Back to Journals » OncoTargets and Therapy » Volume 9
Functional divergence and convergence between the transcript network and gene network in lung adenocarcinoma
Received 21 August 2015
Accepted for publication 10 December 2015
Published 14 January 2016 Volume 2016:9 Pages 335—347
DOI https://doi.org/10.2147/OTT.S94897
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Faris Farassati
Min-Kung Hsu,1 Chia-Lin Pan,2 Feng-Chi Chen1–3
1Department of Biological Science and Technology, National Chiao-Tung University, Hsinchu, 2Institute of Population Health Sciences, National Health Research Institutes, Zhunan, 3School of Dentistry, China Medical University, Taichung, Taiwan
Introduction: Alternative RNA splicing is a critical regulatory mechanism during tumorigenesis. However, previous oncological studies mainly focused on the splicing of individual genes. Whether and how transcript isoforms are coordinated to affect cellular functions remain underexplored. Also of great interest is how the splicing regulome cooperates with the transcription regulome to facilitate tumorigenesis. The answers to these questions are of fundamental importance to cancer biology.
Results: Here, we report a comparative study between the transcript-based network (TN) and the gene-based network (GN) derived from the transcriptomes of paired tumor–normal tissues from 77 lung adenocarcinoma patients. We demonstrate that the two networks differ significantly from each other in terms of patient clustering and the number and functions of network modules. Interestingly, the majority (89.5%) of multi-transcript genes have their transcript isoforms distributed in at least two TN modules, suggesting regulatory and functional divergences between transcript isoforms. Furthermore, TN and GN modules share only ~50%–60% of their biological functions. TN thus appears to constitute a regulatory layer separate from GN. Nevertheless, our results indicate that functional convergence and divergence both occur between TN and GN, implying complex interactions between the two regulatory layers. Finally, we report that the expression profiles of module members in both TN and GN shift dramatically yet concordantly during tumorigenesis. The mechanisms underlying this coordinated shifting remain unclear yet are worth further explorations.
Conclusion: We show that in lung adenocarcinoma, transcript isoforms per se are coordinately regulated to conduct biological functions not conveyed by the network of genes. However, the two networks may interact closely with each other by sharing the same or related biological functions. Unraveling the effects and mechanisms of such interactions will significantly advance our understanding of this deadly disease.
Keywords: lung adenocarcinoma, transcriptome analysis, gene network, module, alternative splicing, transcriptional regulation
Introduction
The human genome contains tens of thousands of genes. The large number of genes suggests sophisticated gene–gene interactions and high levels of regulatory coordination. Indeed, the dynamics of the human gene network has been suggested to reflect cellular processes and disease status.1,2 In a gene network, individual genes frequently form densely connected “modules”, which have been reported to signify functional associations and regulatory relatedness.1,3,4 Gene networks can convey rich information regarding abnormalities as well as normal cellular functions. Accordingly, network analysis has been widely applied in oncological studies.5–7
The vast majority of network studies have been focused on the gene level. However, >90% of human genes are known to be alternatively spliced, generating multiple transcript/protein isoforms of similar or distinct functions.8 At least 5% of human genes can produce protein isoforms that conduct different biological functions.9,10 Furthermore, individual transcript isoforms have been proposed to constitute a critical layer in the human regulome separate from the transcriptional regulations of complete genes.11 Gene-centered network analyses lack such transcript-level resolution, and thus may miss clues important for molecular pathogenesis.
Alternative splicing is strictly regulated both spatially and temporally. Dysregulation of alternative splicing is closely related to a variety of human diseases.12–14 Importantly, the abnormalities in alternative splicing have been suggested to contribute significantly to tumorigenesis.7,14 While this issue has attracted increasing attention, the effects and mechanisms of splicing dysregulation in tumorigenesis remain largely unclear.15 Furthermore, most of the previous studies on alternative splicing have been directed to a limited number of cancer-related genes16,17 or the functional implications of individual transcript isoforms.15 Transcriptome-scale network analyses have remained scarce. Given the functional divergences between transcript isoforms, transcript-based and gene-based networks (designated as “TN” and “GN”, respectively) are expected to differ considerably from each other in function, regulation, and biological significance in tumorigenesis. By comparing the networks at these two molecular levels (gene and transcript) in tumor and normal tissues, we may be able to not only delineate the splicing regulome but also explore the regulatory associations between splicing and transcription during tumorigenesis.
Here, we report a comparative study between GN and TN in paired tumor–normal tissues derived from 77 lung adenocarcinoma patients. We show that TN exhibits different topological and functional properties from GN. Interestingly, a considerable proportion of transcript isoforms of the same genes are inferred to serve distinct molecular functions potentially important for the tumorigenesis of lung adenocarcinoma. Importantly, the two networks show both convergence and divergence in biological functions, indicating complex interactions between the two regulatory layers. Furthermore, we demonstrate that in both of the networks, the expression profiles of module members shift dramatically yet concordantly during tumorigenesis. Our results suggest that the network of transcript isoforms constitutes an important regulatory layer that is separate from yet functionally intertwined with the gene network in lung adenocarcinoma.
Methods
Data source and processing
The RNA-sequencing (RNA-seq) dataset (GSE40419) of lung adenocarcinoma was downloaded from the Gene Expression Omnibus database.18 The dataset contained RNA-seq data derived from paired normal and tumor tissues from 77 Korean patients.
The raw data retrieved from Gene Expression Omnibus (in Sequence Read Archive format) were converted to the FASTQ format by using fastq-dump. The RNA-seq reads were then mapped to the human reference genome (GRCh37; Ensemble Version 75) by using STAR19 with default parameters. The expression levels (in Fragments Per Kilobase of transcript per Million mapped reads [FPKM]) of genes and transcripts were generated separately by Cufflinks.20 To ensure data quality, two types of genes and the corresponding transcripts were excluded:
- The genes that had more than one FPKM value (12 genes).
- The genes that did not include all of the annotated transcript isoforms (ie, some of the annotated transcripts were just “absent”, rather than being assigned a zero FPKM value) according to Cufflinks results (776 genes).
In addition, we required that the FPKM values be ≥1 in all of the 77 normal or tumor tissues. Finally, 70,131 transcript isoforms of 10,510 genes were retained for subsequent analyses.
Network construction
The gene-based and transcript-based networks were constructed by using Weighted Gene Correlation Network Analysis (WGCNA).21 According to the WGCNA manual, the FPKM values were log-transformed by log2(FPKM +1). The transformed FPKM values were then input to WGCNA for calculation of the Pearson’s coefficients of correlation for all gene/transcript pairs. The network connectivity was weighted based on these coefficients. The FPKM values of normal and tumor tissues of the same molecular level (gene or transcript) were input together to WGCNA so that the modules could be compared between the two tissue types. The “β” parameter of WGCNA, which was used to determine adjacency, was set to be 6 according to data distribution (Figure S1).
The Gene Ontology (GO) annotation system was selected for functional analysis by WGCNA. The GO term of an individual transcript isoform was considered as the same as that of the corresponding gene. The statistical significance of GO term enrichment was Bonferroni-corrected.
Estimation of transcript module entropy
The transcript module entropy (Ei) was used to quantify the dispersion of transcript isoforms of gene i in the TN modules. Ei was defined as:
|
|
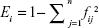
where fij indicated the proportion of transcript isoform of gene i that belonged to TN module j, and n indicated the total number of TN modules assigned to the transcript isoforms of gene i.
The theoretical maximum of Ei (Max(Ei)) was estimated by:
|
|
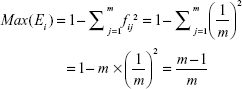
where m was the number of transcript isoforms of gene i as annotated by the Ensemble Database (Version 75). Note that single-transcript genes were excluded from this analysis.
Correlations between module members and known cancer-related genes
The expressions of the genes involved in three cancer-related pathways annotated by Kyoto Encyclopedia of Genes and Genomes (KEGG) were singled out, including the “cancer pathway”, “non-small-cell lung cancer (NSCLC) pathway”, and “small-cell lung cancer (SCLC) pathway”. The eigengene for the genes in each pathway was derived using the principal component analysis module of the R package. An eigengene was defined as the first principal component of the expression profiles of the analyzed genes.22 The correlations between module members and each of the three eigengenes were evaluated to determine whether the identified modules were functionally related to tumorigenesis. The cutoff P-value for statistical significance was Bonferroni-adjusted to 0.002 and 0.0007 for GN and TN modules, respectively, to account for multiple testing (0.05/# modules; 0.05/27=0.002 for GN; 0.05/68=0.0007 for TN).
Results
Functional divergences and convergences between the gene network and transcript network in lung adenocarcinoma
We used WGCNA21 to construct GNs and TNs of lung adenocarcinoma (described in the “Methods” section). The expression levels of individual transcript isoforms were calculated by using Cufflinks, which assigned RNA-seq reads to transcript isoforms according to a likelihood model.20 The expression levels (in FPKM) of tumor and normal tissues were together submitted to WGCNA but separately for genes and transcripts so that the modules could be compared between the two tissue types and between the two molecular levels. WGCNA identifies modules according to hierarchical clustering of the genes/transcripts and a dissimilarity measure-based cutoff value.21 This approach has been widely applied and proved to be powerful in detecting functionally related genes.23–27
Intuitively, transcript isoforms are merely “subsets” of the corresponding genes. Transcript-based analyses thus should yield results fairly similar to those derived from gene-based analyses. Interestingly, this proposition was not supported by our results. We first examined whether the expression profiles of genes and transcripts could concordantly reflect the genetic relationships among the 77 patients. Our results indicated that the gene-based and transcript-based tree topologies differed considerably from each other (Figure 1). To quantify the difference between gene-based and transcript-based patient clustering, we used FastTree28 to compare the topologies of the two patient trees. Figure 1A shows that for normal tissues, only ten out of the 74 splits (colored circles and star) were shared between the gene-based and the transcript-based tree. Similarly, for tumor tissues, only eleven of the 74 splits were shared between the two trees (Figure 1B). However, a large split covering 71 of the patients (the star in Figure 1A) was shared between gene-based and transcript-based trees in normal tissues, which was not observed in tumor tissues. The dissimilarities appeared to imply dissociation of transcript-centric regulations from gene-level regulations, possibly due to the functional/regulatory divergences between isoforms.29,30 Furthermore, the gene–transcript divergence seemed to be larger in tumor tissues than in normal tissues.
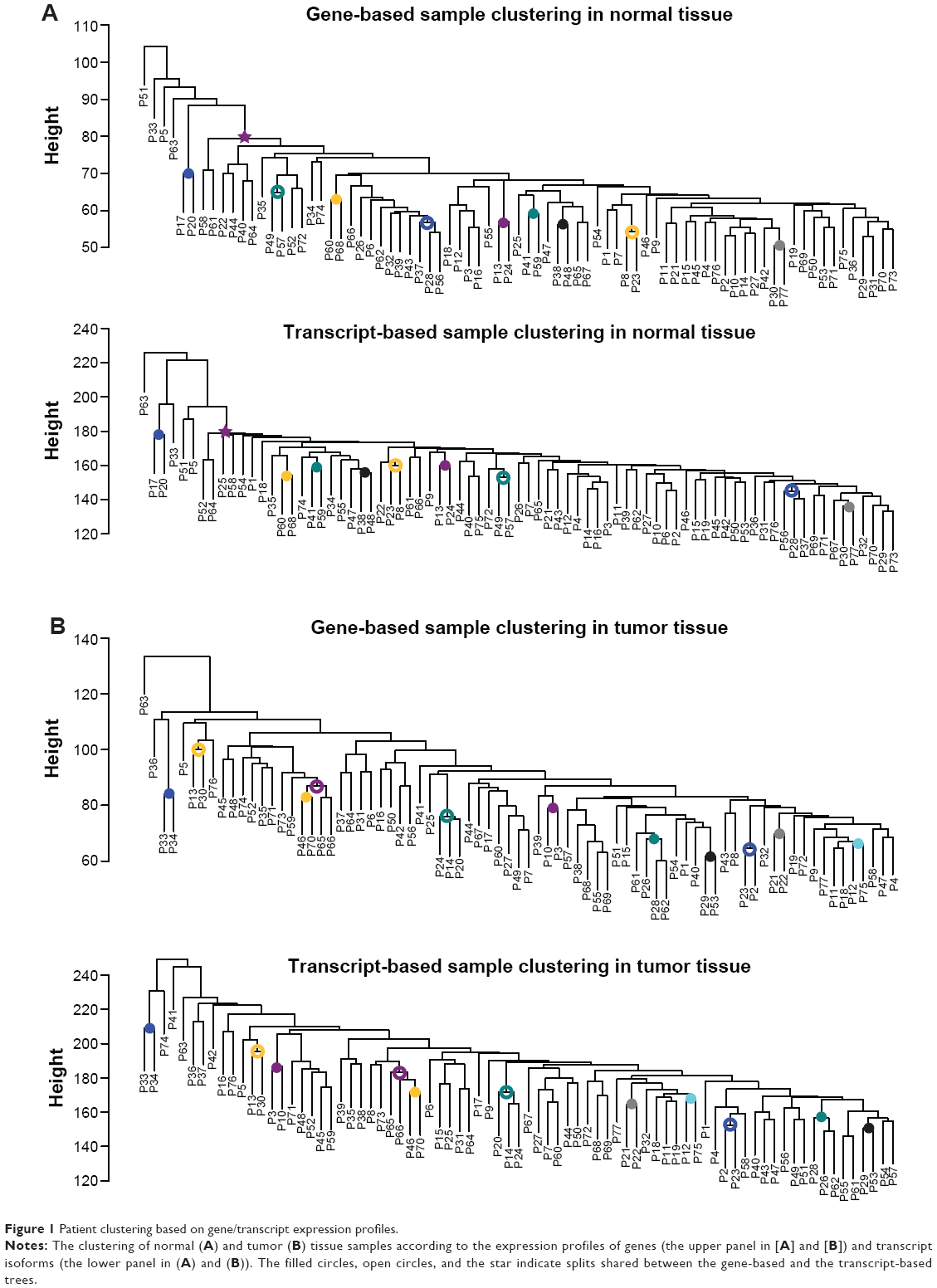 | Figure 1 Patient clustering based on gene/transcript expression profiles. |
Next, we compared the modules derived from GN and those derived from TN. We reasoned that if the expression profiles of transcript isoforms closely resembled those of the corresponding genes, the number of TN modules should be close to that of GN modules. Otherwise, TN modules should outnumber GN modules. Our results indicated that GN and TN contained 27 and 68 modules, respectively. This disparity in module number indicated that first, the expression profiles of individual transcript isoforms diverged considerably from those of the corresponding genes. Second, transcript isoform-specific regulations were fairly common in the transcriptome of lung adenocarcinoma. Third, transcript isoforms might convey significant functional versatility not achievable by single-transcript genes alone.
To investigate whether TN and GN modules serve different biological functions, we conducted GO analyses for these modules. Thirteen of the GN modules and 18 of the TN modules were found to be enriched for 108 and 137 GO terms, respectively (Tables S1 and S2). The majority of these modules corresponded to multiple GO terms. These multifunctional modules were related to such important functions as cell cycle, immune response, and regulation of cell migration (Tables S1 and S2). Interestingly, only 68 GO terms were shared between GN and TN modules. These 68 GO terms accounted for 63.0% and 49.6% of the GO terms enriched for GN and TN modules, respectively. This observation indicated that GN and TN modules functionally overlapped with each other by sharing half or more of the GO terms. However, the divergences between the two module groups were substantial, accounting for ~40%–50% of the GO terms. This interpretation, nevertheless, should be taken with caution because different GO terms may be in fact functionally related. For example, TN module #42 was enriched for the GO terms “chemokine activity” and “cytokine activity”, which were absent from the GO terms enriched for GN modules. But such GO terms as “immune response” and “regulation of immune system process” were found to be enriched for the “black” GN module. This observation implied potential functional convergences between GN and TN modules despite apparent divergences.
Meanwhile, a closer inspection revealed that transcript isoforms from different genes could be convened to conduct functions not conducted by the corresponding genes. For instance, TN module #16 was enriched for the GO terms “cell motility” and “cell migration”. This TN module contained 951 transcripts, which corresponded to 674 genes distributed in 27 GN modules of fairly diversified functions (Table S3). Overall, our results suggested complex regulatory relationships between GN and TN, where functional divergences and convergences were intertwined.
Transcript isoforms of the same genes are usually distributed in different network modules
The observations that (1) TN modules outnumbered GN module and (2) TN and GN modules functionally diverged from each other suggested that transcript isoforms of the same genes might have been clustered into different TN modules. Indeed, our results indicated that 89.5% (7,418/8,284) of the analyzed multiple-transcript genes had their transcript isoforms distributed in at least two modules. To quantify the level of isoform distribution, we calculated the “transcript module entropy” (Ei) for each analyzed gene. Ei represented the entropy of isoform distribution in TN modules for a given gene (described in the “Methods” section). A larger Ei value indicated a wider module distribution of transcript isoforms of a gene. Of note, theoretically Ei should increase with the number of transcript isoforms of a gene. For comparison, we calculated the theoretical maxima of Ei for genes with different numbers of isoforms. An Ei value equal to the theoretical maximum indicated that all of the transcript isoforms of the interested gene were assigned to different TN modules. And an Ei value of zero indicated that all of the transcript isoforms of the interested gene belonged to the same TN module.
It was expected that most, if not all, of the transcript isoforms of a gene belonged to the same module. The Ei values should thus be close to zero in the majority of cases. However, Figure 2 shows that, in general, the average Ei value increased with the number of transcript isoforms for both coding (blue bars) and noncoding genes (red bars), with the Ei values of coding genes slightly higher than those of the corresponding noncoding genes in most of the cases. Interestingly, for coding genes, the ratio of observed-to-maximal Ei value fell between 0.7 and 0.8 regardless of the number of transcript isoforms. For noncoding genes, the ratio seemed to be increasing with the number of transcript isoforms. Notably, however, the numbers of noncoding genes in this analysis were fairly small (inset table in Figure 2). The results for noncoding genes, especially for those with larger isoform numbers (>5), should be taken with caution.
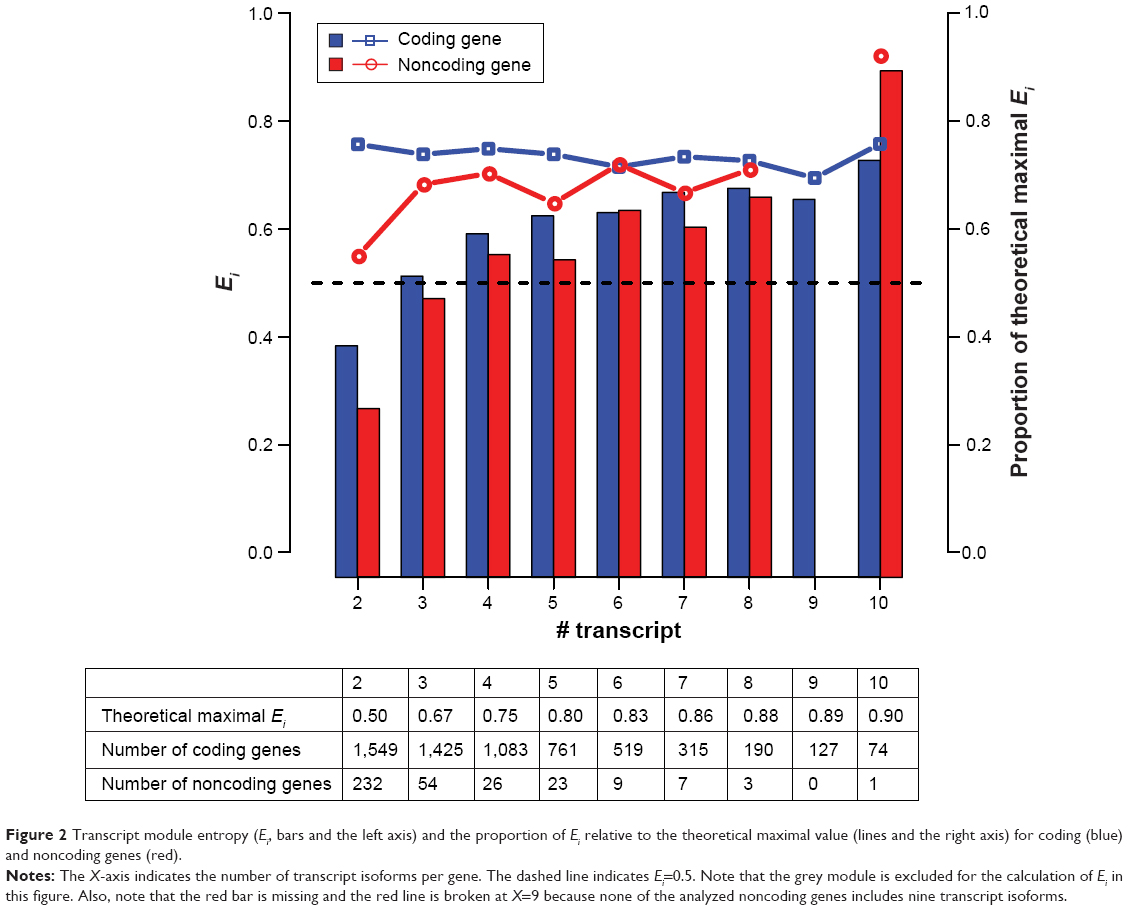 | Figure 2 Transcript module entropy (Ei, bars and the left axis) and the proportion of Ei relative to the theoretical maximal value (lines and the right axis) for coding (blue) and noncoding genes (red). |
Of note, in the above analysis, we excluded the “gray module”, which included the transcript isoforms not assigned to any other modules. This specific module was a mixture of either biological “noises” or transcript isoforms with unique expression profiles. The exclusion of the grey module could have affected the results because it would have been counted as a de facto module in the calculation of Ei values. To evaluate the influence of the grey module, we put this module back for the calculation of the Ei values. Figure S2 shows that when the grey module was included, the average Ei values appeared to reach the maximum at isoform number =6 for both coding and noncoding genes. Furthermore, for coding genes, the ratio of observed-to-maximal Ei value decreased with the number of isoforms, whereas for noncoding genes, this ratio peaked at isoform number =6. These observations suggested that coding genes with more transcript isoforms tended to have a larger fraction of the isoforms assigned to the grey module. But this was not true for noncoding genes. Again, the numbers of analyzed noncoding genes were relatively small, and the results thereof must be taken with caution.
Overall, the above observations implied that the expression profiles (and likely functions) of transcript isoforms of the same genes, at least those in the TN modules, diverged substantially from each other. And this between-isoform divergence was slightly larger in coding genes than in noncoding genes. Interestingly, the functional versatility of a gene (as measured by Ei) appeared to increase with the number of transcript isoforms if the grey module was excluded. This observation implied that regulatory flexibility of a gene could be amplified by an increase in the number of transcript isoforms.
The module members in both of the gene- and transcript-based networks are significantly correlated with known cancer genes in expression profile
To clarify whether the GN and TN modules were associated with patient features (sex, age at diagnosis, smoking history, and cancer stage), we measured the correlation between the network modules and each of the features. We also evaluated the correlations between the module members and the “eigengenes” derived from the known cancer genes as annotated in the KEGG Pathway Database (described in the “Methods” section). The eigengene of a gene group is the eigenvector of the expression profile matrix of the member genes.21 Note that the cutoff P-value for statistical significance was Bonferroni-adjusted to P<0.002 and P<0.0007 for GN and TN modules, respectively, to account for multiple testing (described in the “Methods” section). Our result showed that for normal tissues, the GN modules were correlated with none of the patient features or cancer-related gene groups (Figure 3A). In comparison, for tumor tissues, the “brown” and “salmon” modules were positively correlated, but the grey module was negatively correlated with sex (Figure 3B). Note that male and female, respectively, was assigned the value of “1” and “2” for the calculation of coefficient of correlation. The correlations with the KEGG cancer pathway genes were positive for the “blue”, “darkgreen”, “royalblue”, “red”, and “grey” modules but negative for the “yellow” module. Meanwhile, the correlations with SCLC-related genes were positive for the “magenta”, “purple”, and “red” modules but negative for the “yellow” module (Figure 3B). These observations suggested that the GN modules could indeed encompass genes that were important for the tumorigenesis of lung cancer.
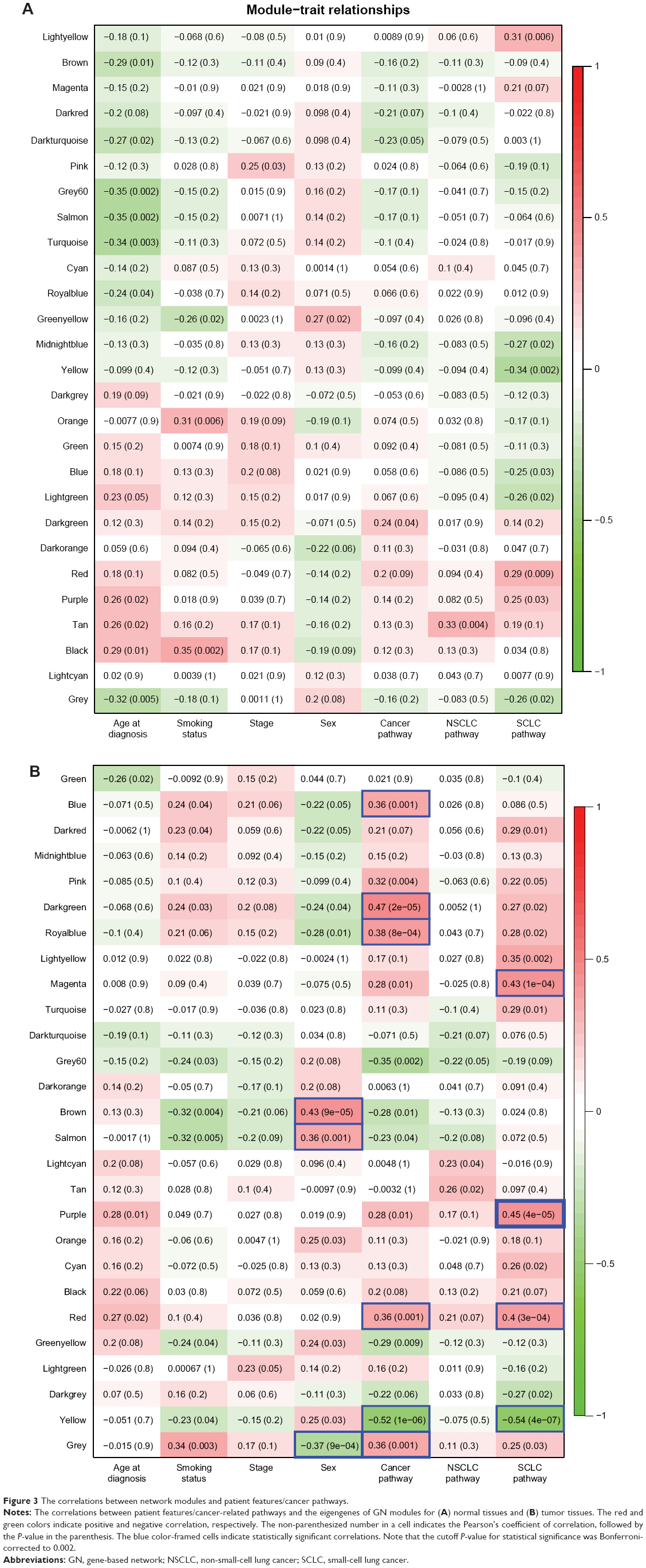 | Figure 3 The correlations between network modules and patient features/cancer pathways. |
For TN modules, on the other hand, in normal tissues, none of the modules were significantly correlated with any patient feature or cancer-related gene groups except for module #37, which was positively correlated with age (Figure S3). In tumor tissues, the TN modules were correlated with none of the patient features except for sex, which was correlated with TN modules #21, #29, and #10. Interestingly, the correlations between TN module members and KEGG cancer pathway genes were significantly negative for modules #32, 28, 26, 20, and 54 but significantly positive for modules #37, 43, 56, 36, 67, 58, 39, 41, 42, and 40. Meanwhile, TN modules #26, 20, and 54 were negatively correlated with SCLC-related genes in tumor tissues (Figure S3). These observations implied that a considerable proportion (15/68) of the TN modules in tumor tissues was involved in the tumorigenesis of lung cancer and cancer cell development in general.
We then tried to investigate whether the expressions of the network modules were associated with the tumor/normal status. To this end, we calculated the correlations between the module eigengene (eigentranscript) and the member genes (transcripts) of the interested module in view of expression profile separately for tumor and normal tissues. For comparison, we also calculated the correlations between a selected patient feature (smoking history) and the expressions of the module members. Figure 4 shows that in three example TN modules (A: #16, B: #24, and C: #14) in tumor tissues (but not in normal tissues), the correlations with module eigentranscript (X-axis) either split into two clusters (Figure 4A and B) or moved toward one extreme (Figure 4C), which were not observed along the Y-axis (correlations with smoking history). Similar trends were also observed for the GN modules (Figure S4). These observations suggested that during the tumorigenesis of lung adenocarcinoma, the expression profiles of the module member genes/transcripts tended to “move” toward both extremes in view of similarity to those of the eigengenes/eigentranscripts. Such movements implied higher-level regulations that coordinated the changes in expression profiles of the module members in both GN and TN. Interestingly, in Figure 4, only module #16 of the three modules was found to be enriched for biological functions that were important for tumorigenesis (Table S2).
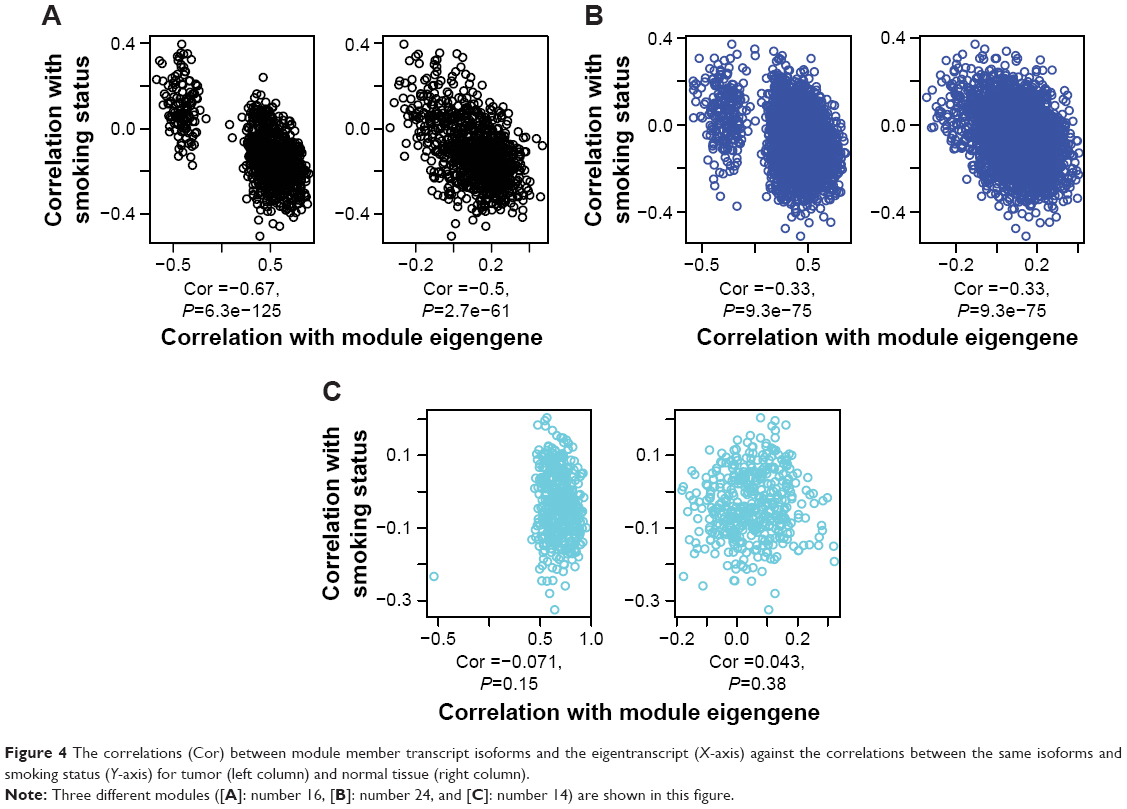 | Figure 4 The correlations (Cor) between module member transcript isoforms and the eigentranscript (X-axis) against the correlations between the same isoforms and smoking status (Y-axis) for tumor (left column) and normal tissue (right column). |
Discussion
Alternative splicing has been credited as a major regulatory mechanism in tumorigenesis.14 For instance, Hong et al suggested that fine-grained regulations (eg, allele-specific splicing) could yield sub-genic level variations that were linked to the pathogenesis of lung cancer.31 Meanwhile, isoform-level expressions have been shown to correlate well with the prognosis and subtyping of glioblastoma.32 Alterations in splicing patterns in cancer driver genes have also been reported to occur repeatedly in a variety of cancer types.33 Nevertheless, none of these studies addressed how different splicing events are coordinated on a transcriptome-wide scale, or how splicing regulations interact with transcriptional regulations during cancer progression. Meanwhile, network-based cancer studies are abundant. These network studies were based on gene–gene, protein–protein, or regulatory interactions.33–37 Each type of biological network conveys biological insights from a different perspective. Studies on isoform-based networks have remained rare, thus representing an important missing piece in the cancer regulome atlas.
In this study, we report the first comparative analysis between TN and GN in lung adenocarcinoma. We demonstrate that TN differs substantially from GN in view of deciphering the genetic relationships among patients (Figure 1), module distribution of transcript isoforms (Figure 2), and the biological functions of network modules (Tables S1 and S2). These differences suggest that TN and GN constitute two separate layers in the cancer regulome. However, it is worth noting that, as we pointed out in the “Results” section, the two regulatory layers could conduct the same or related functions. Two possible reasons may explain this functional convergence. First, the transcript isoforms of the same genes may conduct virtually the same biological functions. In this case, alternative splicing would convey no functional versatility to the genes of interest. The second possibility is that TN and GN work synergistically to implement biological functions. In this latter case, there should be higher levels of regulations to direct such synergies. These regulatory mechanisms, though currently unclear, are worth further explorations.
One unexpected observation is that the majority (89.5%) of the analyzed genes had their transcript isoforms clustered into different TN modules. Intuitively, most of the same-gene transcript isoforms should conduct similar functions, which should be reflected in the similarity in expression profiles. This proposition is nevertheless unsupported in most of the cases in our analysis. To be sure, similarity in expression profile does not equal to similarity in functionality. Yet our results show that, for instance, an isoform of Gene A shares similar expression profiles with an isoform of Gene B but not with other isoforms of Gene A. This observation points to a possibility of cross-gene coordination of transcript isoforms. We concede that such “similarities” in expression profile could have occurred simply by chance, which might partly explain why many of the TN modules are not significantly enriched for any GO terms. Nevertheless, the GO terms enriched for TN modules but not for GN modules (Tables S1 and S2) indicate that at least part of the inferred transcript-level coordination might be true. Our result is actually consistent with the recent finding that subfunctionalization of splicing isoforms is widespread in vertebrate genomes.38
The entropy (Ei) analysis also suggests prevalent functional divergence between transcript isoforms in the transcriptome of lung adenocarcinoma. In general, Ei increases with the number of isoforms for both coding and noncoding genes when the grey module is excluded (Figure 2). However, the inclusion of the grey module in the analysis results in a peak of Ei value at isoform number =6 for both gene groups, and a decreasing observed-to-maximal Ei ratio toward high isoform numbers in coding genes (Figure S2). These observations suggest that a considerable proportion of the isoforms of high-isoform number coding genes are assigned to the grey module. The grey module contains member transcripts of unclassifiable expression profiles. We cannot exclude the possibility that the grey module contains functionally relevant transcript isoforms. Nevertheless, if we conservatively consider all of the grey module members as noises and exclude them from our analysis, the transcript module entropy still increases toward high isoform numbers. These observations imply that as the number of isoforms increases, biological “noises” and functional versatility both increase. And the marginal increase in functional versatility might decrease as the number of isoforms rises.
One example of isoforms distributing in different TN modules was observed for the well-known oncogene Myc (ENSG00000136997).39 Myc has five isoforms according to ENSEMBL (Version 75) annotations. Two of the isoforms (ENST00000377970 and ENST00000524013) have their coding sequences at least 75% longer than the other three (ENST00000259523, ENST00000517291, and ENST00000520751). The two long isoforms differ from each other by only two amino acids. Interestingly, these two isoforms belong to the same TN module (#43), whereas the three short ones belong to the grey module. Furthermore, the two long isoforms are the dominant forms of Myc. Together, the two account for a median of 97.7% and 98.1% of Myc gene expression in tumor and normal tissue, respectively. These observations imply that TN modules can indeed cluster together functionally similar isoforms, and set aside potentially nonfunctional isoforms into the grey module. Another example is SPP1 (ENSG00000118785), which has been reported to promote metastasis.40–42 SPP1 contains ten isoforms, including four coding isoforms (ENST00000237623, ENST00000395080, ENST00000360804, and ENST00000508233) and six noncoding isoforms (ENST00000509659, ENST00000509334, ENST00000513981, ENST00000508002, ENST00000504310, and ENST00000505146). Interestingly, the four coding isoforms were clustered to TN module #42, whereas the other six noncoding isoforms belonged to the grey module. This observation suggests that TN modules could help differentiate isoforms of different functionalities. Meanwhile, for FUCA2 (ENSG00000001036), a gene implicated in Helicobacter pylori-caused gastric cancer, the grouping pattern was different. FUCA2 contains three coding isoforms (ENST00000002165, ENST00000438118, and ENST00000451668) and one noncoding isoform (ENST00000367585). The noncoding isoform belonged to the grey module. However, the three coding isoforms were clustered to two TN modules: #24 (ENST00000438118 and ENST00000451668) and #37 (ENST00000002165). ENST00000002165 has a longer coding region and a dominant expression level, accounting for a median of 87.1% and 88.0% of the gene expression in normal and tumor tissue, respectively. Interestingly, although the two short coding isoforms were as lowly expressed as the noncoding isoform (collectively accounting for 8.3% and 6.2% of gene expression in normal and tumor tissue, respectively), they were clustered to a non-grey module (#24). This observation again supports that TN modules can appropriately group functionally divergent isoforms of the same genes.
We also report an interesting observation that in the tumorigenesis of lung adenocarcinoma, the expression profiles of module member genes (transcripts) tend to either converge to or deviate from those of the eigengenes (eigentranscripts) (Figures 4 and S4). This observation suggested that the module eigengenes (eigentranscripts) could well signify the tumor status, and might serve as “virtual biomarkers” to differentiate tumor from normal tissues. Furthermore, the synchronous alterations in expression profiles of the module member genes (transcripts) imply high-level regulatory coordination, which dictates both gene transcription and alternative splicing during tumorigenesis.
Although a number of GN and TN modules are significantly correlated with cancer pathways, we are not certain whether GN or TN modules can better differentiate tumor from normal tissues. To this end, we calculated the “expression value” of the eigengene/eigentranscript for each GN/TN module. This “expression value” is a weighted linear summation of expression levels of the module components. Our results indicated that the expression values of three TN modules (#4, #47, and #48) differed significantly between normal and tumor tissues (Figure S5). By contrast, none of the GN modules yielded an expression value applicable to the tumor–normal differentiation. This observation suggested that certain TN modules could better reflect the regulatory alterations during tumorigenesis than GN modules. This proposition, however, should be taken with caution because the larger sizes of TN modules than GN modules might be accountable for this difference.
Analyzing network modules can help identify potential gene–gene interactions. For instance, both MDM2 and MEK belonged to the yellow GN module (Figure S6). It has been reported that chemical blockade of these two genes could synergistically induce apoptosis in acute myeloid leukemia.43 This synergy may not be readily inferred from current cancer pathway information as shown in Figure S6 but is somehow implied in the modularity of GN. Another exemplar interaction occurs between HIF-α and β-catenin, both clustered to the “royalblue” module. These two genes do not appear to share the same cancer pathway (Figure S6). However, a recent study indicated that miR-622-mediated downregulation of HIF-1α correlated with decreased β-catenin expression,44 suggesting a regulatory relationship between these two genes. These examples show that network analysis can compensate for the insufficiency of current pathway information, potentially leading to novel biological discoveries.
In conclusion, we demonstrate that transcript isoforms per se constitute a separate and important regulatory layer in the tumorigenesis of lung adenocarcinoma. This regulatory layer appears to interact closely with transcriptional regulations to affect cellular functions.
Acknowledgments
This work was supported by the National Health Research Institutes (PH-103-PP06 and PH-104-PP06) and the Ministry of Science and Technology (MOST-103-2311-B-400-003), Taiwan. The computational facilities were partly supported by the Taiwan Bioinformatics Institute Core Facility (MOST-103-2319-B-400-001), with the assistance of the National Center for High-speed Computing of Taiwan. The authors wish to thank Ms Chieh-Hua Lin for technical assistance, and Dr Ben-Yang Liao and Dr Yu-Chieh Liao for constructive comments. This research was presented as a poster at the GIW/InCoB Conference 2015 in Tokyo.
Disclosure
The authors report no conflicts of interest in this work.
References
Karlebach G, Shamir R. Modelling and analysis of gene regulatory networks. Nature Reviews. Molecular Cell Biology. 2008;9(10): 770–780. | ||
del Sol A, Balling R, Hood L, Galas D. Diseases as network perturbations. Current Opinion in Biotechnology. 2010;21(4):566–571. | ||
Hartwell LH, Hopfield JJ, Leibler S, Murray AW. From molecular to modular cell biology. Nature. 1999;402(6761 Suppl):C47–C52. | ||
Carlson MR, Zhang B, Fang Z, Mischel PS, Horvath S, Nelson SF. Gene connectivity, function, and sequence conservation: predictions from modular yeast co-expression networks. BMC Genomics. 2006;7:40. | ||
Wang J, Zuo Y, Man YG, et al. Pathway and network approaches for identification of cancer signature markers from omics data. Journal of Cancer. 2015;6(1):54–65. | ||
Pal S, Gupta R, Davuluri RV. Alternative transcription and alternative splicing in cancer. Pharmacology & Therapeutics. 2012;136(3): 283–294. | ||
Oltean S, Bates DO. Hallmarks of alternative splicing in cancer. Oncogene. 2014;33(46):5311–5318. | ||
Wang ET, Sandberg R, Luo S, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–476. | ||
Lopez AJ. Alternative splicing of pre-mRNA: developmental consequences and mechanisms of regulation. Annual Review of Genetics. 1998;32:279–305. | ||
Lee Y, Rio DC. Mechanisms and regulation of alternative pre-mRNA splicing. Annual Review of Biochemistry. 2015;84:291–323. | ||
Blencowe BJ. An exon-centric perspective. Biochemistry and Cell Biology = Biochimie et Biologie Cellulaire. 2012;90(5):603–612. | ||
Biamonti G, Catillo M, Pignataro D, Montecucco A, Ghigna C. The alternative splicing side of cancer. Seminars in Cell & Developmental Biology. 2014;32:30–36. | ||
Venables JP. Aberrant and alternative splicing in cancer. Cancer Research. 2004;64(21):7647–7654. | ||
Ladomery M. Aberrant alternative splicing is another hallmark of cancer. International Journal of Cell Biology. 2013;2013:463786. | ||
Tsai YS, Dominguez D, Gomez SM, Wang Z. Transcriptome-wide identification and study of cancer-specific splicing events across multiple tumors. Oncotarget. 2015;6(9):6825–6839. | ||
Liu J, Lee W, Jiang Z, et al. Genome and transcriptome sequencing of lung cancers reveal diverse mutational and splicing events. Genome Research. 2012;22(12):2315–2327. | ||
Eswaran J, Horvath A, Godbole S, et al. RNA sequencing of cancer reveals novel splicing alterations. Scientific Reports. 2013;3:1689. | ||
Seo JS, Ju YS, Lee WC, et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Research. 2012;22(11):2109–2119. | ||
Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. | ||
Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology. 2010; 28(5):511–515. | ||
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. | ||
Langfelder P, Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC Systems Biology. 2007;1:54. | ||
Liu R, Guo CX, Zhou HH. Network-based approach to identify prognostic biomarkers for estrogen receptor-positive breast cancer treatment with tamoxifen. Cancer Biology & Therapy. 2015;16(2):317–324. | ||
Deng SP, Zhu L, Huang DS. Mining the bladder cancer-associated genes by an integrated strategy for the construction and analysis of differential co-expression networks. BMC Genomics. 2015;16 Suppl 3:S4. | ||
Udyavar AR, Hoeksema MD, Clark JE, et al. Co-expression network analysis identifies Spleen Tyrosine Kinase (SYK) as a candidate oncogenic driver in a subset of small-cell lung cancer. BMC Systems Biology. 2013;7 Suppl 5:S1. | ||
Clarke C, Madden SF, Doolan P, et al. Correlating transcriptional networks to breast cancer survival: a large-scale coexpression analysis. Carcinogenesis. 2013;34(10):2300–2308. | ||
Yang Y, Han L, Yuan Y, Li J, Hei N, Liang H. Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nature Communications. 2014;5:3231. | ||
Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Molecular Biology and Evolution. 2009;26(7):1641–1650. | ||
Li HD, Menon R, Omenn GS, Guan Y. The emerging era of genomic data integration for analyzing splice isoform function. Trends in Genetics: TIG. 2014;30(8):340–347. | ||
Eksi R, Li HD, Menon R, et al. Systematically differentiating functions for alternatively spliced isoforms through integrating RNA-seq data. PLoS Computational Biology. 2013;9(11):e1003314. | ||
Hong S, Chen X, Jin L, Xiong M. Canonical correlation analysis for RNA-seq co-expression networks. Nucleic Acids Research. 2013; 41(8):e95. | ||
Pal S, Bi Y, Macyszyn L, Showe LC, O’Rourke DM, Davuluri RV. Isoform-level gene signature improves prognostic stratification and accurately classifies glioblastoma subtypes. Nucleic Acids Research. 2014;42(8):e64. | ||
Sebestyen E, Zawisza M, Eyras E. Detection of recurrent alternative splicing switches in tumor samples reveals novel signatures of cancer. Nucleic Acids Research. 2015;43(3):1345–1356. | ||
Chen C, Hu Y, Li L. NRP1 is targeted by miR-130a and miR-130b, and is associated with multidrug resistance in epithelial ovarian cancer based on integrated gene network analysis. Molecular Medicine Reports. 2016;13(1):188–196. | ||
Zhu J, Wang S, Zhang W, et al. Screening key microRNAs for castration-resistant prostate cancer based on miRNA/mRNA functional synergistic network. Oncotarget. 2015;6(41):43819–43830. | ||
Azevedo H, Moreira-Filho CA. Topological robustness analysis of protein interaction networks reveals key targets for overcoming chemotherapy resistance in glioma. Scientific Reports. 2015;5:16830. | ||
Gustafsson M, Gawel DR, Alfredsson L, et al. A validated gene regulatory network and GWAS identifies early regulators of T cell-associated diseases. Science Translational Medicine. 2015;7(313):313ra178. | ||
Lambert MJ, Cochran WO, Wilde BM, Olsen KG, Cooper CD. Evidence for widespread subfunctionalization of splice forms in vertebrate genomes. Genome Research. 2015;25(5):624–632. | ||
Kress TR, Sabo A, Amati B. MYC: connecting selective transcriptional control to global RNA production. Nature Reviews. Cancer. 2015;15(10): 593–607. | ||
Chuang CY, Chang H, Lin P, et al. Up-regulation of osteopontin expression by aryl hydrocarbon receptor via both ligand-dependent and ligand-independent pathways in lung cancer. Gene. 2012;492(1): 262–269. | ||
Boldrini L, Donati V, Dell’Omodarme M, et al. Prognostic significance of osteopontin expression in early-stage non-small-cell lung cancer. British Journal of Cancer. 2005;93(4):453–457. | ||
Donati V, Boldrini L, Dell’Omodarme M, et al. Osteopontin expression and prognostic significance in non-small cell lung cancer. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research. 2005;11(18):6459–6465. | ||
Zhang W, Konopleva M, Burks JK, et al. Blockade of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase and murine double minute synergistically induces apoptosis in acute myeloid leukemia via BH3-only proteins Puma and Bim. Cancer Research. 2010;70(6):2424–2434. | ||
Cheng CW, Chen PM, Hsieh YH, et al. Foxo3a-mediated overexpression of microRNA-622 suppresses tumor metastasis by repressing hypoxia-inducible factor-1alpha in erk-responsive of lung cancer. Oncotarget. Epub 2015. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.