DMP-039 complex caused obvious cell apoptosis (Figure 4E). With the assistance of Annexin V/PI staining, the apoptotic cell rate for the mBim/DMP-039 complex group was 31.86%, while those of the pBim/CLP complex and DMP-039 groups were 25.21% and 6.24% (P < 0.05) (Figure 4E). Additionally, decreased Bcl-2 protein levels were detected by Western blotting following treatment with the mBim/DMP-039 complex, while the levels of caspase-9/cleaved caspase-9 and cytochrome c were obviously elevated (Figure 4F). These results suggest that the mBim/DMP-039 complex can efficiently suppress C26 cell growth by inducing mitochondria-dependent apoptosis.
The mBim/DMP-039 Complex Suppresses the C26 Cancer Model Through Local Administration
We then verified the treatment effect of the mBim/DMP-039 complex by local administration. The therapeutic effect of the mBim/DMP-039 complex was first evaluated in a C26 colon cancer abdominal cavity metastasis model. Mice were intraperitoneally treated with NS, DMP-039, pBim (5 μg)/CLP complex, or mBim (10 μg)/DMP-039 complex, and we observed fewer tumor nodules (white arrows, Figure 5A and B) in the mBim/DMP-039 complex group, which exhibited a strong anti-cancer effect. The abdominal tumor nodules were then harvested, and statistical analysis was performed.
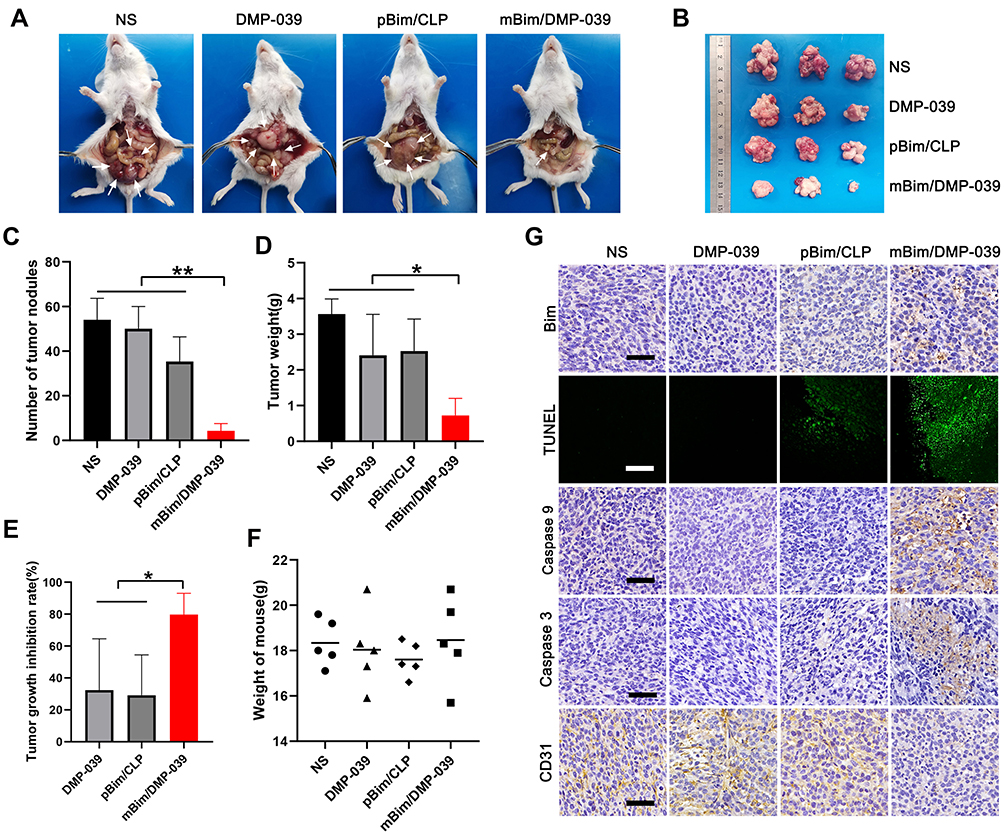 |
Figure 5 The mBim/DMP-039 complex inhibited abdominal cavity metastatic tumor growth in vivo. (A) Images of representative mice from each treatment group. Tumor nodules (partial) are indicated by white arrows. (B) Tumor nodules harvested from each group. (C) The average number of tumor nodules in each group (**P < 0.01). (D) The average tumor weight in each group (*P < 0.05). (E) The tumor growth inhibition rate in each group (*P < 0.05). (F) The body weights of mice from each group before euthanasia. (G) Immunohistochemical evaluation of tumor tissues from each group and H&E analysis of main organ tissues from the mBim/DMP-039 complex group (scale bars: 50 μm). |
The same trend was observed for the average number of tumor nodules in the mBim/DMP-039 complex group (5 ± 2), which was less than that in the NS group (54 ± 7; P < 0.05), DMP-039 group (50 ± 8; P < 0.05), and pBim/CLP complex group (35 ± 9; P < 0.05) (Figure 5C). Similarly, a lower average tumor weight was calculated in the mBim/DMP-039 complex group (0.72 ± 0.38 g) (Figure 5D), while those of the NS, DMP-039, and pBim/CLP complex groups were 3.56 ± 0.35 g (P < 0.05), 2.41 ± 0.93 g (P < 0.05), and 2.52 ± 0.74 g (P < 0.05), respectively. This resulted in a 79.71% inhibition rate in the mBim/DMP-039 complex group (P < 0.05) (Figure 5E). In addition, the mouse body weights in all groups showed no significant change (Figure 5F).
We then studied the therapeutic mechanism of the mBim/DMP-039 complex by immunohistochemistry (IHC). As shown in Figure 5G, as compared with the NS, DMP-039, and pBim/CLP groups, we observed more positive Bim protein signals in the mBim/DMP-039 complex group, which indicated that mBim can be expressed in tumor tissue by the DMP-039 delivery system. Because the Bim gene can induce cancer cell apoptosis, we evaluated the activities of apoptosis-related proteins in tumor tissue. Much stronger signals from TUNEL-positive cells were observed in the mBim/DMP-039 complex group as compared to the other groups. Additionally, there were strong positive signals from caspase-9 and caspase-3 proteins in tumor sections of the mBim/DMP-039 complex group, suggesting obvious apoptosis induction. However, CD31 staining demonstrated that microvessel formation was obviously inhibited after treatment with the mBim/DMP-039 complex, suggesting strong anti-angiogenetic effects. Additionally, there were no obvious pathology changes or serious adverse effects in the main organs of treated mice, which resolved concerns regarding the safety of the in vivo therapy (Figure 6A). These results suggest that DMP-039-delivered mBim can effectively inhibit the growth of C26 abdominal metastatic tumors by multiple mechanisms.
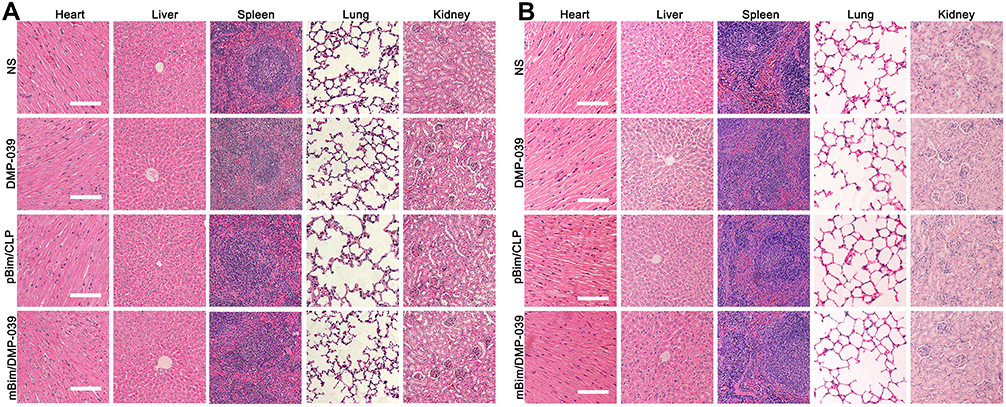 |
Figure 6 H&E analysis in vivo. (A) H&E analysis of the main organ tissues in the C26 abdominal cavity metastatic model (scale bars: 50 μm). (B) H&E analysis of the main organ tissues in the C26 subcutaneous xenograft model (scale bars: 50 μm). |
We then investigated the therapeutic potential of the mBim/DMP-039 complex to treat C26 subcutaneously xenografted mice by intratumoral administration. As shown in Figure 7A, the amounts of subcutaneous tumor tissue in the mBim (10 μg)/DMP-039 complex group were observed to be less than that in other groups, suggesting a strong anti-cancer effect. Subcutaneous tumor tissues were then harvested and statistically analyzed. The tumor growth curves recorded in Figure 7B show that in the mBim/DMP-039 complex group, the growth in average tumor volume was slower than that in the NS (P < 0.05), DMP-039 (P < 0.05), and pBim (5 μg)/CLP complex (P < 0.05) groups. Additionally, a lower level of average tumor weight was calculated in the mBim/DMP-039 complex group (0.15 ± 0.03 g) (Figure 7C), while those of the NS, DMP-039, and pBim/CLP complex groups were 0.38 ± 0.08 g (P < 0.05), 1.34 ± 0.12 g (P < 0.05), and 0.74 ± 0.16 g (P < 0.05), respectively. These results indicate that intratumoral administration of the mBim/DMP-039 complex can efficiently suppress the growth in a C26 subcutaneously xenografted model.
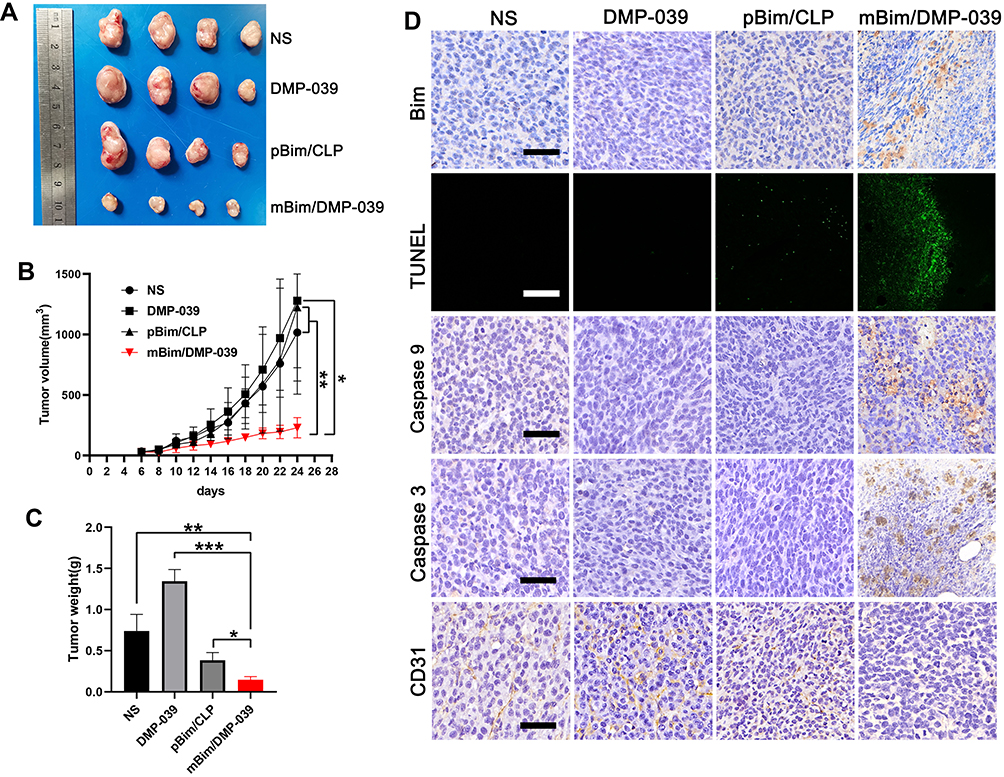 |
Figure 7 The mBim/DMP-039 complex suppressing the C26 subcutaneous xenograft model in vivo. (A) Tumors collected from each group. (B) Tumor growth curves from each treatment group (*P < 0.05, **P < 0.01). (C) Average tumor weights (*P < 0.05, **P < 0.01, ***P < 0.001). (D) Immunohistochemical evaluation of tumor tissue (scale bars: 50 μm). (E) H&E analysis of the main organs from the mBim DMP-039 complex group (scale bars: 50 μm). |
We then studied the therapeutic mechanism of the mBim/DMP-039 complex by IHC. As shown in Figure 7D, relative to the NS, DMP-039, and pBim/CLP complex groups, we observed more positive signals from Bim in the mBim/DMP-039 complex group, which indicated that mBim can be efficiently delivered and expressed in tumor tissue by the DMP-039 delivery system. Because the Bim gene can induce cancer cell apoptosis, more TUNEL-positive cells were observed in the mBim/DMP-039 complex group as compared to the other groups. There were strong positive signals from caspase-9 and caspase-3 proteins in tumor sections from the mBim/DMP-039 complex group, suggesting obvious apoptosis induction. In addition, CD31 staining demonstrated that microvessel formation was obviously inhibited after treatment with the mBim/DMP-039 complex, suggesting strong anti-angiogenic effects. Additionally, there were no obvious pathology changes or serious adverse effects in the main organs of treated mice, which relieved concerns regarding the safety of the in vivo therapy (Figure 6B). These results suggest that the mBim/DMP-039 complex can inhibit the growth of a C26 subcutaneously xenografted tumor by inducing cancer cell apoptosis.
The mBim/DMP-039 Complex Suppresses a C26 Pulmonary Metastasis Model Through Systemic Administration
We further intended to evaluate the therapeutic potential of the mBim/DMP-039 complex through systemic administration. Relative to local treatment, there are extra concerns regarding intravenous administration and the distribution and safety issues of gene delivery systems. Therefore, we first analyzed the in vivo distribution behavior of the DMP-039 nanoparticles. As shown in Figure 8A, starting at 2 min after intravenous injection, high fluorescence intensity was detected in the lungs. At 5 min post-administration, a peak in lung distribution was reached, with a distribution rate of 38%, which was the second-highest percentage after the kidneys. This behavior lasted for 30 min, suggesting that sustained lung accumulation occurred for the systemically administered mBim/DMP-039 complex.
 |
Figure 8 (A) In vivo distribution study of the DMP-039 nanoparticles upon intravenous injection; the fluorescence intensities were detected and calculated. (B) The degradation and metabolic behaviors of the DMP-039 nanoparticles upon intravenous injection. Urine samples were analyzed by HPLC-MS. |
The metabolic behavior of the mBim/DMP-039 complex was also studied. With the assistance of HPLC-MS, the urine and feces samples collected from DMP-039-treated mice were analyzed. As shown in Figure 8B, relative to untreated mice, a strong absorption peak at 2.92 min was detected in urine samples from the DMP-039 group, consistent with that of the mPEG-PCL standard. Furthermore, when observing the mass spectrum of this newly formed peak, as compared to mPEG-PCL standard products, the number of peaks in the DMP-039 group was greater, which might represent the potential degradation products of mPEG-PCL fragments. However, there was no obvious characteristic absorption peak detected in the fecal samples (data not shown). These results suggest that DMP-039 nanoparticles can be degraded and eliminated from the body by urine.
We further investigated the therapeutic potential of the mBim/DMP-039 complex to treat a C26 pulmonary metastatic tumor model by i.v. administration. As shown in Figure 9A, the mBim (12 μg)/DMP-039 complex group exhibited obvious anti-cancer effects after 13 doses. Almost no pathology changes were observed on the surface of lung tissues from the mBim–DMP-039 complex group. On the contrary, the surface of the lung tissue was incomplete and swollen, with a large number of tumor nodules in the NS, DMP-039, and pBim (5 μg)/CLP complex groups.
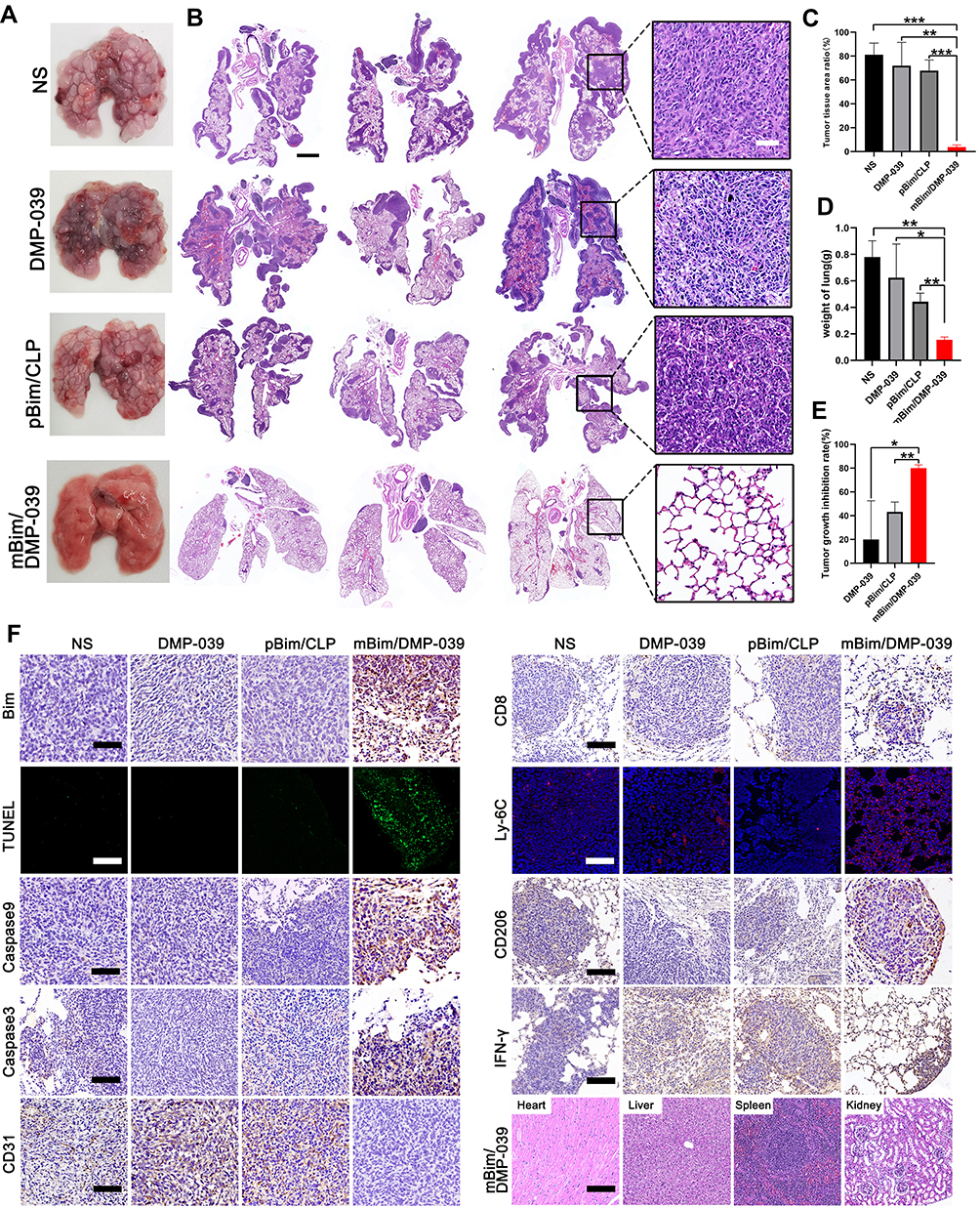 |
Figure 9 Therapeutic effect of the mBim/DMP-039 complex on the C26 pulmonary metastases model upon systemic administration. (A) Representative lung tissues harvested from each group. (B) H&E analysis of lung tissues using a whole lung view (scale bars: 2 mm) and partially enlarged view (scale bars: 50 μm). (C) Average tumor mass area rate for each group (**P < 0.01, ***P < 0.001). (D) Average weight of lungs (*P < 0.05, **P < 0.01). (E) Tumor growth inhibition rate for each group (*P < 0.05, **P < 0.01). (F) Immunohistochemical evaluation of tumor tissues from each group and H&E analysis of the main organ tissues from the mBim/DMP-039 complex group (scale bars: 50 μm). |
We then investigated the interior of these lungs with the assistance of H&E staining. As shown in Figure 9B, large areas of tumors were observed in whole lung sections from the NS, DMP-039, and pBim/CLP complex groups, with massive immune cell infiltration and severe lung alveolar damage. On the contrary, the lung tissues from the mBim/DMP-039 complex group were much healthier, with fewer tumor mass areas (average rate of 3.83% in the tumor mass area vs 67.8% in the pBim/CLP complex group, 72% in the DMP-039 group, and 81.12% in the NS group) (Figure 9C). The average lung weight in the mBim/DMP-039 complex group was 0.16 ± 0.17 g, which was significantly lower than that of the pBim/CLP complex group (0.41 ± 0.05 g), DMP-039 group (0.62 ± 0.21 g), and NS group (0.78 ± 0.1 g) (Figure 9D). This resulted in an inhibition rate of 80.04% in the mBim/DMP-039 complex group (P < 0.05) (Figure 9E). These results suggest that the i.v. injected mBim/DMP-039 complex can efficiently inhibit C26 pulmonary metastatic tumors.
The therapeutic mechanisms were then investigated. As shown in Figure 9F, as compared with the NS, DMP-039, and pBim/CLP complex groups, we observed much higher Bim protein levels in the mBim/DMP-039 complex group, which indicated that mBim can be expressed in tumor tissue by the DMP-039 delivery system. We then studied the activities of apoptosis-related proteins in tumor tissue by using mBim. More TUNEL-positive cells were observed in the mBim/DMP-039 group than in the other groups. There were higher levels of caspase-9 and caspase-3 proteins in the tumor sections of the mBim/DMP-039 complex group, suggesting obvious apoptosis induction. Much higher levels of CD8, Ly-6C, and CD206 were detected in the mBim/DMP-039 complex treatment group than in the other groups, suggesting enhanced infiltration of immune cells, such as T-cells, monocytes, and macrophages.
The strong positive signal of IFN-γ also suggested that there is a potential immune-activation effect by the mBim/DMP-039 complex group. After systemic treatment with the mBim/DMP-039 complex for 13 doses, there were no obvious pathology changes or serious adverse effects in the main organs of treated mice (Figure 9F). Additionally, there were no significant changes or toxicity in the blood circulation, including with respect to MCV, MPV, HGB, MCHC, RBC, PDW, MCH, and Lymph (Figure 10). Our results indicate that i.v. administration of the mBim/DMP-039 complex facilitates strong suppression in tumor growth by inducing apoptosis and immune reactions with a high degree of safety in a C26 lung metastatic tumor model.
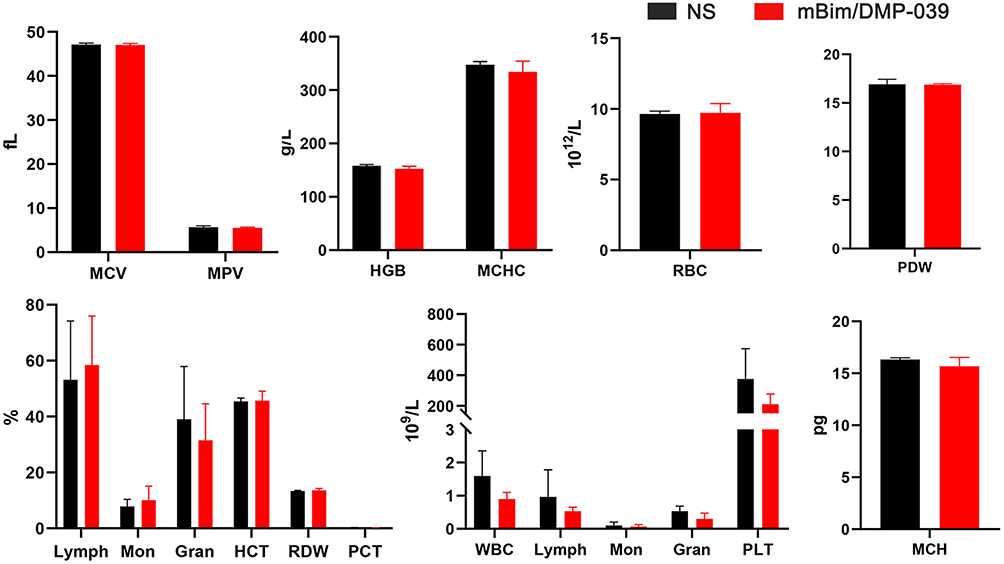 |
Figure 10 Blood routine analysis of mBim/DMP-039 complex-treated mice after intravenous administration. |
Discussion
Currently, mRNA-based gene therapies have exhibited great potential in a variety of applications, including cancer immunotherapy, vaccination, protein substitution, and treatment of genetic diseases.37–39 There are several advantages to mRNA-based therapy.40 For example, unlike plasmid DNA, mRNA does not need to enter the nucleus to be functional. Also, as a gene vector, mRNA does not integrate into the genome. Additionally, mRNA can be easily synthesized by in vitro transcription (IVT) processes, facilitating manufacturing. However, one of the main challenges faced by mRNA-based clinical applications is the success of the delivery system.
These obstacles are highly correlated with the stability, immunogenicity, efficiency, and safety of mRNA cargos. The need to overcome this hurdle thus has promoted studies of mRNA delivery vectors in recent years. For example, Miao et al reported on the use of multi-component nanoparticles to deliver mRNA, which consisted of ionizable lipidoid, DOPE, PEG2000, and cholesterol.41 Capasso Palmiero et al synthesized a poly(β-amino ester) (PBAE) terpolymer to deliver mRNA with a high accumulation in the spleen.42 Hoerr et al reported that protamine-RNA complexes could only protect mRNA in the presence of serum, which led to a strong immune response.43 Choi et al prepared a graphene oxide (GO)-polyethylenimine (PEI25K) complex to deliver mRNA in vitro.44 Although much progress has been made in these mRNA vector studies, limitations still exist in many areas, such as stability, homogeneity, and safety. One explanation for these limitations is that most of those “multi-vector systems” contain multiple components or are complicated in structure, which leads to uncertainty regarding the prepared mRNA formulations.
In this study, we attempted to prepare a single mRNA nano-delivery system by functionally modifying a self-assembled DOTAP-mPEG-PCL hybrid micelle (DMP). The DMP was conjugated with a chimeric CPP, cRGD-R9, forming a DMP-R9-cRGD (DMP-039) nanoparticle (Scheme 2). The prepared DMP-039 nanoparticle was capable of condensing and protecting mRNA, resulting in the promotion of cellular uptake and increased mRNA transfection efficiency in vitro. Our results indicate that these properties of DMP-039 fulfill the demands for a competent mRNA delivery system. More importantly, relative to previously reported systems, DMP-039 is a single-vector system with a simple composition.
The designed DMP-039 cationic nanoparticles interact by negatively charging mRNA for delivery and can also protect mRNA from RNase degradation, resulting in greater stability of protein translation in vivo. At the same time, the toxicity of DMP-039 is low, which renders it a practical mRNA gene vector. At present, there are many frequently used cationic transfection agents, such as Lipofectamine 2000 or cationic PEI. Their abilities in mRNA delivery have also been also evaluated. For example, Huang et al reported that an EGFP/PEI complex triggered a stronger green fluorescence than that of a mRNA and Lipofectamine 2000 complex.45 However, a high transfection rate is associated with the use of these agents, and this results in an unavoidable degree of cell toxicity that affects the safety in vivo.
In our study, the efficiency with which DMP-039 delivered mRNA reached 45%, which was higher than the efficiencies of many multi-vector systems. It is likely that the modification with CPP incorporated into our design contributes to this high efficiency. Here, two CPPs, the cyclic RGD peptide and nona-arginine, were selected to create a chimeric two-block CPP. CPP is widely used in drug delivery systems because of its ability to promote membrane penetration and cargo entry. For example, Cherukula et al reported that cRGD-modified nanoparticles enhanced intracellular uptake with low toxicity during the treatment of C26 cells.46 Yu et al synthesized cRGD-installed unimer polyion complex–assembled gold nanoparticles to deliver E6 siRNA into HeLa tumors, which enhanced the cellular internalization so that subcutaneous tumors could be treated.47 Martínez-Jothar et al prepared a cRGD-functionalized polymeric nanoparticle for endothelial cell targeting in vivo.48
In our study, modification of the cRGD-R9 peptide was shown to greatly promote mRNA internalization by cancer cells. As a nano-sized particle with a comparatively large size, the uptake of the DMP-039/mRNA complex is not easy. However, our results demonstrated that the DMP-039/mRNA complex can trigger multiple uptake pathways, including lipid raft-mediated endocytosis, clathrin-mediated endocytosis, caveolin-mediated endocytosis, and pinocytosis. We further reveal that cRGD also played an important role during entry into the cell membrane. Therefore, the functional modification of the DMP backbone with the chimeric cRGD-R9 peptide greatly contributed to the high efficiency of the DMP-039 nanoparticles, which can be used as a novel single-vector mRNA delivery system.
The systemic administration of therapeutics is always of great interest for drug development. Intravenous injection allows a high concentration of drugs to enter the blood directly after infusion. This is an ideal property for mRNA-based cancer therapy. In recent years, advances have been made by researchers in this area. For example, Islam et al prepared a lipid-PEG shell to coat a PTEN mRNA complex, and this complex was able to significantly suppress the tumor growth of a PCa xenograft model though i.v. administration.49 Lei et al synthesized a protamine/cationic liposome system to deliver IL15 mRNA, and this complex was able to treat colon cancer in a lung metastatic tumor model by systemic administration.50 Pardi et al reported that LNP-supported delivery of modified mRNA resulted in high translation rates for a short time by intravenous administration.51
Despite these previous studies, however, intravenous injection-based gene therapy study is still in the preclinical stage. Regarding successful systemic delivery, difficulties exist in maintaining the stability, immunogenicity, and efficiency of the mRNA itself. Achieving ideal therapeutic effects with high safety is always in demand. In our study, DMP-039-delivered mBim was evaluated for its availability to manage a lung metastasis model when administered systemically. Our results indicated that lung tissue can maintain a complete lung alveolar morphology treatment with the mBim/DMP-039 complex, which induced cancer cell apoptosis and immune cell clearance.
Several factors might contribute to the delivery efficiency of the mBim/DMP-039 complex in vivo. For example, the in vivo distribution results showed that a high fluorescence intensity was mainly detected in the lungs. This suggested that the accumulation in the lungs of the systemically administered mBim/DMP-039 complex was sustained. Our metabolism study also showed that degradation products of the mPEG-PCL fragment absorption peak were detectable in urine samples of the DMP-039 complex, which suggested that DMP-039 can protect nucleic acid from damage by RNase in vivo and facilitate its uptake into cells.
Relative to previously reported mRNA vectors, DMP-039 can interact with mRNA, forming a nano-complex. Thus, via intravenous administration, the DMP-039 complex can protect mRNA from damage in the blood and facilitate accumulation in the lungs. After generating a therapeutic effect, DMP-039 nanoparticles might degrade and be safely eliminated from the body. These advantages of DMP-039 support its inclusion in intravenous injections and therapeutics. Therefore, our findings suggest that DMP-039 modified with CPPs constitutes an available ideal method for a single-vector delivery system with a high level of safety for in vivo gene therapy in clinical cancer applications.
Conclusion
In this work, a novel mRNA-based gene delivery system was created using a DMP nano-backbone and was introduced into CPPs (cRGD and R9). The prepared DMP-039 nanoparticles showed a high mRNA delivery capacity and protected mRNA from RNase. Here, when we introduced the additional suicide gene of mBim, the mBim/DMP-039 complex exhibited strong inhibition and migration therapeutic effects in multiple colon cancer models by inducing mitochondrial apoptosis. Our results indicate that DMP-039 is an available single-vector means for mRNA-based gene therapy in clinical cancer applications.
 |
Scheme 1 The synthesis process for DMP-039 nanoparticles. |
 |
Scheme 2 A schematic view of the formulation and therapeutic processes of the mBim/DMP-039 complex. |
Acknowledgments
This work was supported by the Key Research and Development Program of the Science and Technology Department of Sichuan Province (2020YFS0200), the Science and Technology Plan Project of the Science and Technology Department of Sichuan (20YYJC1516), and the Science Foundation of Chengdu (2020-YF05-00240-SN). We would like to thank Guangzhou Sagene Biotech Co., Ltd. for providing assistance with the illustrations during the preparation of this manuscript. We also thank Accdon for its linguistic assistance during the preparation of this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi:10.3322/caac.21590
2. Miller KD, Nogueira L, Mariotto AB, et al. Cancer treatment and survivorship statistics, 2019. CA Cancer J Clin. 2019;69(5):363–385. doi:10.3322/caac.21565
3. Siegel RL, Miller KD, Goding Sauer A, et al. Colorectal cancer statistics, 2020. CA Cancer J Clin. 2020;70(3):145–164. doi:10.3322/caac.21601
4. Ladabaum U, Dominitz JA, Kahi C, Schoen RE. Strategies for colorectal cancer screening. Gastroenterology. 2020;158(2):418–432. doi:10.1053/j.gastro.2019.06.043
5. Kaleta-Richter M, Kawczyk-Krupka A, Aebisher D, Bartusik-Aebisher D, Czuba Z, Cieślar G. The capability and potential of new forms of personalized colon cancer treatment: immunotherapy and photodynamic therapy. Photodiagnosis Photodyn Ther. 2019;25:253–258. doi:10.1016/j.pdpdt.2019.01.004
6. Luo C, Miao L, Zhao Y, et al. A novel cationic lipid with intrinsic antitumor activity to facilitate gene therapy of TRAIL DNA. Biomaterials. 2016;102:239–248. doi:10.1016/j.biomaterials.2016.06.030
7. Tauriello DVF, Palomo-Ponce S, Stork D, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554(7693):538–543. doi:10.1038/nature25492
8. Zhou M, Liu X, Li Z, Huang Q, Li F, Li CY. Caspase-3 regulates the migration, invasion and metastasis of colon cancer cells. Int J Cancer. 2018;143(4):921–930. doi:10.1002/ijc.31374
9. Chen EX, Jonker DJ, Loree JM, et al. Effect of combined immune checkpoint inhibition vs best supportive care alone in patients with advanced colorectal cancer: the Canadian cancer trials group co.26 study. JAMA Oncol. 2020;6(6):831–838. doi:10.1001/jamaoncol.2020.0910
10. Toor SM, Murshed K, Al-Dhaheri M, Khawar M, Abu Nada M, Elkord E. Immune checkpoints in circulating and tumor-infiltrating CD4(+) T cell subsets in colorectal cancer patients. Front Immunol. 2019;10:2936. doi:10.3389/fimmu.2019.02936
11. Liang Q, Monetti C, Shutova MV, et al. Linking a cell-division gene and a suicide gene to define and improve cell therapy safety. Nature. 2018;563(7733):701–704. doi:10.1038/s41586-018-0733-7
12. Kuo WY, Hwu L, Wu CY, Lee JS, Chang CW, Liu RS. STAT3/NF-κB-regulated lentiviral TK/GCV suicide gene therapy for cisplatin-resistant triple-negative breast cancer. Theranostics. 2017;7(3):647–663. doi:10.7150/thno.16827
13. Chi X, Nguyen D, Pemberton JM, et al. The carboxyl-terminal sequence of bim enables bax activation and killing of unprimed cells. eLife. 2020;9:e44525. doi:10.7554/eLife.44525
14. Hildeman DA, Zhu Y, Mitchell TC, et al. Activated T cell death in vivo mediated by proapoptotic bcl-2 family member bim. Immunity. 2002;16(6):759–767. doi:10.1016/s1074-7613(02)00322-9
15. Qian L, Van Laake LW, Huang Y, Liu S, Wendland MF, Srivastava D. miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J Exp Med. 2011;208(3):549–560. doi:10.1084/jem.20101547
16. Krishnamurthy B, Chee J, Jhala G, et al. BIM deficiency protects NOD mice from diabetes by diverting thymocytes to regulatory T cells. Diabetes. 2015;64(9):3229–3238. doi:10.2337/db14-1851
17. Biswas SC, Shi Y, Vonsattel JP, Leung CL, Troy CM, Greene LA. Bim is elevated in Alzheimer’s disease neurons and is required for beta-amyloid-induced neuronal apoptosis. J Neurosci. 2007;27(4):893–900. doi:10.1523/jneurosci.3524-06.2007
18. Lauer C, Brunner T, Corazza N. The proapoptotic Bcl-2 family member Bim plays a central role during the development of virus-induced hepatitis. J Immunol. 2012;188(2):916–922. doi:10.4049/jimmunol.1101864
19. Lei S, Zhang X, Li J, et al. Current progress in messenger RNA-based gene therapy. J Biomed Nanotechnol. 2020;16(7):1018–1044. doi:10.1166/jbn.2020.2961
20. Sahin U, Muik A, Derhovanessian E, et al. COVID-19 vaccine BNT162b1 elicits human antibody and T(H)1 T cell responses. Nature. 2020;586(7830):594–599. doi:10.1038/s41586-020-2814-7
21. Corbett KS, Flynn B, Foulds KE, et al. Evaluation of the mRNA-1273 vaccine against SARS-CoV-2 in nonhuman primates. N Engl J Med. 2020;383(16):1544–1555. doi:10.1056/NEJMoa2024671
22. Yan H, Hu Y, Akk A, et al. Induction of WNT16 via peptide-mRNA nanoparticle-based delivery maintains cartilage homeostasis. Pharmaceutics. 2020;12(1):73. doi:10.3390/pharmaceutics12010073
23. An D, Frassetto A, Jacquinet E, et al. Long-term efficacy and safety of mRNA therapy in two murine models of methylmalonic acidemia. EBioMedicine. 2019;45:519–528. doi:10.1016/j.ebiom.2019.07.003
24. Feldman RA, Fuhr R, Smolenov I, et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in Phase 1 randomized clinical trials. Vaccine. 2019;37(25):3326–3334. doi:10.1016/j.vaccine.2019.04.074
25. Corbett KS, Edwards DK, Leist SR, et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature. 2020;586(7830):567–571. doi:10.1038/s41586-020-2622-0
26. Monslow MA, Elbashir S, Sullivan NL, et al. Immunogenicity generated by mRNA vaccine encoding VZV gE antigen is comparable to adjuvanted subunit vaccine and better than live attenuated vaccine in nonhuman primates. Vaccine. 2020;38(36):5793–5802. doi:10.1016/j.vaccine.2020.06.062
27. Huang KW, Hsu FF, Qiu JT, et al. Highly efficient and tumor-selective nanoparticles for dual-targeted immunogene therapy against cancer. Sci Adv. 2020;6(3):eaax5032. doi:10.1126/sciadv.aax5032
28. Xu Z, Shen G, Xia X, et al. Comparisons of three polyethyleneimine-derived nanoparticles as a gene therapy delivery system for renal cell carcinoma. J Transl Med. 2011;9(1):46. doi:10.1186/1479-5876-9-46
29. Bell GD, Yang Y, Leung E, Krissansen GW. mRNA transfection by a Xentry-protamine cell-penetrating peptide is enhanced by TLR antagonist E6446. PLoS One. 2018;13(7):e0201464. doi:10.1371/journal.pone.0201464
30. Duan X, Wang P, Men K, et al. Treating colon cancer with a suicide gene delivered by self-assembled cationic MPEG-PCL micelles. Nanoscale. 2012;4(7):2400–2407. doi:10.1039/c2nr30079f
31. Men K, Huang R, Zhang X, et al. Local and systemic delivery of interleukin-12 gene by cationic micelles for cancer immunogene therapy. J Biomed Nanotechnol. 2018;14(10):1719–1730. doi:10.1166/jbn.2018.2593
32. Guidotti G, Brambilla L, Rossi D. Cell-penetrating peptides: from basic research to clinics. Trends Pharmacol Sci. 2017;38(4):406–424. doi:10.1016/j.tips.2017.01.003
33. Vaissière A, Aldrian G, Konate K, et al. A retro-inverso cell-penetrating peptide for siRNA delivery. J Nanobiotechnology. 2017;15(1):34. doi:10.1186/s12951-017-0269-2
34. Guo F, Fu Q, Zhou K, et al. Matrix metalloprotein-triggered, cell penetrating peptide-modified star-shaped nanoparticles for tumor targeting and cancer therapy. J Nanobiotechnology. 2020;18(1):48. doi:10.1186/s12951-020-00595-5
35. Chen L, Fang S, Xiao X, Zheng B, Zhao M. Single-stranded DNA assisted cell penetrating peptide-DNA conjugation strategy for intracellular imaging of nucleases. Anal Chem. 2016;88(23):11306–11309. doi:10.1021/acs.analchem.6b03743
36. Song J, Huang S, Zhang Z, et al. SPA: a peptide antagonist that acts as a cell-penetrating peptide for drug delivery. Drug Deliv. 2020;27(1):91–99. doi:10.1080/10717544.2019.1706669
37. Gleeson FC, Levy MJ, Jackson RA, et al. Endoscopic ultrasound may be used to deliver gene expression signatures using digital mRNA detection methods to immunophenotype pancreatic ductal adenocarcinoma to facilitate personalized immunotherapy. Pancreatology. 2020;20(2):229–238. doi:10.1016/j.pan.2019.12.002
38. Maruggi G, Zhang C, Li J, Ulmer JB, Yu D. mRNA as a transformative technology for vaccine development to control infectious diseases. Mol Ther. 2019;27(4):757–772. doi:10.1016/j.ymthe.2019.01.020
39. Sahu I, Haque A, Weidensee B, Weinmann P, Kormann MSD. Recent developments in mRNA-based protein supplementation therapy to target lung diseases. Mol Ther. 2019;27(4):803–823. doi:10.1016/j.ymthe.2019.02.019
40. Kowalski PS, Rudra A, Miao L, Anderson DG. Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol Ther. 2019;27(4):710–728. doi:10.1016/j.ymthe.2019.02.012
41. Miao L, Li L, Huang Y, et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat Biotechnol. 2019;37(10):1174–1185. doi:10.1038/s41587-019-0247-3
42. Capasso Palmiero U, Kaczmarek JC, Fenton OS, Anderson DG. Poly(β-amino ester)-co-poly(caprolactone) terpolymers as nonviral vectors for mrna delivery in vitro and in vivo. Adv Healthcare Mater. 2018;7(14):e1800249. doi:10.1002/adhm.201800249
43. Hoerr I, Obst R, Rammensee HG, Jung G. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur J Immunol. 2000;30(1):1–7. doi:10.1002/1521-4141(200001)30:1<1::aid-immu1>3.0.co;2-#
44. Choi HY, Lee TJ, Yang GM, et al. Efficient mRNA delivery with graphene oxide-polyethylenimine for generation of footprint-free human induced pluripotent stem cells. J Control Release. 2016;235:222–235. doi:10.1016/j.jconrel.2016.06.007
45. Huang X, Zheng R, Ding F, et al. Efficient delivery of mRNA using crosslinked nucleic acid nanogel as a carrier. ACS Mater Lett. 2020;2(11):1509–1515. doi:10.1021/acsmaterialslett.0c00375
46. Cherukula K, Uthaman S, Park IK. Design of an amphiphilic poly(aspartamide)-mediated self-assembled nanoconstruct for long-term tumor targeting and bioimaging. Molecules. 2019;24(5):885. doi:10.3390/molecules24050885
47. Yu Y, Kim HJ, Mi P, et al. Targeted systemic delivery of siRNA to cervical cancer model using cyclic RGD-installed unimer polyion complex-assembled gold nanoparticles. J Control Release. 2016;244(Pt B):247–256. doi:10.1016/j.jconrel.2016.08.041
48. Martínez-Jothar L, Barendrecht AD, de Graaff AM, et al. Endothelial cell targeting by cRGD-functionalized polymeric nanoparticles under static and flow conditions. Nanomaterials. 2020;10(7):1353. doi:10.3390/nano10071353
49. Islam MA, Xu Y, Tao W, et al. Restoration of tumour-growth suppression in vivo via systemic nanoparticle-mediated delivery of PTEN mRNA. Nat Biomed Eng. 2018;2(11):850–864. doi:10.1038/s41551-018-0284-0
50. Lei S, Zhang X, Men K, et al. Efficient colorectal cancer gene therapy with IL-15 mRNA nanoformulation. Mol Pharm. 2020;17(9):3378–3391. doi:10.1021/acs.molpharmaceut.0c00451
51. Pardi N, Tuyishime S, Muramatsu H, et al. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J Control Release. 2015;217:345–351. doi:10.1016/j.jconrel.2015.08.007
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.