Back to Journals » Infection and Drug Resistance » Volume 12
Functional aspects, phenotypic heterogeneity, and tissue immune response of macrophages in infectious diseases
Authors de Sousa JR, Da Costa Vasconcelos PF, Quaresma JAS ![]()
Received 13 March 2019
Accepted for publication 5 July 2019
Published 22 August 2019 Volume 2019:12 Pages 2589—2611
DOI https://doi.org/10.2147/IDR.S208576
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Joachim Wink
Jorge Rodrigues de Sousa,1,2 Pedro Fernando Da Costa Vasconcelos,2,3 Juarez Antonio Simões Quaresma1–4
1Tropical Medicine Center, Federal University of Pará, Belém, PA, Brazil; 2Evandro Chagas Institute, Ministry of Health, Ananindeua, PA, Brazil; 3Center of Biological and Health Sciences, State University of Pará, Belém, PA, Brazil; 4School of Medicine, São Paulo University, São Paulo, SP, Brazil
Correspondence: Juarez Antonio Simões Quaresma
Tropical Medicine Center, Federal University of Pará, Av. Generalissimo Deodoro 92, Umarizal, Belém 66055-240, PA, Brazil
Email [email protected]
Abstract: Macrophages are a functionally heterogeneous group of cells with specialized functions depending not only on their subgroup but also on the function of the organ or tissue in which the cells are located. The concept of macrophage phenotypic heterogeneity has been investigated since the 1980s, and more recent studies have identified a diverse spectrum of phenotypic subpopulations. Several types of macrophages play a central role in the response to infectious agents and, along with other components of the immune system, determine the clinical outcome of major infectious diseases. Here, we review the functions of various macrophage phenotypic subpopulations, the concept of macrophage polarization, and the influence of these cells on the evolution of infections. In addition, we emphasize their role in the immune response in vivo and in situ, as well as the molecular effectors and signaling mechanisms used by these cells. Furthermore, we highlight the mechanisms of immune evasion triggered by infectious agents to counter the actions of macrophages and their consequences. Our aim here is to provide an overview of the role of macrophages in the pathogenesis of critical transmissible diseases and discuss how elucidation of this relationship could enhance our understanding of the host–pathogen association in organ-specific immune responses.
Keywords: macrophage, macrophage phenotype, macrophage activation, infections, infectious diseases
Introduction
The primary function of the immune system has traditionally been defined as protection against microorganisms, although it plays an equally crucial role in protecting the host against invasion by foreign agents and eliminating affected cells that might compromise organic homeostasis.1–5 The immune system functions through mechanisms involving both humoral and cellular immunity, which generally act in the early and late inborn phases of the response to antigens, respectively.2–5 In this context, the concept of organ-specific tissue immunity, which is related to the functional specificity of each organ mostly arose from the research of Engwerda and Kaye.1 Particularly, in their study in 2000, the authors discussed the immunological mechanisms triggered in response to hepatic infection by parasites of the genus Leishmania.1 This work helped focus attention on the concept of tissue immunity and prompted investigations into its mechanisms in various organs because of the close relationship between the induction routes of the cellular lesions involved in distinct organs in infectious, inflammatory, metabolic, and neoplastic diseases.1
According to the concept of tissue immunity, responses can be categorized into three major groups: immunity of epithelial barriers, complex organ immunity, and organ immunity with immunological privilege.6 In this context, the cells of the immune system acquire functional specializations that are related to the characteristics of the organs where these cells are.7–12 Consequently, investigating local immune responses is crucial to understanding the host–pathogen relationship in infectious diseases.7–12 Among the various cells of the immune system, macrophages constitute a functionally heterogeneous group that act in various stages of the immune response. In addition, they are also involved in the maintenance of homeostasis because they release growth factors and enzymes for tissue renewal and remodeling. Similar to lymphocytes, macrophages display considerable functional heterogeneity and are classified according to both their ability to release cytokines and chemokines and their expression of specific receptors. This heterogeneity probably reflects the plasticity and versatility of macrophages in their response to signals from the microenvironment.13,14
The process of macrophage formation is initiated in the bone marrow, where the differentiation of hematopoietic stem cells generates precursor cells. Subsequently, pre-monocytic lines in the blood produce monocytes, which enter various tissues from the bloodstream to form resident macrophages that perform multiple functions.13–15
Both under normal or pathological conditions, macrophages perform diverse functions in an organism dependent on their location.13 In the liver, macrophages named Kupffer cells are responsible for removing various substances, toxins, and aggressive pathogens.10,16–18 The lung contains alveolar macrophages, which eliminate microorganisms, allergens, and microparticles.19,20 In the brain, macrophages known as microglia provide the main cellular defense of the central nervous system and modulate proliferation, responses, morphological transformation, motility, migration, intracellular communication, and phagocytosis.13,21–25
Macrophages in the lymph nodes are classified as spinal or subcapsular sinusoidal, which capture antigens and present them to B cells.23 In the intestine, macrophages recognize and remove enteric pathogens and maintain the balance necessary for the operation of the microbiota.23,26–28 The spleen contains macrophage lines derived to perform diverse functions. Splenic macrophages are responsible for phagocytizing senescent or altered erythrocytes, capturing antigens, and processing and presenting them to T lymphocytes.23,29,30 Lastly, in the skin, macrophages might be present as Langerhans cells or histiocytes, which primarily function in phagocytosis as well as antigen capture, processing, and presentation.13,21–23,31–33
Macrophage phenotypes
Emerging new concepts have recently highlighted the need for a novel approach to our understanding of macrophage response.34–36 Similar to T lymphocytes, which feature numerous cellular subtypes, macrophages also differentiate into several phenotypes.37,38 Cells belonging to distinct phenotypes are distinguished by expression of groups of markers that define each phenotype. Thorough elucidation of these groups of markers, or panels, has revealed that these phenotypes are distinguished by expression differences in chemokines, chemokine receptors, enzymes, costimulatory molecules, Toll receptors, cytokines, and cytokine receptors.39–43
M1 macrophages express inducible nitric oxide synthase (iNOS), the primary marker that confirms the response of these cells in a specific disease (Table 1).44–46 However, a surface marker that defines the identity of M1 macrophages has not yet been identified.
 |
Table 1 Macrophage phenotypes, stimulating factors, genes, primary markers, and functions described in the scientific literature |
Among the phenotypes of M2 macrophages, M2a, M2b, and M2c macrophages are characterized by the presence of arginase (Arg).47–49 These subpopulations express a cell-surface scavenger receptor that recognizes haptoglobin–hemoglobin complexes used to identify M2 macrophages and is labeled with CD163. Moreover, a previously unrecognized subpopulation of M2 macrophages, M2d, has been described, but the molecules responsible for the differentiation of M0 macrophages into M2d macrophages remain unknown. However, this population participates in the angiogenesis response, producing interleukin (IL)-10 and vascular endothelial growth factor (Table 1).50–52
Macrophages are capable of differentiating into six additional subpopulations: regulatory (Mreg), hematophage-associated (Mha), oxidative (Mox), M3, M4, and M17 macrophages (Table 1).53–56
Mreg macrophages respond to the stimulation of Treg cells and are responsible for inducing the repair response and inhibiting triggering of mechanisms that produce pro-inflammatory cytokines in tissue lesions.57–59 Mox macrophages differ from the other cell types in expressing oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine and producing HO-1 and sulfiredoxin-1.60–62 Mox macrophages are also associated with triggering of oxidative responses (Table 1).60–62
The recently discovered M3 macrophages are known as commuting macrophages. The first study discussing the emergence of this previously unknown lineage showed that their appearance depended on the switching response between M1 and M2 macrophage pathways (Table 1).63
M4 macrophages, similar to M3 macrophages, represent a group of newly identified cells that have attracted much attention in research because of the response mechanisms they can develop.48,64 M4 macrophages differentiate in the presence of colony-stimulating factor (CSF) and (CXCL4). The markers of these cells are CD206, CD68, metalloproteinase 7 (MMP7), MMP12, and calcium-binding protein A8 (S100A8); the cells produce IL-6, TNF-α, and CCL18 and CCL20.65,66 M4 macrophages highly express the low-density lipoprotein (LDL) receptor, and in chronic lesions, these cells form a large contingent of foam cells that in the inflammatory process (Table 1).67–69
Lastly, the concept of cell emergence in the case of M17 macrophages is new. One of the earliest studies supporting this hypothesis showed that cell formation can be induced by corticosteroids, granulocyte-macrophage (GM)-CSF, or IL-10, and a key feature is the cells that respond to IL-17 become resistant to apoptosis (Table 1).70
Markers of M1, M2, and M4 macrophages
With advances in technology, discovery of the transcriptome has allowed for identification of several genes related to the phenotypic modulation of macrophages.71,72
This transcriptomic characterization of the phenotypes M1, M2, and M4 is a topic that needs to be addressed.73,74 For instance, the integrated network established by a combination of factors typically characterizes a complex phenomenon where real-time polymerase chain reaction (PCR) validation, for example, shows that the expression of CD38, Gpr18, and Fpr2 is exclusive to the M1 phenotype.75 The importance of this relationship is the fact that these newly identified markers are essential to cell activation and the oxidative stress mechanism.76
Without disregarding findings previously confirmed by other techniques, the transcriptome study reaffirmed the expression of classical markers that affect not only cell activation but also differentiation and development of the pro-inflammatory response. Therefore, IFN-γ, TNF-α, LPS, IFN-β, iNOS, IL-1β, IL-6, IL-12, B7, B7.1, MCP3, IP-10 or CXCL10, CD16, CD32, CD62, CTLA8, and α-chain still represent a set of fundamental markers that characterize activation of the classical pathway (M1).77–79 In situations where the stimulus is mediated by LPS + IFN-γ, there is an increase in expression of pSTAT1, pSTAT6, Socs1, NF-kB Nfkbiz, IRF5, IRF1, TNF, II6, II27, IL23a, II12a, CCL5, CXCL9, CXCL10, CXCL11, NOS2, GBPI, CCR7, and CD40 on M1 macrophages (Table 1).80
In humans, a very peculiar phenomenon occurs by polarization of the response between M1 and M2 macrophages, attributable to the M1 phenotype expression of Wnt family member 5A (WNTA5A) and glycerol-3-phosphate dehydrogenase 2 (GPD2) and modulation of oncogenesis and mitochondrial stress. In addition, another important discovery made using transcriptome analysis and in vitro experimental models is that CD120b, Toll-like receptor 2 (TLR2), SLAM family member 7 (SLAMF7), and chemotactic factors, such as CXCL8, CCL5, CCL2, and IL-6, regulate the M1 phenotype matrix.81 Considerable data support that polarization via the M1 phenotype classically conjugates the result, such as stimulation mainly by IFN-γ, LPS, and TNF-α.82 However, an interesting approach has shown that the M1 phenotype is dependent on transient receptor potential cation channel subfamily C member 3 (TRPC3) to activate calcium/calmodulin-dependent protein kinase II alpha (CAMK2) and STAT1, and activation of intracellular signaling mechanisms that influence the production of apoptotic markers.83
As for the M2 phenotype, regulation of the alternative pathway results in interaction of different stimuli that modulate gene expression in different cellular environments. For example, in mice, the M (IL-4) regulates expression of pStat6, pStat1, ifr4, Socs2, ccl17, Ccl22, Ccl24, and Arg1. In humans, modulation of IRF4, SOCS1, GATA3, CCL13, CCL17, CCL17, CCL18, MRC1, STAB1, MARCO, CD163, FN, TGFB1, MMP1, MMP12, TG, F13A1, TGM2, ADORA3, TGFBR2, IL17RB, ALOX15, and CD200R occurs (Table 1).80,84
Expression of Arg1 is classical evidence of the presence of the repair phenotype. In addition, cytokines such as IL-4 and IL-13 are required to establish cell differentiation. The transcriptome can aggregate a standard of analysis for the M2 phenotype characterized mainly by the expression of M130, CD163, CD206, MRC1, CCR7, МСР-3, FceRII, CD23, GPR86, GPR105, TLR-2, P2Y8, P2Y11, P2Y12, dectin-1, DC-SIGN, CD209, DCIR (CLECSF6), CLACSF13, FIZZ1, ST2 (SR-A, М60), CD184, TRAIL, STAT6, IL-27Ra, Chil3, Retnla, Ppar-γ, ARG1, TGF-β, IL-4, IL-5, IL-10, and IL-13 (Table 1).85,86
It is noteworthy that transcriptome analysis has enabled elucidation of mechanisms underlying the modulation of IL-4 intracellular signaling by the induction of zinc finger TFs, Egr2, and c-Myc expression. As a final outcome, ingenuity pathway analysis shows that expression of these markers is directly involved in cell cycle progression in tumorigenesis.34
How the M2 phenotype converts to M2a, M2b, or M2c depends highly on the stimuli provided.87,88 The M2a phenotype is generally observed after exposure to IL-4/IL-13, which induces this phenotype during the process of immunoregulation, tissue repair, and tumor progression.34 Transcriptome analysis reveals modulation of M2a, which shows that the response triggered by this cell is quite ambiguous and mainly involves polarization of the response between M1 and M2 macrophages. This is because the M2a phenotype can activate genes that regulate pro- and anti-inflammatory responses. Confirmation of such a situation is centered on the exacerbated expression of cytochrome c oxidase subunit 5A (COX5a) and WNT5b.89 In contrast, previous reports have documented that the M2a phenotype cannot process an adequate response to produce pro-inflammatory cytokines and mediate an efficient microbicidal effect.
The response mediated by the M2c phenotype is basically induced by the stimulus provided by IL-10/transforming growth factor (TGF)-β, where post-differentiation transcriptome analysis indicates that this phenotype strongly regulates the expression of genes, such as CD163, MMP8, TIMP1, VCAM, SERPINA1, MARCO, PLOD2, PCOCLE2, and F5. Furthermore, the results of responses triggered by the production of these markers are directly associated with angiogenesis, remodeling of the extracellular matrix, and phagocytosis (Table 1).90
Regarding the M4 phenotype, the modulatory effect correlates strongly with the CXCL-4-mediated response that directly influences cell differentiation.48,91,92 This was confirmed mainly by approaches investigating the development of atherosclerosis where the appearance of foamy macrophages with overexpressed genes, such as GM-CSF, CCL18, CD86, TNFSF10, ALOX5, IL1RN,and AIF1, induced production of pro-inflammatory mediators. In addition, development of the immune response in the processing and presentation of antigens by this cell triggers a regulatory phenomenon leading to overexpression of CD86, HLA-DRB1, HLA-DRB3, HLA-DRB4, and HLA-DQA1.93 Interestingly, in the activation of an intracellular cascade that aims to regulate metabolism by accumulation of lipids, the cell overexpresses APOC2, APOE, and SORL1.93 The purpose of the M4 phenotype study expanded to a set of interpretations, including the overexpression of chemotaxis-inducing genes (CCL3, CCL7, and CCR1) and cell adhesion where the cell overexpresses ITGAV, ITGA6, and ITGB8B.93
The importance and relevance of the M4 phenotype in understanding atherogenesis indicate that M4 macrophages strongly express genes responsible for inducing proteases that degrade the extracellular matrix such as MMP7, MMP8, and MMP12. The results of the pathophysiological analysis of atherosclerosis investigated using transcriptome analysis shows that these cells express genes that facilitate the formation of foam cells such as CD36, MSR1, and ABCG1 by accumulation of LDL, which when metabolized, causes oxidative reactions that impair not only phagocytosis but also the destruction of microorganisms (Table 1).93,94
Transduction signal and polarization of macrophage M1, M2, and M4 macrophages
Investigation of the spectral role of macrophages began at the end of the 19th century, more precisely in 1883 with elaboration of the phagocytosis theory.95 In 1905, Metchnikoff showed that phagocytic cells are able to destroy bacteriain infection persistent.96 From that moment, the concept of macrophage activation began to be constructed. In 1964, the idea was further expounded, and between 1966 and 1971, lymphocytes were identified as the major antigen-specific cells responsible for macrophage microbicidal activation.96–98 The discovery of IFN-γ in 1983 was the determinant for the proposal of the classical pathway of macrophage activation.99 It is important to note that in 1986, the identification of the two subpopulations of T lymphocytes (Th1 and Th2) represented further advances underlying the emergence of the spectral macrophage concept.100 In 1992, the concept of M2 macrophages began to be constructed, and in 2000, Mills et al101,102 proposed the classification of M1 and M2 macrophages based on the response mediated by Th1 and Th2 lymphocytes. Between 2002 and 2006, the concepts of activation, heterogeneity, and plasticity of M1 and M2 macrophages were amplified given the diversity of a set of experimental models, which heralded the appearance of M2 subtypes M2a, M2b, and others.103–106 However, the theoretical basis of the response behavior of macrophages indicates a complex relationship, mainly with polarization. In 2010, Biswas and Mantovani107,108 showed a response paradigm between M1 and M2 macrophages. Finally, during the last few years, technological advances have led to deeper discussions on the polarization of response of M1 and M2 macrophages. In 2014, Murray et al80 attempted to standardize terminology regarding macrophage polarization concepts from the responses obtained in several experimental models. However, problems still exist at the conceptual level, particularly regarding the stabilization of macrophage subpopulations through their participation in pathophysiological, pathological, and immunological mechanisms (Figure 1).109–111
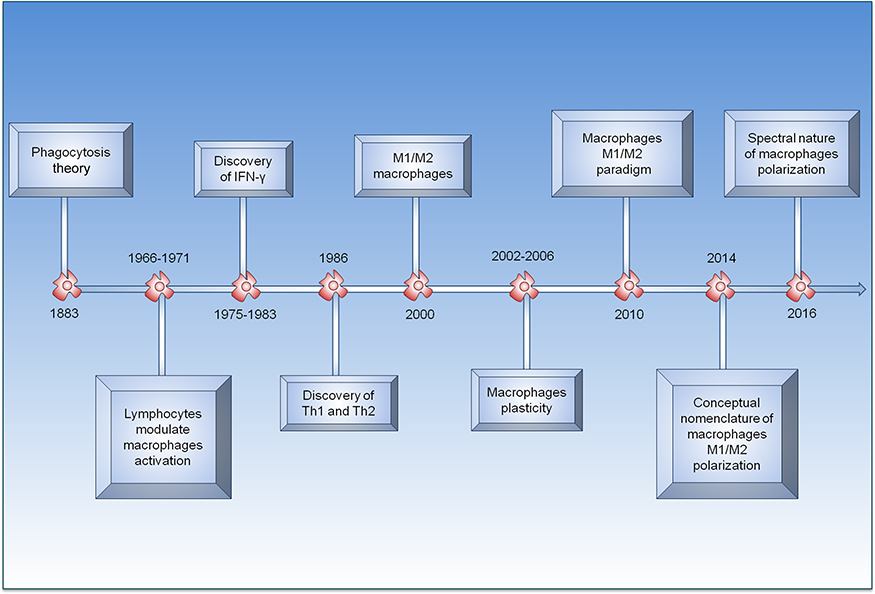 |
Figure 1 Timeline of main events marking development of concept of polarization of macrophages. |
Following the identification of the M2 phenotype, studies began to emerge demonstrating that two distinct types of macrophages were present, which were each associated with a distinct activation pathway. In the classical pathway, the main cells are M1 macrophages, also known as inflammatory macrophages and microbicides. In the alternative pathway, also known as the reparative pathway, cells responsible for the repair response are the M2 macrophages.112–114
Considering the versatility of macrophages, their response mechanisms in destroying microorganisms appear to include aspects of the immune response that remain incompletely elucidated. However, current evidence further strengthens their relationship with components of the innate and adaptive immune systems.115,116
In the innate immune response, when macrophages exposed to certain stimuli, such as LPS-peptide complexes, are recognized by TLRs and CD14, signaling cascades might be initiated that generate reactive oxygen and nitrogen species and inflammatory cytokines such as IFN-α and IFN-β.117–119
During the humoral response, phagocytosis of certain microorganisms might be mediated by recognition of the Fc portion.120 Specific costimulatory molecules act as a key step in enhancing phagocytosis, antigen presentation, and endocytosis. These molecules belong to the family of receptors known as scavenger and mannose receptors.121
During the innate response, certain components of the complement system regulate polarization of the macrophage response. In the inflammatory pathway, some complements, such as C3a, C5a, C5b, and C9, might induce production of inflammatory mediators. In the regenerative pathway, the presence of opsonized apoptotic bodies with fragments of complement C1q or C3b might stimulate M2 macrophages to produce Arg and anti-inflammatory cytokines.122
The classical signal-transduction response scheme described for both M1 and M2 macrophages follows a pathway starting from the binding of a protein or some other component to a receptor, which initiates a signaling cascade involving signal transduction through phosphorylation of transcription factors, migration of the factors to the nucleus, activation of genes, and production of molecules such as cytokines, enzymes, and costimulatory molecules.21,123–125
In the classical pathway, we highlight three routes that are essential to the modulation of the microbicidal activity of M1 macrophages. The first is the binding of IFN-γ or TNF-α to their respective receptors (IFN-γR or TNF-αR), whose main event is the development of intracellular cascade based on the activation of JAK1 and JAK2 and STAT1 or STAT2 responsible for inducing the production of MHCII, IL-12, and iNOS.126–128 We cannot fail to mention the importance of GM-CSF in mediating NOS2 production. MHC class II, IL-12b, which is dependent on the activation of JAK2 and STAT5 and phosphorylation of IRF5.129,130 In addition to a set of immunological strategies mediated by response of M1 macrophages, TLR4 recognizes LPS, one of the major components produced by gram-negative bacteria, which activates NF-κB, AP1, and the production of TNF-α, IL-1β, IL-6, and IFN-γ (Figure 2).131–133
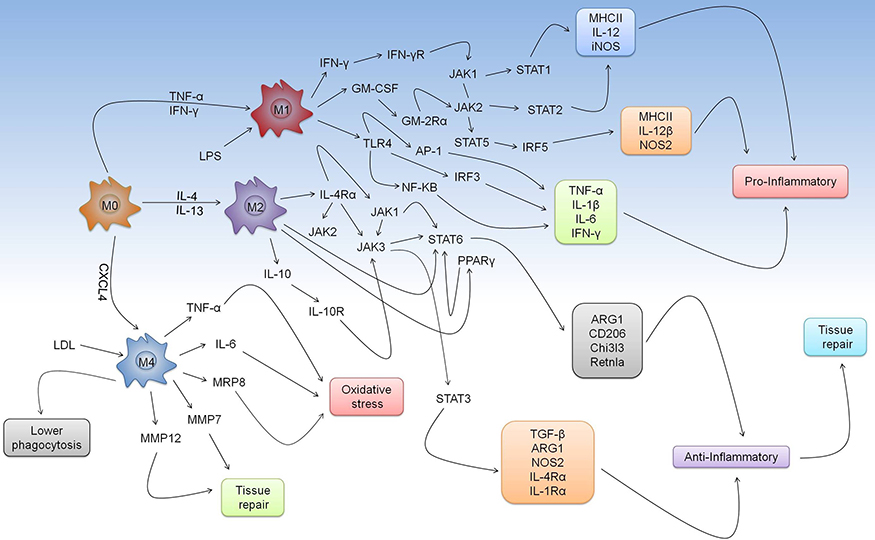 |
Figure 2 Mechanism of response mediated by M1, M2, and M4 macrophages. Macrophage differentiation spectrum includes markers that modulate development of certain phenotypes. Depending on the stimulus, cytokines, such as TNF-α and IFN-γ, may influence the process of shifting the M0 macrophage to M1. In this scenario, we highlight the performance of IFN-γ, GM-CSF, and TLR4 that influence development of intracellular cascade, which aims to produce of a series of markers to enhance the pro-inflammatory response in several aspects of classical pathway. In contrast, the antagonistic relationship between the duality of M1/M2 macrophages shows that IL-4 and IL-13 cytokines are mainly responsible for inducing differentiation of macrophages from M0 to M2. In the alternative pathway, activation of IL-4Rα or IL-10R shows that IL-4, IL-13, or IL-10 directly modulate development of anti-inflammatory responses and tissue repair. Through pathways of polarization, we emphasized the significance of M4 macrophages in the modulation of oxidative stress response as well as tissue repair. This relationship is particularly important in pathophysiology of atherosclerosis, where formation of foam cells due to LDL accumulation and low phagocytosis implies a regulatory phenomenon that triggers the production of TNF-α, IL-6, MRP8, MMP7, and MMP12. |
As an alternative pathway, M2 macrophages respond to stimulation by IL-4, IL-10, or IL-13 to mediate the development of the repair phenotype. By binding to their respective receptors, these cytokines can trigger activation of repair signaling cascades that modulate the activation and phosphorylation of JAK1-3, STAT3, STAT6, peroxisome proliferator-activated receptor γ (PPARγ) or phosphoinositide 3-K (PI3K) in order to induce production of Arg1, CD206, chitinase-like 3 (Chi313), reticulon-like A (Rtnla), TGF-β, NOS2, IL-4Rα, and IL-1Rα (Figure 2).134,135
M4 macrophages have attracted attention in the research community due to their relationship to M0 macrophages. Specifically, their exposure to CXCL4 was shown to lead to the production of CD68, IL-6, TNF-α, S100A8, MMP7, and MMP12.136,137 M4 macrophages are highly enriched in atherosclerotic lesions, where the cells exhibit increased expression of LDL receptors.138–140 This phenomenon causes accumulation of fat inside phagocytes, leading to the development of atheroma plaques and, consequently, oxidative lesions.138–140 Although more comprehensive studies must be conducted to further elucidate the roles of M4 macrophages in the pathogenesis of diseases, the functions are probably related to an immunosuppressive environment targeting infectious pathogens (Figure 2).141–143
Antagonistic phenomena and duality of response between inos and arg1 and its relationship with M1 and M2 phenotype in infectious diseases
Macrophages are one of the primary cell types that exert microbicidal activity against several groups microorganisms via polarization and duality of the response between the M1 and M2 phenotypes.144–146 Therefore, an intriguing relationship is established in infectious diseases due to what happens intracellularly.144–146 Classically, iNOS and Arg1 are characteristic and determinant markers for modulating the response of either M1 (iNOS) or M2 (Arg1) macrophages.147–149 The resulting effects of these enzymes demonstrate that both compete for the same substrate (arginine) in order to induce the production of NO (M1) or proliamines and proline (M2). This antagonistic relation causes a phenomenon of duality that facilitates the destruction or survival of microorganisms.149–151 In light of this phenomenon, we highlight the evolution of the understanding of this ambiguous relationship mainly in leprosy, tuberculosis, and leishmaniasis.
In the case of bacteria, this relationship shows that contradictory events are associated with the response mechanism triggered by both enzymes. In leprosy, the in situ expression of iNOS is curiously predominant in the lepromatous form of the disease, whereas the immune response is directed more toward an immunosuppressive profile. Interestingly, in lepromatous leprosy, iNOS correlates positively with the expression of natural resistance-associated macrophage protein 1 (NRAMP1), IL-22, and STAT3, indicating that the M1 phenotype is present in the tissue environment, generating reactive metabolites an attempt to eliminate the infection. Given that in this clinical form the anti-inflammatory response is more incisive, the development of the M2 phenotype fundamentally participates in inactivating the microbicidal response linked to the M1 phenotype. It is precisely because of this that the tissue damage is much more obvious, and the associated response includes the exacerbated expression of Arg1, IL-4, IL-10, TGF-β, and FGFβ.152–154
With regard to Mycobacterium tuberculosis, the response mediated by iNOS is more complex. Several seminal studies have shown that, as in leprosy, in tuberculosis iNOS exerts microbicidal activity.155,156 However, one of the final products generated in the metabolic cascade of iNOS favors the development of an intracellular signaling mechanism that controls inflammation. In this context, NO is able to inhibit the expression of NF-κB, one of the major transcription factors responsible for inducing the production of pro-inflammatory cytokines such as IFN-γ and TNF-α.157 Controversial events in the immunopathogenesis of M. tuberculosis infection are crucial in the interpretation of iNOS and Arg1 responses when the tissue microenvironment in tuberculoid granulomas are analyzed. The duality of the polarization of the relationship between M1 and M2 macrophages in both human and monkey granulomas, the anti-inflammatory phenotype (CD163+iNOS+Arg1high) is located in more external areas, whereas in the internal portion, the pro-inflammatory phenotype (CD11C+CD68+CD163dimiNOS+eNOS+Arg1low) is predominant. Therefore, in the same tissue environment, the presence of the M1 and M2 phenotype is crucial to maintaining the balance of the immune response and tissue damage.158
A study focused on the hypoxic granuloma regions in tuberculosis demonstrated that Arg1 expression seems to control M. tuberculosis growth when NOS2 is ineffective. In this case, enzyme expression correlates negatively with the proliferation of T cells.149
Against leishmaniasis, stimuli presented to the same cells may or may not trigger the microbicidal response.159,160 Therefore, what is pertinent to the performance of TNF-α or IL-4 directly influences the formation of the M1/M2 phenotype. In vitro, for example, TNF-α inhibits IL-4-induced Arg1 expression in bone marrow-derived macrophages. The consequences of this inactivation directly affect the phosphorylation and translocation of STAT6 to mediate production of Arg1.161 In vivo Leishmania infections mainly show that in C57BL/6WT, the control of the cellular and microbicidal stress response is regulated by NOS2 and Arg1 with a predominance of NOS2 and NO. In the case of C57BL/6WT TNF−/- BALB/c, this change favors pathogen survival as well as non-phenotype change and a balanced expression of NOS2 and Arg1.161 In Tie2Cre±Argfl/fl mice, the amastigote forms of Leishmania major are destroyed in activated macrophages where the inflammatory response is accentuated. In light of this, we conclude that the expression of TNF-α influences change in the cellular phenotype and potentiates the response of M1 macrophages in situ by Arg1 deletion, histone acetylation, and NOS2 and NO production. Interestingly, deletion of Arg1 can restore control of parasite proliferation in animals that evolved resistance.161
Mechanisms of immune evasion used by intracellular pathogens against macrophage responses
Macrophages have failed to develop an effective response against certain intracellular pathogens.162,163 The reason for this failure has been a long-standing question in this field. Thus, numerous studies have sought to elucidate the pathway through which the behavior of an infected cell is altered. Regardless of whether a cell is infected by viruses, bacteria, protozoa, or fungi, the pathogen in each case uses the immune system to modulate the response and, thereby, survive inside the cell.163–165
Bacteria and protozoa
One of the key issues arising from the discussion of this subject is understanding the response that PRRs can induce through the recognition of PAMPs of certain microorganisms. Classically, receptors such as those in the Toll family agglutinate a variable range from TLR1 to TLR13.166–168 Therefore, in the case of mycobacteria, the importance of TLR2, TLR4, and TLR9 in the development of the microbicidal response is paramount since the regulation of the intracellular cascade involves the binding of MyD88 and Mal, also known as TIRAP, which recruits IRAK1/4, TRAF6, and TRICA1 to induce the production of mitogen-activated protein kinase (MAPK) and NF-κB.169–171 As a final outcome, NF-kB is responsible for inducing the production of pro-inflammatory cytokines such as TNF-α, IFN-γ, IL-1β, and IL-6 that participate in the activation of macrophages and enzymes such as iNOS, which is responsible the generation of ROS and destruction of the microorganism.126,172,173 However, the immune evasion strategies adopted by mycobacteria include a series of events aimed at inactivating this mechanism. With regard to M. tuberculosis, TLR2 is the main receptor by which mycobacteria modulate immune escape. Therefore, the interaction between TLR2, ESAT-6, and CFP-10 drastically suppresses the immune response of macrophages by inhibiting NF-κB activation, iNOS, and NO production.174 Nevertheless, recognition events of PAMPs show that M. tuberculosis mutants produce sulfoglycolipids that antagonize the activation of both TLR2 and NF-κB.175 Interestingly, in this same dynamic, the bacterium can subvert the TLR2-MyD88 pathway and facilitate its translocation into the cytosol. The consequences of this modulation include the direct involvement of H37Rv strains and the escape of the phagosome or phagolysosome.176 It is worth mentioning that immune evasion via modulation of TLR2 by the recombinant lipoprotein Rv1016c, induces apoptosis of macrophages by increasing annexin expression.177 Consequently, this relationship becomes beneficial because in addition to Rv1016c inhibition of MHC-II expression, TLR2-dependent apoptosis may promote the release of residual bacilli into apoptotic cells and, thereby, decrease the recognition of CD4+ T cells, impairing surveillance in chronic disease conditions (Figure 3).177
 |
Figure 3 Immune evasion and strategies adopted by Mycobacterium tuberculosis, Leishmania, and Trypanosomiasis cruzi to evade microbicidal response of macrophages. Because it is a sequence of events that trigger destruction of microorganisms, immune escape strategies that inhibit microbicidal responses are as varied as possible. However, we seek to centralize an understanding of the crucial points that facilitate a pathogen’s survival in the environment. For M. tuberculosis, it is noteworthy that TLR2 is the main receptor by which the immune escape correlates with ESAT-6 and CFP-10 response, whereas inhibitory modulation mainly affects the inflammatory cascade associated with NF-κB and to iNOS. Since sufoglycolipids inhibit activation of TLR2 and NF-κB, this not only compromises the microbicidal response but also facilitates the survival and proliferation of the bacillus. Through evolution of mycobacterial destruction mechanisms, adaptation of pathogens to evade the immune system has generated certain strains such as H37Rv that modulate phagosome maturation and facilitate translocation of bacillus to the cytosol. In addition, a recombinant lipoprotein derived from M. tuberculosis, such as RV1016c, regulates the mechanism of apoptosis through the annexins. This is crucial to facilitating proliferation of the bacillus in the tissue environment because RV1016c inhibits MHC II expression, compromising the CD4 T cell response and immune surveillance. Considering that the cytokine response is fundamental to induce the activation of macrophages, as well as the microbicidal response, M. tuberculosis inhibits the action of IFN-γ by inhibiting autophagy through responses of IL-6 and IL-27. Expansion of this scenario negatively regulates Atg12-atg5 and positive intracellular cascade involving JAK/PIK3/AKt/mTOR/Mcl1. Implications of such events are inhibitive of phagolysosome maturation and, consequently, the microbicidal response of the cell, which favors pathogen survival in phagocytes. With regard to protozoa, we highlight Leishmania, which, in addition to using the cells of the phagocytic system as a reservoir, also uses innumerable strategies of immune evasion to survive. Therefore, Leishmania major modulates TLR2 responses to induce SOCS1 and 3 to inhibit not only intracellular cascades of NF-κB but also the response of IFN-γ and TNF-α. For Leishmania donovani, by ubiquitination of TRAF 6 through A20, the response of NK-κB is compromised as well as the production of IL-6 and IL-12. Interestingly, L. major modulates immunosuppressive responses to facilitate its survival, and L. donovani follows the same path by mainly regulating expression of SOCS1 and 3 to avoid oxidative stress and catastrophic effects that may occur with apoptosis. Despite this dynamic, Leishmania amazonensis can induce macrophages to produce IFN-β to generate SOD in order to create an environment conducive for the parasite to evade the immune system, survive, and multiply. Considering that vesicular traffic is fundamental for phagosome maturation, either protozoa or M. tuberculosis manipulates macrophages to maintain Rab5 expression increased to establish early phagosome fusion with lysosome, as well as formation of the calcium-calcineurin complex to inhibit the production of cathepsin D and Rab7 and to facilitate the survival of the pathogens. The effect of evasion of Leishmania with T. cruzi is very peculiar in the case of leishmaniasis, autophagy is explored to eliminate promastigotes forms and reduce responses of T cells. In contrast, T. cruzi uses autophagy as a central point to invade phagocytic cells and multiplies whether at basal levels or not. Interestingly, LC3 protein serves as the gateway to mediate this process. |
Concerning protozoa, one of the main parasites that compromise macrophage activity is Leishmania.163 L. major negatively modulates TLR2 receptor activity to increase the recruitment of SOCS1 and SOCS3, which inhibit the production of NF-kB, IFN-γ, and TNF-α in order to establish pathogen survival.178 In Leishmania donovani, the amastigote forms and TLR2 suppress the inhibitor of NF-kB kinase (IKK)-NF-kB complex and the positive regulation of the enzyme deubiquitinating A20.15,179 As a result, this process facilitates the degradation of TRAF6 and inhibition of the TNF-α and IL-12 response. As a survival strategy, L. donovani also prevents apoptosis triggered by oxidative stress, inducing SOCS1 and SOCS3 production.180 Interestingly, that in Leishmania amazonensis, the intracellular regulatory effect anchored in the action of protein kinase triggers production of IFN-β, which in turn, increases expression of superoxide dismutase 1 (SOD1), inhibiting destruction of the parasite and facilitating its proliferation (Figure 3).181
Regarding Trypanosomiasis cruzi, TLRs play a crucial role in resistance to the protozoan infection in macrophages, where GPIs and mucins modulate the behavioral difference among TLR2, TLR4, TLR7, and TLR9. In fact, this relationship mainly affects the production of Th1 cytokines. Notably, in TLR2-deficient mice, a potent pro-inflammatory response was observed, mainly induced by IFN-γ.181,182
One of the central questions surrounding the strategies of immune evasion by M. tuberculosis, Leishmania, or T. cruzi is the mechanism underlying phagosome maturation modulation.183–185 This has direct implications on the biogenesis of the phagolysosome. To survive in a hostile environment, these microorganisms have developed a series of mechanisms aimed at neutralizing one of the cell’s primary lines of defense. It starts at the point of inhibition of the inflammatory cytokine response (IFN-γ or TNF-α), where this relationship influences the endosome immaturity.186–188 Incidentally, Rab family proteins are actively involved in this process. In this context, Rab5 and 7 are directly related to the early and late fusion of the endosome and lysosome, respectively. The stage of immaturity of the phagosome is organized in this duality of interaction between early and late fusion.189–191 Therefore, microorganisms initially maintain Rab5 expression in order to facilitate recruitment of coronin 1a protein and the development of the calcium-calcineurin signaling pathway whose final outcome is the loss of fusion of the phagosome with the lysosome (Figure 3).192–194 The implications of this catastrophic cellular effect include the inhibition of late markers such as Rab7 and the loss of various hydrolases such as cathepsin D. Another final point that dramatically impacts the immune escape of pathogens is inhibition of PI3K, which deregulates the gallbladder traffic in the Golgi apparatus (Figure 3).195–197
Virus
Currently, the action of macrophages has not been shown to be directly related to the antiviral response. This is attributed to the fact that TLR3, TLR7, TLR8, RIG-I, and MDA5 are largely produced by the cell. In this context, the modulation of viral replication starts from information that is passed to the cytoplasm where RIG-1 and MDA5 recognize single-stranded RNA viruses to facilitate the formation of the IKK complex (α, β, and γ). Consequently, the phosphorylation and migration of NF-κB and IRF3 and 7 to the nucleus activate genes responsible for inducing the production of IFN-α and β (Figure 4).198–200 However, the strategies used by the virus modulate the immune evasion mechanism, whereas non-structural NSs of DENV, WNV, ZIKV, and HCV flaviviruses inhibit activation of RIG-I and MDA5 as well as a series of intracellular markers during the inactivation of molecules such as STING, MAVS, and NF-κB. This mechanism directly affects the inhibition of ISGs, Mx, OS, PKR, and the production of IFN-α and β. In the case of type 1 IFN, intracellular signaling triggered by receptor-cytokine binding influences concomitant activation and phosphorylation of JAK1, JAK2, STAT1, and STAT2, as well as the formation of STAT/IRF complexes that primarily activate genes associated with the production of inflammatory mediators responsible for inhibiting viral replication (Figure 4).201–203 Assuming that negative regulation coupled to non-structural proteins of flavivirus or HCV is crucial for viral survival, the immune leakage, in this case, mainly inhibits intracellular signaling pathways (Figure 4).
 |
Figure 4 Antiviral response and immune escape mechanism triggered by hepatitis C virus (HCV) and flaviviruses DENV, ZIKV, and WNV. Among PRRs, RLRs are extremely important in development of antiviral responses mainly associated with actions of IFN-α and β. Among the receptors, we highlight RIG-I and MDA5 that recognize PAMPs either HCV or flaviviruses to activate MAVS and STING, which in turn, provoke the formation of TBK1/IKKε and NEMO/IKK-α/β complex. This mechanism has the final product for both phosphorylation of IRF7 and IRF3 and the release of KF-κB p50/p65 subunit that migrates to the nucleus to activate genes responsible for production of inflammatory cytokines and IFN-type 1. In the case of IFN-α and β, IFNR activation recruits JAK1 and TYR2, which when phosphorylated, trigger formation of an intracellular cascade that culminates in formation of the STAT1/STAT2/IRF9 complex, which migrates to the nucleus, activating genes associated with the antiviral response such as Mx, OAS, PKR, and ISGs. To evade the immune response, HCV or flaviviruses induce the production of non-structural proteins that inhibit response of several markers at points considered essential for the development of the intracellular cascade, facilitating the viral replication process. A priori WNV can establish primordial control of antiviral responses at the level of inhibition of RIG-I and MDA5. In another follow-up, ZIKV, DENV, WNW, or HCV through NS1, NS2A, NS2B, NS3, NS4A, NS4B, or NS5 inhibit TBK1/IKKε complex formation, IRF3 and 7 phosphorylation, and NF-κB activation. In the response by IFN-type 1, NSEN of DENV and WNV inhibited activation of JAK1/TYR2. HCV NS5A inhibits activation of STAT1 and 2. Among various immune escape mechanisms, autophagy is critical for flavivirus to mediate biogenesis of viral replication, mainly because of changes in pH and endocytosis. In this sequence, it is worth noting that NS2B and NS4A/B proteins can manipulate autophagy to alter membrane curvature in the Golgi apparatus to facilitate viral replication. However, in another follow-up, NS2B/3 cleaves FAM134B into endoplasmic reticulum (ER) to suppress autophagolysosome formation and reticulophage, a form of selective autophagy, to facilitate viral replication. As a result, NS4A induces cells to produce PIK3, which impair the conversion of LC3I to LC3II and inhibit autophagy. |
Autophagy
Just as the cell death process directly impacts the immune evasion strategies triggered by microorganisms, autophagy is used to mediate pathogen replication at various stages of development.204–206 Interestingly, the apoptotic-like Leishmania uses autophagy to reduce the elimination of the promastigote forms in macrophages and diminish the response of T cells. It is worth noting that Annexin V, light chain 3 (LC3), beclin 1, and mechanistic target of rapamycin (mTOR) participate actively in this mechanism.207 On the other hand, T. cruzi uses autophagy to subvert the lysosomal exocytic process and invade host cells under basal conditions. This occurs frequently because of recruitment of the LC3 protein.208 Interestingly, when the process is induced by starvation or rapamycin, autophagolysosome formation and LC3 expression is increased. This also results in large-scale formation of the parasitoid vacuole. As a form of escape, the trypomastigote form resides in the autophagolysosome and escapes into the cytosol, increasing the autophagic cell response (Figure 3).209
For M. tuberculosis, the suppression of autophagy to increase the survival of mycobacteria serves as a potential regulatory effect. In this context, cytokines such as IL-6 and IL-27 inhibit IFN-γ-induced autophagy. In this process, expression of IL-6 or IL-27 considerably reduces the maturation of the autophagolysosome from the negative regulation of Atg12-Atg5 and positive effects of complex JAK/PI3K/Akt/mTOR and Mcl-1 (Figure 3).210,211
Concerning the modulation of the autophagic cascade for flavivirus, autophagy seems to be a crucial factor in mediating the biogenesis of viral replication.212–214 This relationship overlaps in a dynamic where the internalization of viruses at endocytosis facilitates the change in pH and, consequently, the release of viral RNA (vRNA) into the cytoplasm. The events of autophagy manipulation are mainly modulated by non-structural proteins, such as NS2B, NS4A, and NS4B.215–217 Interestingly, at the beginning of the infection, both NS4A and NS4B manipulate the cell to mediate membrane curvature and induce autophagy. This process mobilizes the membranes and lipids to establish a proviral event that facilitates viral replication. This directly impacts regulation of the Golgi apparatus because NS2B/3 induces the production of proteases that trigger the cleavage of FAM134B, one of the main receptors involved in endoplasmic reticulum (ER)-phagy.218 Finally, it is worth mentioning that in this dynamic, NS4A can deregulate the PI3K pathway, which, in turn, impairs the conversion of the LC3-I protein to LC3-II (Figure 4).215,219
Tissue response of macrophages to infectious agents
Macrophages are a population of cells that play critical roles in maintaining homeostasis and in the pathogenesis of inflammatory, infectious, and neoplastic diseases.220 Macrophages are found in both blood and tissues, and despite their common origin in the bone marrow, tissue macrophages appear to exhibit an embryogenic development that is independent of that of blood macrophages or even of macrophages that migrate from the blood into tissues.221–223
Macrophages perform a wide spectrum of functions in various tissues, including the removal of dead cells, as well as tissue remodeling and repair, which are associated with an elaborate response to foreign agents and infectious or noninfectious aggressors.224–226 Given this, macrophages are crucial for the development of the organ-specific immune response, which acts on innate and adaptive immunity, frequently serving as a bridge between these two types of responses.2,227,228
During an immune response, macrophages can function to maintain tissue homeostasis as well as contribute to the development of tissue lesions that are ultimately the result of a lack of control of the tissue immune response against aggressors. These aggressors concurrently trigger mechanisms to escape the action of macrophages, thus leading to a lack of control of the cellular response associated with tissue injury.229–234 This phenomenon is observed both in non-infectious and infectious diseases caused by viruses, bacteria, fungi, protozoa, and helminths.
Macrophages can acquire microbicidal properties after stimulation by microbial products, acting in both innate and adaptive immunity mechanisms. One of the main strategies used by microorganisms to escape this macrophage activity involves altering the response profile of macrophages.185,235–252 The activation of a response profile mediated by M1 macrophages is commonly associated with a protective tissue environment and has been described for infections by pathogens such as Helicobacter pylori, M. tuberculosis, Mycobacterium leprae, Salmonella typhi, and Chlamydia.253–258 M1 macrophages elicit an effective immune response against S. typhi and H. pylori, and in the response against H. pylori, the induction of iNOS associated with the M1 profile is closely related to the occurrence of gastric cancer.259–264 Early and acute stages of M. tuberculosis infections are mediated by M1 macrophages. In the later stages and chronic disease, the infection is associated with responses mediated by M2 macrophages, whose weak microbicidal activity and ability to eliminate bacteria are linked to the occurrence of the lesions observed during disease evolution.265–269 In leprosy, macrophages are the primary cells that exert microbicidal activity. Thus, the role of macrophages in the response to M. leprae has been widely described and is correlated with the expression of certain cytokines that are classical representatives of the cellular immune response, such as TNF-α and IFN-γ.270–272
In leprosy, when TNF-α and IFN-γ bind to their specific receptors, the behavior of M0 macrophages is altered. Furthermore, after undergoing phenotypic modification, the M0 macrophages become inflammatory, producing inflammatory cytokines and enzymes such as iNOS, which produces NO and, consequently, reactive oxygen radicals, leading to the destruction of the bacteria.273–275 In contrast, M2 and M4 macrophages are observed mainly in lepromatous lesions, and both cell types present expression profiles associated with immunosuppressive cytokines. Therefore, these cells are likely to be related to more diffuse and severe aspects of the disease. These features are also observed in tegumentary leishmaniasis, where M2 macrophages are related mainly to diffuse and anergic aspects of the disease.276–278
In viral diseases, the macrophage response might be subverted to a certain extent by the numerous escape mechanisms that viral agents use to resist the host immune response.279–289 Nevertheless, viruses generally trigger a response mediated by M1 macrophages that might contribute to the severity of the disease. In hepatitis C, chronic infection can lead to cell damage and cirrhosis, and the virus can induce an immune response, targeting the viral protein NS3, mediated by IL-12 and TNF-α produced by M1 macrophages. This process creates a pro-inflammatory environment that contributes to the induction of cell injury. A similar process occurs in the liver of patients infected with dengue virus and yellow fever, where cytokines, such as TNF-α and TGF-β released by Kupffer cells, contribute to the development of a pro-inflammatory and proapoptotic environment in tissue hepatic necrosis, which leads to massive apoptosis of hepatocytes because of the proapoptotic action of TGF-β.290,291 Viral influenza infections have been linked to lung injury induced by M1 macrophage-associated cytokines, including TNF-α, IFN-γ, and IL-6. Furthermore, these lung lesions might be attenuated in coinfection by Staphylococcus aureus, which is normally present in the airway mucosal microbiota, because of the bacterium’s ability to stimulate the expression of M2 macrophages.292–294 Conversely, in the pathogenesis of coronavirus and cytomegalovirus infections, the lesions that develop are related to the induction of an inflammatory environment mediated by M2 macrophages, which indicates that viral agents can still benefit from the induction of specific tissue environments related to the Th2 response and, consequently, tissue repair and induction of tissue fibrosis.287,295–299
In mice experimentally infected with Schistosoma japonica, liver fibrosis was associated with the presence of M2 macrophages, which exhibited increased regulation of Notch1/Jagged1 signals, and the blockade of this expression reversed the M2 phenotype and, subsequently, the associated fibrosis.300 In another study, induction of the M2 phenotype in Schistosoma mansoni infections elicited a low-grade chronic inflammatory response that was associated with insulin resistance in obese patients.301
Leishmania infection development depends on both the immune response pattern and the Leishmania species. Thus, as described for leprosy, the clinical evolution of leishmaniasis depends on the interaction of the infectious agent and M1 or M2 macrophages, with the M2 subpopulation related to the process of disease progression.302,303 Lastly, in rats experimentally infected with Toxoplasma gondii, peritoneal macrophages in the pre-gestational phase were primarily composed of M1 macrophages, whereas in the intra-gestational phase, the M2 phenotype dominated. This is related to the immune response patterns mediated by Th2 lymphocytes, which are predominant during gestation.304
Conclusion and perspective
The immune mechanisms inherent to the macrophage response in various organs and tissues are distinct to the pathogen–host interaction. Furthermore, these mechanisms are strongly influenced by both the infectious agents that induce a specific phenotype and the tissue characteristics of the organs where the cells are located. This phenomenon clearly demonstrates the diverse nature and importance of the response in the evolution of specific tissue lesions. Elucidation of the involvement of each phenotypic macrophage subpopulations in infections in various organs and tissues would undoubtedly enhance our understanding of the complex mechanisms of interactions between pathogens and the immune system and their consequence in the genesis of tissue lesions in infectious diseases.
Acknowledgment
This work was funded by the Council for Scientific and Technological Development – CNPq/Brazil (Grant numbers 302553/2015-0 and 116427/2016-7) and Coordination of Superior Level Staff Improvement - CAPES/Brazil. Program of support for qualified production – PAPQ/UFPA.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Engwerda CR, Kaye PM. Organ-specific immune responses associated with infectious disease. Immunol Today. 2000;21:73–78.
2. Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol. 2015;16:343–353. doi:10.1038/ni.3123
3. Villani A-C, Sarkizova S, Hacohen N. Systems immunology: learning the rules of the immune system. Annu Rev Immunol. 2018;36:813–842. doi:10.1146/annurev-immunol-042617-053035
4. Davis MM, Tato CM, Furman D. Systems immunology: just getting started. Nat Immunol. 2017;18:725–732.
5. Sattler S. The role of the immune system beyond the fight against infection. Adv Exp Med Biol. 2017;1003:3–14. doi:10.1007/978-3-319-57613-8_1
6. Quaresma JAS, Sotto MN, Balato A. Inflammatory and immune-mediated cutaneous diseases. Mediators Inflamm. 2017;2017:1–2. doi:10.1155/2017/6793968
7. Vijayan A, Rumbo M, Carnoy C, Sirard J-C. Compartmentalized antimicrobial defenses in response to flagellin. Trends Microbiol. 2018;26:423–435. doi:10.1016/j.tim.2017.10.008
8. Rodríguez-Cortés A, Carrillo E, Martorell S, et al. Compartmentalized immune response in leishmaniasis: changing patterns throughout the disease. PLoS One. 2016;11:e0155224. doi:10.1371/journal.pone.0155224
9. Quiding-Järbrink M, Granström G, Nordström I, Holmgren J, Czerkinsky C. Induction of compartmentalized B-cell responses in human tonsils. Infect Immun. 1995;63:853–857.
10. Spengler U, Lechmann M, Irrgang B, Dumoulin FL, Sauerbruch T. Immune responses in hepatitis C virus infection. J Hepatol. 1996;24:20–25.
11. Newman LS, Bobka C, Schumacher B, et al. Compartmentalized immune response reflects clinical severity of beryllium disease. Am J Respir Crit Care Med. 1994;150:135–142. doi:10.1164/ajrccm.150.1.8025739
12. McDermott AJ, Huffnagle GB. The microbiome and regulation of mucosal immunity. Immunology. 2014;142:24–31. doi:10.1111/imm.12231
13. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi:10.1038/nri3073
14. Chow A, Brown BD, Merad M. Studying the mononuclear phagocyte system in the molecular age. Nat Rev Immunol. 2011;11:788–798. doi:10.1038/nri3087
15. Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi:10.1038/nri978
16. Quaresma JA, Barros VL, Pagliari C, et al. Hepatocyte lesions and cellular immune response in yellow fever infection. Trans R Soc Trop Med Hyg. 2007;101:161–168. doi:10.1016/j.trstmh.2006.02.019
17. Zannetti C, Roblot G, Charrier E, et al. Characterization of the inflammasome in human Kupffer cells in response to synthetic agonists and pathogens. J Immunol. 2016;197:356–367. doi:10.4049/jimmunol.1502301
18. Lee JC, Lee SS, Schlesinger KJ, Richter GW. Detection of protein subunits of ferritin in situ in cells by immunofluorescence. Am J Pathol. 1974;75:473–487.
19. Yamasaki K, Eeden SFV. Lung macrophage phenotypes and functional responses: role in the pathogenesis of COPD. Int J Mol Sci. 2018;19:E582. doi:10.3390/ijms19020582
20. Arora S, Dev K, Agarwal B, Das P, Syed MA. Macrophages: their role, activation and polarization in pulmonary diseases. Immunobiology. 2018;223:383–396. doi:10.1016/j.imbio.2017.11.001
21. Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–761. doi:10.1038/nri3088
22. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762–774. doi:10.1038/nri3070
23. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–455. doi:10.1038/nature12034
24. London A, Cohen M, Schwartz M. Microglia and monocyte-derived macrophages: functionally distinct populations that act in concert in CNS plasticity and repair. Front Cell Neurosci. 2013;7:34. doi:10.3389/fncel.2013.00034
25. Chavez-Galán L, Olleros ML, Vesin D, Garcia I. Much more than M1 and M2 macrophages, there are also CD169+ and TCR+ macrophages. Front Immunol. 2015;6:263–278. doi:10.3389/fimmu.2015.00263
26. Liu Y-H, Ding Y, Gao -C-C, Li L-S, Wang Y-X, Xu J-D. Functional macrophages and gastrointestinal disorders. World J Gastroenterol. 2018;24:1181–1195. doi:10.3748/wjg.v24.i11.1181
27. Joeris T, Müller-Luda K, Agace WW, Mowat AM. Diversity and functions of intestinal mononuclear phagocytes. Mucosal Immunol. 2017;10:845–864. doi:10.1038/mi.2017.22
28. Grainger JR, Konkel JE, Zangerle-Murray T, Shaw TN. Macrophages in gastrointestinal homeostasis and inflammation. Pflugers Arch. 2017;469:527–539. doi:10.1007/s00424-017-1958-2
29. Ganz T. Macrophages and iron metabolism. Microbiol Spectr. 2016;4:5.
30. Steiniger BS. Human spleen microanatomy: why mice do not suffice. Immunology. 2015;145:334–346. doi:10.1111/imm.12469
31. Mowat AM, Scott CL, Bain CC. Barrier-tissue macrophages: functional adaptation to environmental challenges. Nat Med. 2017;23:1258–1270. doi:10.1038/nm.4430
32. Yanez DA, Lacher RK, Vidyarthi A, Colegio OR. The role of macrophages in skin homeostasis. Pflugers Arch. 2017;469:455–463. doi:10.1007/s00424-017-1953-7
33. Minutti CM, Knipper JA, Allen JE, Zaiss DM. Tissue-specific contribution of macrophages to wound healing. Semin Cell Dev Biol. 2017;61:3–11. doi:10.1016/j.semcdb.2016.08.006
34. Atri C, Guerfali FZ, Laouini D. Role of human macrophage polarization in inflammation during infectious diseases. Int J Mol Sci. 2018;19:1801. doi:10.3390/ijms19061801
35. Sjoelund V, Smelkinson M, Nita-lazar A. Phosphoproteome profiling of the macrophage response to different Toll-like receptor ligands identifies differences in global phosphorylation dynamics. J Proteome Res. 2014;13:5185–5197. doi:10.1021/pr5002466
36. Liu K, Zhao E, Ilyas G, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy. 2015;11:271–284. doi:10.1080/15548627.2015.1009787
37. Weirather J, Hofmann UDW, Beyersdorf N, et al. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. 2014;115:55–67. doi:10.1161/CIRCRESAHA.115.303895
38. Eligini S, Crisci M, Bono E, et al. Human monocyte-derived macrophages spontaneously differentiated in vitro show distinct phenotypes. J Cell Physiol. 2013;228:1464–1472. doi:10.1002/jcp.24390
39. Schmall A, Al-Tamari HM, Herold S, et al. Macrophage and cancer cell cross-talk via CCR2 and CX3CR1 is a fundamental mechanism driving lung cancer. Am J Respir Crit Care Med. 2015;191:437–447. doi:10.1164/rccm.201406-1137OC
40. Makita N, Hizukuri Y, Yamashiro K, Murakawa M, Hayashi Y. IL-10 enhances the phenotype of M2 macrophages induced by IL-4 and confers the ability to increase eosinophil migration. Int Immunol. 2015;27:131–141. doi:10.1093/intimm/dxv003
41. Fernando MR, Reyes JL, Iannuzzi J, Leung G, McKay DM. The pro-inflammatory cytokine, interleukin-6, enhances the polarization of alternatively activated macrophages. PLoS One. 2014;9:e94188. doi:10.1371/journal.pone.0094188
42. Said EA, Al-Reesi I, Al-Riyami M, et al. Increased CD86 but not CD80 and PD-L1 expression on liver CD68+ cells during chronic HBV infection. PLoS One. 2016;11:e0158265. doi:10.1371/journal.pone.0158265
43. Kumar P, Tyagi R, Das G, Bhaskar S. Mycobacterium indicus pranii and mycobacterium bovis BCG lead to differential macrophage activation in toll-like receptor-dependent manner. Immunology. 2014;143:258–268. doi:10.1111/imm.12319
44. Zhu Y, Zhang L, Lu Q, et al. Identification of different macrophage subpopulations with distinct activities in a mouse model of oxygen-induced retinopathy. Int J Mol Med. 2017;40:281–292. doi:10.3892/ijmm.2017.3022
45. Klar AS, Michalak-Mićka K, Biedermann T, Simmen-Meuli C, Reichmann E, Meuli M. Characterization of M1 and M2 polarization of macrophages in vascularized human dermo-epidermal skin substitutes in vivo. Pediatr Surg Int. 2018;34:129–135. doi:10.1007/s00383-017-4179-z
46. Davis MJ, Tsang TM, Qiu Y, et al. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in cryptococcus neoformans infection. MBio. 2013;4:e00264–e00313. doi:10.1128/mBio.00264-13
47. Canton J, Neculai D, Grinstein S. Scavenger receptors in homeostasis and immunity. Nat Rev Immunol. 2013;13:621–634. doi:10.1038/nri3515
48. Colin S, Chinetti-Gbaguidi G, Staels B. Macrophage phenotypes in atherosclerosis. Immunol Rev. 2014;262(1):153–166. doi:10.1111/imr.12218
49. De Paoli F, Staels B, Chinetti-Gbaguidi G. Macrophage phenotypes and their modulation in atherosclerosis. Circ J. 2014;78:1775–1781. doi:10.1253/circj.CJ-14-0621
50. Rőszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm. 2015;2015:816460. doi:10.1155/2015/125380
51. Wang Q, Ni H, Lan L, Wei X, Xiang R, Wang Y. Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res. 2010;20:701–712. doi:10.1038/cr.2010.52
52. Ferrante CJ, Pinhal-Enfield G, Elson G, et al. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Rα) signaling. Inflammation. 2013;36:921–931. doi:10.1007/s10753-012-9579-6
53. Kadl A, Meher AK, Sharma PR, et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res. 2010;107:737–746. doi:10.1161/CIRCRESAHA.109.215715
54. Kalish S, Lyamina S, Manukhina E, Malyshev Y, Raetskaya A, Malyshev I. M3 macrophages stop division of tumor cells in vitro and extend survival of mice with Ehrlich ascites carcinoma. Med Sci Monit Basic Res. 2017;23:8–19. doi:10.12659/MSMBR.902285
55. Zizzo G, Cohen PL. IL-17 stimulates differentiation of human anti-inflammatory macrophages and phagocytosis of apoptotic neutrophils in response to IL-10 and glucocorticoids. J Immunol. 2013;190:5237–5246. doi:10.4049/jimmunol.1203017
56. Erbel C, Wolf A, Lasitschka F, et al. Prevalence of M4 macrophages within human coronary atherosclerotic plaques is associated with features of plaque instability. Int J Cardiol. 2015;186:219–225. doi:10.1016/j.ijcard.2015.03.151
57. Riquelme P, Tomiuk S, Kammler A, et al. IFN-γ-induced iNOS expression in mouse regulatory macrophages prolongs allograft survival in fully immunocompetent recipients. Mol Ther. 2013;21:409–422. doi:10.1038/mt.2012.168
58. Riquelme P, Haarer J, Kammler A, et al. TIGIT+ iTregs elicited by human regulatory macrophages control T cell immunity. Nat Commun. 2018;9:2858. doi:10.1038/s41467-018-05167-8
59. Riquelme P, Amodio G, Macedo C, et al. DHRS9 is a stable marker of human regulatory macrophages. Transplantation. 2017;101:2731–2738. doi:10.1097/TP.0000000000001588
60. Hristodorov D, Mladenov R, Huhn M, Barth S, Thepen T. Macrophage-targeted therapy: CD64-based immunotoxins for treatment of chronic inflammatory diseases. Toxins Basel. 2012;4:676–694. doi:10.3390/toxins4090676
61. Bories GFP, Leitinger N. Macrophage metabolism in atherosclerosis. FEBS Lett. 2017;591:3042–3060. doi:10.1002/1873-3468.12786
62. Marques L, Negre-Salvayre A, Costa L, Canonne-Hergaux F. Iron gene expression profile in atherogenic Mox macrophages. Biochim Biophys Acta - Mol Basis Dis. 2016;1862:1137–1146. doi:10.1016/j.bbadis.2016.03.004
63. Malyshev I, Malyshev Y. Current concept and update of the macrophage plasticity concept: intracellular mechanisms of reprogramming and M3 macrophage “switch” phenotype. Biomed Res Int. 2015;2015:1–22. doi:10.1155/2015/341308
64. Medbury HJ, Williams H, Fletcher JP. Clinical significance of macrophage phenotypes in cardiovascular disease. Clin Transl Med. 2014;3:63. doi:10.1186/s40169-014-0042-1
65. Butcher MJ, Galkina EV. Phenotypic and functional heterogeneity of macrophages and dendritic cell subsets in the healthy and atherosclerosis-prone aorta. Front Physiol. 2012;3:1–13. doi:10.3389/fphys.2012.00044
66. Bekkering S, Joosten LA, van der Meer JW, Netea MG, Riksen NP. The epigenetic memory of monocytes and macrophages as a novel drug target in atherosclerosis. Clin Ther. 2015;37:914–923. doi:10.1016/j.clinthera.2015.01.008
67. Hoeksema MA, Stöger JL, de Winther MP. Molecular pathways regulating macrophage polarization: implications for atherosclerosis. Curr Atheroscler Rep. 2012;14:254–263. doi:10.1007/s11883-012-0240-5
68. Gleissner CA. Macrophage phenotype modulation by CXCL4 in atherosclerosis. Front Physiol. 2012;3:1. doi:10.3389/fphys.2012.00001
69. Van Overmeire E, Laoui D, Keirsse J, Van Ginderachter JA, Sarukhan A. Mechanisms driving macrophage diversity and specialization in distinct tumor microenvironments and parallelisms with other tissues. Front Immunol. 2014;5:1–16. doi:10.3389/fimmu.2014.00127
70. de la Paz Sánchez-Martínez M, Blanco-Favela F, Mora-Ruiz MD, Chávez-Rueda AK, Bernabe-García M, Chávez-Sánchez L. IL-17-differentiated macrophages secrete pro-inflammatory cytokines in response to oxidized low-density lipoprotein. Lipids Health Dis. 2017;16:196. doi:10.1186/s12944-017-0565-8
71. Das A, Yang C-S, Arifuzzaman S, et al. High-resolution mapping and dynamics of the transcriptome, transcription factors, and transcription co-factor networks in classically and alternatively activated macrophages. Front Immunol. 2018;9:22. doi:10.3389/fimmu.2018.00022
72. Ramsey SA, Vengrenyuk Y, Menon P, et al. Epigenome-guided analysis of the transcriptome of plaque macrophages during atherosclerosis regression reveals activation of the wnt signaling pathway. PLoS Genet. 2014;10:e1004828. doi:10.1371/journal.pgen.1004541
73. Walter W, Alonso-Herranz L, Trappetti V, et al. Deciphering the dynamic transcriptional and post-transcriptional networks of macrophages in the healthy heart and after myocardial injury. Cell Rep. 2018;23:622–636. doi:10.1016/j.celrep.2018.03.029
74. Karunakaran D, Rayner KJ. Macrophage miRNAs in atherosclerosis. Biochim Biophys Acta - Mol Cell Biol Lipids. 2016;1861:2087–2093. doi:10.1016/j.bbalip.2016.02.006
75. Jablonski KA, Amici SA, Webb LM, et al. Novel markers to delineate murine M1 and M2 macrophages. PLoS One. 2015;10:e0145342. doi:10.1371/journal.pone.0145342
76. Biagioli M, Carino A, Cipriani S, et al. The bile acid receptor GPBAR1 regulates the M1/M2 phenotype of intestinal macrophages and activation of GPBAR1 rescues mice from murine colitis. J Immunol. 2017;199:718–733. doi:10.4049/jimmunol.1700183
77. Novoselov VV, Sazonova MA, Ivanova EA, Orekhov AN. Study of the activated macrophage transcriptome. Exp Mol Pathol. 2015;99:575–580. doi:10.1016/j.yexmp.2015.09.014
78. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi:10.12703/P
79. Lisi L, Ciotti GM, Braun D, et al. Expression of iNOS, CD163 and ARG1 taken as M1 and M2 markers of microglial polarization in human glioblastoma and the surrounding normal parenchyma. Neurosci Lett. 2017;645:106–112. doi:10.1016/j.neulet.2017.02.076
80. Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi:10.1016/j.immuni.2014.06.008
81. Beyer M, Mallmann MR, Xue J, et al. High-resolution transcriptome of human macrophages. PLoS One. 2012;7:e45466. doi:10.1371/journal.pone.0045466
82. Genard G, Lucas S, Michiels C. Reprogramming of tumor-associated macrophages with anticancer therapies: radiotherapy versus chemo- and immunotherapies. Front Immunol. 2017;8:828. doi:10.3389/fimmu.2017.00828
83. Kumarasamy S, Solanki S, Atolagbe OT, Joe B, Birnbaumer L, Vazquez G. Deep transcriptomic profiling of M1 macrophages lacking Trpc3. Sci Rep. 2017;7:3–8. doi:10.1038/srep39867
84. Ren R, Tyryshkin K, Graham CH, Koti M, Robert Siemens D. Comprehensive immune transcriptomic analysis in bladder cancer reveals subtype specific immune gene expression patterns of prognostic relevance. Oncotarget. 2017;8:70982–71001. doi:10.18632/oncotarget.v8i41
85. Chistiakov DA, Myasoedova VA, Revin VV, Orekhov AN, Bobryshev YV. The impact of interferon-regulatory factors to macrophage differentiation and polarization into M1 and M2. Immunobiology. 2018;223:101–111. doi:10.1016/j.imbio.2017.10.005
86. Zhang N, Bao Y-J, Tong AH-Y, et al. Whole transcriptome analysis reveals differential gene expression profile reflecting macrophage polarization in response to influenza A H5N1 virus infection. BMC Med Genomics. 2018;11:20. doi:10.1186/s12920-018-0335-0
87. Roszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm. 2015;2015:16–18. doi:10.1155/2015/125380
88. Tang L, Zhang H, Wang C, Li H, Zhang Q, Bai J. M2A and M2C macrophage subsets ameliorate inflammation and fibroproliferation in acute lung injury through interleukin 10 pathway. Shock. 2017;48:119–129. doi:10.1097/SHK.0000000000000820
89. Derlindati E, Cas AD, Montanini B, et al. Transcriptomic analysis of human polarized macrophages: more than one role of alternative activation? PLoS One. 2015;10:e0119751. doi:10.1371/journal.pone.0119751
90. Lurier EB, Dalton D, Dampier W, et al. Transcriptome analysis of IL-10stimulated M2c macrophages by next-generation sequencing. Immunobiology. 2017;222:847–856. doi:10.1016/j.imbio.2016.10.009
91. Vinchi F, Muckenthaler MU, Da Silva MC, Balla G, Balla J, Jeney V. Atherogenesis and iron: from epidemiology to cellular level. Front Pharmacol. 2014;5:94. doi:10.3389/fphar.2014.00094
92. Chistiakov DA, Bobryshev YV, Orekhov AN. Changes in transcriptome of macrophages in atherosclerosis. J Cell Mol Med. 2015;19:1163–1173. doi:10.1111/jcmm.12658
93. Gleissner CA, Shaked I, Little KM, Ley K. CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J Immunol. 2010;184:4810–4818. doi:10.4049/jimmunol.0902016
94. Rojas J, Salazar J, Martínez MS, et al. Macrophage heterogeneity and plasticity: impact of macrophage biomarkers on atherosclerosis. Scientifica Cairo. 2015;2015:1–17. doi:10.1155/2015/851252
95. Metchnikoff E. Untersuchungen ueber die mesodermalen Phagocyten einiger Wirbeltiere. Biologisches Centralblatt. 1883;3:560–565.
96. Liu Y-C, Zou X-B, Chai Y-F, Yao Y-M. Macrophage polarization in inflammatory diseases. Int J Biol Sci. 2014;10:520–529. doi:10.7150/ijbs.8879
97. Bloom BR, Bennett B. Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science. 1966;153:80–82. doi:10.1126/science.153.3731.80
98. Nathan CF, Karnovsky ML, David JR. Alterations of macrophage functions by mediators from lymphocytes. J Exp Med. 1971;133:1356–1376. doi:10.1084/jem.133.6.1356
99. Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med. 1983;158:670–689. doi:10.1084/jem.158.3.670
100. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–2357.
101. Stein M. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176:287–292. doi:10.1084/jem.176.1.287
102. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 Paradigm. J Immunol. 2000;164:6166–6173. doi:10.4049/jimmunol.164.12.6166
103. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–555. doi:10.1016/S1471-4906(02)02302-5
104. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi:10.1016/j.it.2004.09.015
105. Stout RD, Suttles J. Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J Leukoc Biol. 2004;76:509–513. doi:10.1189/jlb.0504272
106. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi:10.1172/JCI57313
107. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–896. doi:10.1038/ni.1937
108. Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22:231–237. doi:10.1016/j.coi.2010.01.009
109. Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. 2016;44:439–449. doi:10.1016/j.immuni.2016.02.024
110. Murray PJ. Macrophage Polarization. Annu Rev Physiol. 2017;79:541–566. doi:10.1146/annurev-physiol-022516-034339
111. Gordon S, Plüddemann A. Tissue macrophages: heterogeneity and functions. BMC Biol. 2017;15:1–18. doi:10.1186/s12915-017-0392-4
112. Raggi F, Pelassa S, Pierobon D, et al. Regulation of human macrophage M1-M2 polarization balance by hypoxia and the triggering receptor expressed on myeloid cells-1. Front Immunol. 2017;8:1–18. doi:10.3389/fimmu.2017.01097
113. Bertani FR, Mozetic P, Fioramonti M, et al. Classification of M1/M2-polarized human macrophages by label-free hyperspectral reflectance confocal microscopy and multivariate analysis. Sci Rep. 2017;7:1–9. doi:10.1038/s41598-017-08121-8
114. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44:450–462. doi:10.1016/j.immuni.2016.02.015
115. Novak ML, Koh TJ. Macrophage phenotypes during tissue repair. J Leukoc Biol. 2013;93:875–881. doi:10.1189/jlb.1012512
116. Mills CD. Anatomy of a discovery: M1 and M2 macrophages. Front Immunol. 2015;6:1–12. doi:10.3389/fimmu.2015.00212
117. Da Silva TA, Zorzetto-Fernandes ALV, Cecílio NT, Sardinha-Silva A, Fernandes FF, Roque-Barreira MC. CD14 is critical for TLR2-mediated M1 macrophage activation triggered by N-glycan recognition. Sci Rep. 2017;7:1–14. doi:10.1038/s41598-017-07397-0
118. Baumann CL, Aspalter IM, Sharif O, et al. CD14 is a coreceptor of Toll-like receptors 7 and 9. J Exp Med. 2010;207:2689–2701. doi:10.1084/jem.20101111
119. Rajaiah R, Perkins DJ, Ireland DDC, Vogel SN. CD14 dependence of TLR4 endocytosis and TRIF signaling displays ligand specificity and is dissociable in endotoxin tolerance. Proc Natl Acad Sci. 2015;112:8391–8396. doi:10.1073/pnas.1424980112
120. Gallo P, Gonçalves R, Mosser DM. The influence of IgG density and macrophage Fc (gamma) receptor cross-linking on phagocytosis and IL-10 production. Immunol Lett. 2011;133:301–314.
121. Linehan SA, Martinez-Pomares L, Gordon S. Mannose receptor and scavenger receptor: two macrophage pattern recognition receptors with diverse functions in tissue homeostasis and host defense. Adv Exp Med Biol. 2000;479:1–14. doi:10.1007/0-306-46831-X_1
122. Bohlson SS, O’Conner SD, Hulsebus HJ, Ho MM, Fraser DA. Complement, C1Q, and C1q-related molecules regulate macrophage polarization. Front Immunol. 2014;5:1–7. doi:10.3389/fimmu.2014.00402
123. Li H, Jiang T, Li MQ, Zheng XL, Zhao GJ. Transcriptional regulation of macrophages polarization by microRNAs. Front Immunol. 2018;9:1–12.
124. Roy S, Schmeier S, Arner E, et al. Redefining the transcriptional regulatory dynamics of classically and alternatively activated macrophages by deepCAGE transcriptomics. Nucleic Acids Res. 2015;43:6969–6982. doi:10.1093/nar/gkv646
125. Lyroni K, Patsalos A, Daskalaki MG, et al. Epigenetic and transcriptional regulation of IRAK-M expression in macrophages. J Immunol. 2017;198:1297–1307. doi:10.4049/jimmunol.1600009
126. Salim T, Sershen CL, May EE. Investigating the role of TNF-α and IFN-γ activation on the dynamics of iNOS gene expression in LPS stimulated macrophages. PLoS One. 2016;11:e0153289. doi:10.1371/journal.pone.0153289
127. Wesemann DR, Benveniste EN. STAT-1α and IFN-γ as modulators of TNF-α signaling in macrophages: regulation and functional implications of the TNF receptor 1: STAT-1α complex. J Immunol. 2003;171:5313–5319. doi:10.4049/jimmunol.171.10.5313
128. Rauch I, Müller M, Decker T. The regulation of inflammation by interferons and their STATs. Jak-Stat. 2013;2:e23820. doi:10.4161/jkst.23820
129. Ushach I, Zlotnik A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. J Leukoc Biol. 2016;100:481–489. doi:10.1189/jlb.3RU0316-144R
130. Hamilton TA, Zhao C, Pavicic PG, Datta S. Myeloid colony-stimulating factors as regulators of macrophage polarization. Front Immunol. 2014;5:1–7. doi:10.3389/fimmu.2014.00554
131. Lombardo E, Alvarez-Barrientos A, Maroto B, Boscá L, Knaus UG. TLR4-mediated survival of macrophages is MyD88 dependent and requires TNF-α autocrine signalling. J Immunol. 2007;178:3731–3739. doi:10.4049/jimmunol.178.6.3731
132. Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem. 1999;274:10689–10692. doi:10.1074/jbc.274.16.10689
133. He X, Qian Y, Li Z, et al. TLR4-upregulated IL-1β and IL-1RI promote alveolar macrophage pyroptosis and lung inflammation through an autocrine mechanism. Sci Rep. 2016;6:1–11.
134. Porta C, Riboldi E, Ippolito A, Sica A. Molecular and epigenetic basis of macrophage polarized activation. Semin Immunol. 2015;27:237–248. doi:10.1016/j.smim.2015.10.003
135. Galván-Peña S, O’Neill LAJ. Metabolic reprograming in macrophage polarization. Front Immunol. 2014;5:1–7.
136. Erbel C, Tyka M, Helmes CM, et al. CXCL4-induced plaque macrophages can be specifically identified by co-expression of MMP7+S100A8+ in vitro and in vivo. Innate Immun. 2015;21:255–265. doi:10.1177/1753425914526461
137. Oksala NKJ, Seppälä I, Rahikainen R, et al. Synergistic expression of histone deacetylase 9 and matrix metalloproteinase 12 in M4 macrophages in advanced carotid plaques. Eur J Vasc Endovasc Surg. 2017;53:632–640. doi:10.1016/j.ejvs.2017.02.014
138. Leitinger N, Schulman IG. Phenotypic polarization of macrophages in atherosclerosis. Arterioscler Thromb Vasc Biol. 2013;33:1120–1126. doi:10.1161/ATVBAHA.112.300173
139. Liberale L, Dallegri F, Montecucco F, Carbone F. Pathophysiological relevance of macrophage subsets in atherogenesis. Thromb Haemost. 2017;117:7–18. doi:10.1160/TH16-08-0593
140. Chinetti-Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol. 2015;12:10–17. doi:10.1038/nrcardio.2014.173
141. Taghavie-Moghadam PL, Butcher MJ, Galkina EV. The dynamic lives of macrophage and dendritic cell subsets in atherosclerosis. Ann N Y Acad Sci. 2014;1319:19–37. doi:10.1111/nyas.2014.1319.issue-1
142. Getz GS, Reardon CA. Atherogenic lipids and macrophage subsets. Curr Opin Lipidol. 2015;26:357–361. doi:10.1097/MOL.0000000000000217
143. Taleb S. L’inflammation dans l’athérosclérose. Arch Cardiovasc Dis. 2016;109:708–715. doi:10.1016/j.acvd.2016.04.002
144. Guler R, Parihar SP, Savvi S, et al. IL-4Rα-dependent alternative activation of macrophages is not decisive for mycobacterium tuberculosis pathology and bacterial burden in mice. PLoS One. 2015;10:1–14. doi:10.1371/journal.pone.0121070
145. Mandell MA, Beverley SM. Continual renewal and replication of persistent Leishmania major parasites in concomitantly immune hosts. Proc Natl Acad Sci. 2017;114:E801–E810. doi:10.1073/pnas.1619265114
146. Cuervo H, Pineda MA, Aoki MP, Gea S, Fresno M, Gironès N. Inducible nitric oxide synthase and arginase expression in heart tissue during acute Trypanosoma cruzi infection in mice: arginase I is expressed in infiltrating CD68+ macrophages. J Infect Dis. 2008;197:1772–1782. doi:10.1086/590464
147. Rath M, Müller I, Kropf P, Closs EI, Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol. 2014;5:532. doi:10.3389/fimmu.2014.00532
148. Zhou D, Huang C, Lin Z, et al. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal. 2014;26:192–197. doi:10.1016/j.cellsig.2013.11.004
149. Duque-Correa MA, Kühl AA, Rodriguez PC, et al. Macrophage arginase-1 controls bacterial growth and pathology in hypoxic tuberculosis granulomas. Proc Natl Acad Sci U S A. 2014;111:E4024–32. doi:10.1073/pnas.1408839111
150. Muraille E, Leo O, Moser M. Th1/Th2 paradigm extended: macrophage polarization as an unappreciated pathogen-driven escape mechanism? Front Immunol. 2014;5:1–12. doi:10.3389/fimmu.2014.00603
151. Murray PJ, Wynn TA. Obstacles and opportunities for understanding macrophage polarization. J Leukoc Biol. 2011;89:557–563. doi:10.1189/jlb.0710409
152. de Sousa JR, de Sousa RPM, de Souza Aarão TL, Dias LB, Oliveira Carneiro FR, Simões Quaresma JA. Response of iNOS and its relationship with IL-22 and STAT3 in macrophage activity in the polar forms of leprosy. Acta Trop. 2017;171:74–79. doi:10.1016/j.actatropica.2017.03.016
153. Pereira-Suárez AL, Alvarado-Navarro A, Barrietos-García JG, Estrada-Chávez C, Muñoz-Valle JF, Fafutis-Morris M. Differential expression of solute carrier family 11a member 1 and inducible nitric oxide synthase 2 in skin biopsies from leprosy patients. Indian J Dermatol Venereol Leprol. 2015;81:594–599. doi:10.4103/0378-6323.168345
154. de Sousa JR, de Sousa RPM, de Souza Aarão TL, et al. In situ expression of M2 macrophage subpopulation in leprosy skin lesions. Acta Trop. 2016;157:108–114. doi:10.1016/j.actatropica.2016.01.008
155. Davis AS, Vergne I, Master SS, Kyei GB, Chua J, Deretic V. Mechanism of inducible nitric oxide synthase exclusion from mycobacterial phagosomes. PLoS Pathog. 2007;3:1887–1894. doi:10.1371/journal.ppat.0030186
156. Cunningham-Bussel A, Zhang T, Nathan CF. Nitrite produced by mycobacterium tuberculosis in human macrophages in physiologic oxygen impacts bacterial ATP consumption and gene expression. Proc Natl Acad Sci. 2013;110:E4256–E4265. doi:10.1073/pnas.1316894110
157. Braverman J, Stanley SA. Nitric oxide modulates macrophage responses to mycobacterium tuberculosis infection through activation of HIF-1α and repression of NF-κB. J Immunol. 2017;199:1805–1816. doi:10.4049/jimmunol.1700515
158. Mattila JT, Ojo OO, Kepka-Lenhart D, et al. Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. J Immunol. 2013;191:773–784. doi:10.4049/jimmunol.1300113
159. Osorio EY, Travi BL, Da Cruz AM, Saldarriaga OA, Medina AA, Melby PC. Growth factor and Th2 cytokine signaling pathways converge at STAT6 to promote arginase expression in progressive experimental visceral leishmaniasis. PLoS Pathog. 2014;10:11–14. doi:10.1371/journal.ppat.1004165
160. Muleme HM, Reguera RM, Berard A, et al. Infection with arginase-deficient leishmania major reveals a parasite number-dependent and cytokine-independent regulation of host cellular arginase activity and disease pathogenesis. J Immunol. 2009;183:8068–8076. doi:10.4049/jimmunol.0900838
161. Schleicher U, Paduch K, Debus A, et al. TNF-mediated restriction of arginase 1 expression in myeloid cells triggers type 2 NO synthase activity at the site of infection. Cell Rep. 2016;15:1062–1075. doi:10.1016/j.celrep.2016.04.001
162. Bernard Q, Smith AA, Yang X, et al. Plasticity in early immune evasion strategies of a bacterial pathogen. Proc Natl Acad Sci U S A. 2018;115:E3788–E3797. doi:10.1073/pnas.1718595115
163. Ren Y, Khan FA, Pandupuspitasari NS, Zhang S. Immune evasion strategies of pathogens in macrophages: the potential for limiting pathogen transmission. Curr Issues Mol Biol. 2017;21:21–40.
164. Yang C, Wang J, Zou L. Innate immune evasion strategies against cryptococcal meningitis caused by Cryptococcus neoformans. Exp Ther Med. 2017;14:5243–5250. doi:10.3892/etm.2017.5220
165. Flávia Nardy A, Freire-De-Lima CG, Morrot A. Immune evasion strategies of trypanosoma cruzi. J Immunol Res. 2015;2015:178947. doi:10.1155/2015/178947
166. Vijay K. Toll-like receptors in immunity and inflammatory diseases: past, present, and future. Int Immunopharmacol. 2018;59:391–412. doi:10.1016/j.intimp.2018.03.002
167. Fieber C, Janos M, Koestler T, et al. Innate immune response to streptococcus pyogenes depends on the combined activation of TLR13 and TLR2. PLoS One. 2015;10:1–20. doi:10.1371/journal.pone.0119727
168. Bordon Y. Innate immunity: TLR13, unlucky, but just for some. Nat Rev Immunol. 2012;12:618–619. doi:10.1038/nri3284
169. Maglione PJ, Simchoni N, Cunningham-Rundles C. Toll-like receptor signaling in primary immune deficiencies. Ann N Y Acad Sci. 2015;1356:1–21. doi:10.1111/nyas.12763
170. Chandler CE, Ernst RK. lipids: powerful modifiers of the innate immune response. F1000Research. 2017;6:1334. doi:10.12688/f1000research.10493.2
171. Kawai T, Akira S. Signaling to NF-κB by Toll-like receptors. Trends Mol Med. 2007;13:460–469. doi:10.1016/j.molmed.2007.09.002
172. Liu S, Jia H, Hou S, et al. Recombinant Mtb9.8 of mycobacterium bovis stimulates TNF-α and IL-1β secretion by RAW264.7 macrophages through activation of NF-κB pathway via TLR2. Sci Rep. 2018;8:1928. doi:10.1038/s41598-018-20433-x
173. Sharma S, Sharma M, Roy S, Kumar P, Bose M. Mycobacterium tuberculosis induces high production of nitric oxide in coordination with production of tumour necrosis factor-α in patients with fresh active tuberculosis but not in MDR tuberculosis. Immunol Cell Biol. 2004;82:377–382. doi:10.1111/j.0818-9641.2004.01245.x
174. Seghatoleslam A, Hemmati M, Ebadat S, Movahedi B, Mostafavi-Pour Z. Macrophage immune response suppression by recombinant mycobacterium tuberculosis antigens, the ESAT-6, CFP-10, and ESAT-6/CFP-10 fusion proteins. Iran J Med Sci. 2016;41:296–304.
175. Blanc L, Gilleron M, Prandi J, et al. Mycobacterium tuberculosis inhibits human innate immune responses via the production of TLR2 antagonist glycolipids. Proc Natl Acad Sci. 2017;114:11205–11210. doi:10.1073/pnas.1707840114
176. Rahman MA, Sobia P, Gupta N, Van Kaer L, Das G. Mycobacterium tuberculosis subverts the TLR-2 - MyD88 pathway to facilitate its translocation into the cytosol. PLoS One. 2014;9:e0086886. doi:10.1371/journal.pone.0086886
177. Su H, Zhu S, Zhu L, et al. Recombinant lipoprotein Rv1016c derived from mycobacterium tuberculosis is a TLR-2 ligand that induces macrophages apoptosis and inhibits MHC II antigen processing. Front Cell Infect Microbiol. 2016;6:1–13. doi:10.3389/fcimb.2016.00147
178. de Veer MJ, Curtis JM, Baldwin TM, et al. MyD88 is essential for clearance OF Leishmania major: possible role for lipophosphoglycan and Toll-like receptor 2 signaling. Eur J Immunol. 2003;33:2822–2831. doi:10.1002/(ISSN)1521-4141
179. Srivastav S, Kar S, Chande AG, Mukhopadhyaya R, Das PK. Leishmania donovani exploits host deubiquitinating enzyme A20, a negative regulator of TLR signaling, to subvert host immune response. J Immunol. 2012;189:924–934. doi:10.4049/jimmunol.1102845
180. Srivastav S, Ball WB, Gupta P, Giri J, Ukil A, Das PK. Leishmania donovani prevents oxidative burst-mediated apoptosis of host macrophages through selective induction of suppressors of cytokine signaling SOCSproteins. J Biol Chem. 2014;289:1092–1105. doi:10.1074/jbc.M113.496323
181. Khouri R, Bafica A, Silva MDPP, et al. IFN-beta impairs superoxide-dependent parasite killing in human macrophages: evidence for a deleterious role of SOD1 in cutaneous leishmaniasis. J Immunol. 2009;182:2525–2531. doi:10.4049/jimmunol.0802775
182. Gravina HD, Antonelli L, Gazzinelli RT, Ropert C. Differential use of TLR2 and TLR9 in the regulation of immune responses during the infection with trypanosoma cruzi. PLoS One. 2013;8:e63100. doi:10.1371/journal.pone.0063100
183. Cardoso MS, Reis-Cunha JL, Bartholomeu DC. Evasion of the immune response by trypanosoma cruzi during acute infection. Front Immunol. 2016;6:1–15. doi:10.3389/fimmu.2015.00659
184. Welin A, Raffetseder J, Eklund D, Stendahl O, Lerm M. Importance of phagosomal functionality for growth restriction of mycobacterium tuberculosis in primary human macrophages. J Innate Immun. 2011;3:508–518. doi:10.1159/000325297
185. Semini G, Aebischer T. Phagosome proteomics to study Leishmania’s intracellular niche in macrophages. Int J Med Microbiol. 2017;pii:S1438-4221(17)30332–6. doi:10.1016/j.ijmm.2017.09.003
186. Pieters J. Mycobacterium tuberculosis and the macrophage: maintaining a balance. Cell Host Microbe. 2008;3:399–407. doi:10.1016/j.chom.2008.05.006
187. Lodge R, Diallo TO, Descoteaux A. Leishmania donovani lipophosphoglycan blocks NADPH oxidase assembly at the phagosome membrane. Cell Microbiol. 2006;8:1922–1931. doi:10.1111/cmi.2006.8.issue-12
188. Osuna A, Gamarro F, Castanys S, Ruiz-Perez LM. Inhibition of lysosomal fusion by trypanosoma cruzi in peritoneal macrophages. Int J Parasitol. 1986;16:629–632. doi:10.1016/0020-7519(86)90031-7
189. Wilkowsky SE, Isola ELD, Barbieri MA, Stahl PD. Regulation of trypanosoma cruzi invasion of nonphagocytic cells by the endocytically active GTPases dynamin, Rab5, and Rab7. Biochem Biophys Res Commun. 2002;291:516–521. doi:10.1006/bbrc.2002.6437
190. Mottola G. The complexity of Rab5 to Rab7 transition guarantees specificity of pathogen subversion mechanisms. Front Cell Infect Microbiol. 2014;4:5–8. doi:10.3389/fcimb.2014.00180
191. Singh SB, Tandon R, Krishnamurthy G, Vikram R, Sharma N, Basu SK. Rab5-mediated endosome-endosome fusion regulates hemoglobin endocytosis in leishmania donovani. Embo J. 2003;22:5712–5722. doi:10.1093/emboj/cdg557
192. Jayachandran R, Sundaramurthy V, Combaluzier B, et al. Survival of mycobacteria in macrophages is mediated by coronin 1-dependent activation of calcineurin. Cell. 2007;130:37–50. doi:10.1016/j.cell.2007.04.043
193. Gogulamudi VR, Dubey ML, Kaul D, Atluri VSR, Sehgal R. Downregulation of host tryptophan–aspartate containing coat (TACO) gene restricts the entry and survival of Leishmania donovani in human macrophage model. Front Microbiol. 2015;6:1–10. doi:10.3389/fmicb.2015.00946
194. Maganto-Garcia E, Punzon C, Terhorst C, Fresno M. Rab5 activation by toll-like receptor 2 is required for trypanosoma cruzi internalization and replication in macrophages. Traffic. 2008;9:1299–1315. doi:10.1111/tra.2008.9.issue-8
195. Pires D, Marques J, Pombo JP, et al. Role of cathepsins in mycobacterium tuberculosis survival in human macrophages. Sci Rep. 2016;6:1–13. doi:10.1038/srep32247
196. Banerjee S, Bose D, Chatterjee N, et al. Attenuated Leishmania induce pro-inflammatory mediators and influence leishmanicidal activity by p38 MAPK dependent phagosome maturation in Leishmania donovani co-infected macrophages. Sci Rep. 2016;6:1–14. doi:10.1038/srep22335
197. Borges EC, Machado EMM, Garcia ES, Azambuja P. Trypanosoma cruzi: effects of infection on cathepsin D activity in the midgut of rhodnius prolixus. Exp Parasitol. 2006;112:130–133. doi:10.1016/j.exppara.2005.09.008
198. Chen S, Wu Z, Wang M, Cheng A. Innate immune evasion mediated by flaviviridae non-structural proteins. Viruses. 2017;9:1–19. doi:10.3390/v9100291
199. Ye J, Zhu B, Fu ZF, Chen H, Cao S. Immune evasion strategies of flaviviruses. Vaccine. 2013;31:461–471. doi:10.1016/j.vaccine.2012.11.015
200. Ashhurst TM, Vreden C, Munoz-Erazo L, et al. Antiviral macrophage responses in flavivirus encephalitis. Indian J Med Res. 2013;138:632–647.
201. Errett JS, Suthar MS, McMillan A, Diamond MS, Gale M. The essential, nonredundant roles of RIG-I and MDA5 in detecting and controlling West Nile virus infection. J Virol. 2013;87:11416–11425. doi:10.1128/JVI.01488-13
202. Fan C, Zhang Y, Zhou Y, et al. Up-regulation of A20/ABIN1 contributes to inefficient M1 macrophage polarization during Hepatitis C virus infection Hepatitis viruses. Virol J. 2015;12:1–10. doi:10.1186/s12985-015-0379-0
203. Carlin AF, Vizcarra EA, Branche E, et al. Deconvolution of pro- and antiviral genomic responses in zika virus-infected and bystander macrophages. Proc Natl Acad Sci U S A. 2018;115:E9172–E9181. doi:10.1073/pnas.1807690115
204. Sun J, Desai M, Fang R. The role of autophagy in microbial infection and immunity. ImmunoTargets Ther. 2015;4:13–26. doi:10.2147/ITT.S76720
205. Moraco AH, Kornfeld H. Cell death and autophagy in tuberculosis. Semin Immunol. 2014;26:497–511. doi:10.1016/j.smim.2014.10.001
206. Gong L, Devenish RJ, Prescott M. Autophagy as a macrophage response to bacterial infection. IUBMB Life. 2012;64:740–747. doi:10.1002/iub.1095
207. Crauwels P, Bohn R, Thomas M, et al. Apoptotic-like Leishmania exploit the host´s autophagy machinery to reduce T-cell-mediated parasite elimination. Autophagy. 2015;11:285–297. doi:10.1080/15548627.2014.998904
208. Romano PS, Arboit MA, Vázquez CL, Colombo MI. The autophagic pathway is a key component in the lysosomal dependent entry of Trypanosoma cruzi into the host cell. Autophagy. 2009;5:6–18. doi:10.4161/auto.5.1.7160
209. Romano PS, Cueto JA, Casassa AF, Vanrell MC, Gottlieb RA, Colombo MI. Molecular and cellular mechanisms involved in the Trypanosoma cruzi/host cell interplay. IUBMB Life. 2012;64:387–396. doi:10.1002/iub.1095
210. Dutta RK, Kathania M, Raje M, Majumdar S. IL-6 inhibits IFN-γ induced autophagy in Mycobacterium tuberculosis H37Rv infected macrophages. Int J Biochem Cell Biol. 2012;44:942–954. doi:10.1016/j.biocel.2012.02.021
211. Sharma G, Dutta RK, Khan MA, et al. IL-27 inhibits IFN-γ induced autophagy by concomitant induction of JAK/PI3 K/Akt/mTOR cascade and up-regulation of Mcl-1 in mycobacterium tuberculosis H37Rv infected macrophages. Int J Biochem Cell Biol. 2014;55:335–347. doi:10.1016/j.biocel.2014.08.022
212. Blázquez AB, Escribano-Romero E, Merino-Ramos T, Saiz JC, Martín- Acebes MA. Stress responses in flavivirus-infected cells: activation of unfolded protein response and autophagy. Front Microbiol. 2014;5:1–7. doi:10.3389/fmicb.2014.00547
213. Vicenzi E, Pagani I, Ghezzi S, et al. Subverting the mechanisms of cell death: flavivirus manipulation of host cell responses to infection. Biochem Soc Trans. 2018;46:609–617. doi:10.1042/BST20170399
214. Ghosh Roy S, Sadigh B, Datan E, Lockshin RA, Zakeri Z. Regulation of cell survival and death during Flavivirus infections. World J Biol Chem. 2014;5:93–105.
215. McLean JE, Wudzinska A, Datan E, Quaglino D, Zakeri Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J Biol Chem. 2011;286:22147–22159. doi:10.1074/jbc.M110.192500
216. Datan E, Roy SG, Germain G, et al. Dengue-induced autophagy, virus replication and protection from cell death require ER stress (PERK) pathway activation. Cell Death Dis. 2016;7:e2127. doi:10.1038/cddis.2015.409
217. Lennemann NJ, Coyne CB. Dengue and Zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy. 2017;13:322–332. doi:10.1080/15548627.2016.1265192
218. Ma X, Parson C, Ding W-X. Regulation of the homeostasis of hepatic endoplasmic reticulum and cytochrome P450 enzymes by autophagy. Liver Res. 2018;42:645–648.
219. Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol. 2017;198:1006–1014. doi:10.4049/jimmunol.1601515
220. Pereira-Lopes S, Tur J, Calatayud-Subias JA, Lloberas J, Stracker TH, Celada A. NBS1 is required for macrophage homeostasis and functional activity in mice. Blood. 2015;126:2502–2510. doi:10.1182/blood-2015-04-637371
221. Gundra UM, Girgis NM, Ruckerl D, et al. Alternatively activated macrophages derived from monocytes and tissue macrophages are phenotypically and functionally distinct. Blood. 2014;123:e110–e122. doi:10.1182/blood-2013-08-520619
222. Krishnasamy K, Limbourg A, Kapanadze T, et al. Blood vessel control of macrophage maturation promotes arteriogenesis in ischemia. Nat Commun. 2017;8:952. doi:10.1038/s41467-017-00953-2
223. Stremmel C, Schuchert R, Wagner F, et al. Yolk sac macrophage progenitors traffic to the embryo during defined stages of development. Nat Commun. 2018;9:75. doi:10.1038/s41467-017-02492-2
224. Yoon KW. Dead cell phagocytosis and innate immune checkpoint. BMB Rep. 2017;50:496–503. doi:10.5483/BMBRep.2017.50.10.147
225. Bosurgi L, Cao YGG, Cabeza-Cabrerizo M, et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science. 2017;356:1072–1076. doi:10.1126/science.aai8132
226. Liang J, Jung Y, Tighe RM, et al. A macrophage subpopulation recruited by CC chemokine ligand-2 clears apoptotic cells in noninfectious lung injury. Am J Physiol Cell Mol Physiol. 2012;302:L933–L940. doi:10.1152/ajplung.00256.2011
227. A-Gonzalez N, Quintana JA, García-Silva S, et al. Phagocytosis imprints heterogeneity in tissue-resident macrophages. J Exp Med. 2017;214:1281–1296. doi:10.1084/jem.20161375
228. Mora-Bau G, Platt AM, van Rooijen N, Randolph GJ, Albert ML, Ingersoll MA. Macrophages subvert adaptive immunity to urinary tract infection. PLoS Pathog. 2015;11:1–23. doi:10.1371/journal.ppat.1005044
229. Zhao Y, Zou W, Du J, Zhao Y. The origins and homeostasis of monocytes and tissue-resident macrophages in physiological situation. J Cell Physiol. 2018;233:6425–6439. doi:10.1002/jcp.v233.10
230. Vogel DYS, Vereyken EJF, Glim JE, et al. Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J Neuroinflammation. 2013;10:1–12. doi:10.1186/1742-2094-10-151
231. Edholm E-S, Rhoo KH, Robert J. Evolutionary aspects of macrophages polarization. Results Probl Cell Differ. 2017;62:3–22. doi:10.1007/978-3-319-54090-0_1
232. Laskin DL, Sunil VR, Gardner CR, Laskin JD. Macrophages and tissue injury: agents of defense or destruction? Annu Rev Pharmacol Toxicol. 2011;51:267–288. doi:10.1146/annurev.pharmtox.010909.105812
233. Hirayama D, Iida T, Nakase H. The phagocytic function of macrophage-enforcing innate immunity and tissue homeostasis. Int J Mol Sci. 2018;19:92. doi:10.3390/ijms19010092
234. Sindrilaru A, Peters T, Wieschalka S, et al. An unrestrained proinflammatory M1 mac population induced by iron imparis wound healing in mice and humans. J Clin Invest. 2011;121:985–997. doi:10.1172/JCI57873
235. Elsegeiny W, Marr KA, Williamson PR. Immunology of cryptococcal infections: developing a rational approach to patient therapy. Front Immunol. 2018;9:651. doi:10.3389/fimmu.2018.00651
236. Rusek P, Wala M, Druszczyńska M, Fol M. Infectious agents as stimuli of trained innate immunity. Int J Mol Sci. 2018;19:E456. doi:10.3390/ijms19020456
237. Couturier J, Lewis DE. HIV persistence in adipose tissue reservoirs. Curr HIV/AIDS Rep. 2018;15:60–71. doi:10.1007/s11904-018-0378-z
238. Lannes N, Eppler E, Etemad S, Yotovski P, Filgueira L. Microglia at center stage: a comprehensive review about the versatile and unique residential macrophages of the central nervous system. Oncotarget. 2017;8:114393–114413. doi:10.18632/oncotarget.23106
239. Teng O, Ang CKE, Guan XL. Macrophage-bacteria interactions-a lipid-centric relationship. Front Immunol. 2017;8:1836. doi:10.3389/fimmu.2017.01836
240. Soulat D, Bogdan C. Function of macrophage and parasite phosphatases in Leishmaniasis. Front Immunol. 2017;8:1838. doi:10.3389/fimmu.2017.01838
241. Di Pietro M, Filardo S, Falasca F, Turriziani O, Sessa R. Infectious agents in atherosclerotic cardiovascular diseases through oxidative stress. Int J Mol Sci. 2017;18:E2459. doi:10.3390/ijms18112459
242. Fedele G, Schiavoni I, Adkins I, Klimova N, Sebo P. Invasion of dendritic cells, macrophages and neutrophils by the Bordetella adenylate cyclase toxin: a subversive move to fool host immunity. Toxins. 2017;9:E293. doi:10.3390/toxins9100293
243. Terziroli Beretta-Piccoli B, Mainetti C, Peeters M-A, Laffitte E. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131–146. doi:10.1007/s12016-017-8666-8
244. Stocks CJ, Schembri MA, Sweet MJ, Kapetanovic R. For when bacterial infections persist: toll-like receptor-inducible direct antimicrobial pathways in macrophages. J Leukoc Biol. 2018;103:35–51. doi:10.1002/JLB.4RI0917-358R
245. Rathnaiah G, Zinniel DK, Bannantine JP, et al. Pathogenesis, molecular genetics, and genomics of Mycobacterium avium subsp., the etiologic agent of Johne’s disease. Front Vet Sci. 2017;4:187. doi:10.3389/fvets.2017.00127
246. Dalli J, Serhan CN. Pro-resolving mediators in regulating and conferring macrophage function. Front Immunol. 2017;8:1400. doi:10.3389/fimmu.2017.01400
247. Muraille E, Leo O, Moser M. TH1/TH2 paradigm extended: macrophage polarization as an unappreciated pathogen-driven escape mechanism? Front Immunol. 2014;5:603. doi:10.3389/fimmu.2014.00603
248. Ishii K, Kawakami K. Up-to-date findings in the host defence mechanism to cryptococcus infection. Med Mycol J. 2014;55:J107–J114.
249. Stijlemans B, Guilliams M, Raes G, Beschin A, Magez S, De Baetselier P. African trypanosomosis: from immune escape and immunopathology to immune intervention. Vet Parasitol. 2007;148:3–13. doi:10.1016/j.vetpar.2007.05.005
250. Yamamoto T. Bacterial strategies for escaping the bactericidal mechanisms by macrophage. Yakugaku Zasshi. 2006;126:1235–1243. doi:10.1248/yakushi.126.1235
251. Mitsuyama M. Escape mechanism of intracellular parasitic bacteria and prospect of new approach to infection control. Nihon Rinsho. 2001;59:1013–1021.
252. Bremermann HJ. Mechanism of HIV persistence: implications for vaccines and therapy. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;9:459–483.
253. Sica A, Erreni M, Allavena P, Porta C. Macrophage polarization in pathology. Cell Mol Life Sci. 2015;72:4111–4126. doi:10.1007/s00018-015-1995-y
254. Patel U, Rajasingh S, Samanta S, Cao T, Dawn B, Rajasingh J. Macrophage polarization in response to epigenetic modifiers during infection and inflammation. Drug Discov Today. 2017;22:186–193. doi:10.1016/j.drudis.2016.08.006
255. Mills CD. Anatomy of a discovery: m1 and m2 macrophages. Front Immunol. 2015;6:212. doi:10.3389/fimmu.2015.00212
256. Mills CD. M1 and M2 macrophages: oracles of health and disease. Crit Ver Immunol. 2012;32:463–488. doi:10.1615/CritRevImmunol.v32.i6.10
257. Tomioka H, Tatano Y, Maw WW, Sano C, Kanehiro Y, Shimizu T. Characteristics of suppressor macrophages induced by mycobacterial and protozoal infections in relation to alternatively activated M2 macrophages. Clin Dev Immunol. 2012;2012:1–19. doi:10.1155/2012/635451
258. Mège JL, Mehraj V, Capo C. Macrophage polarization and bacterial infections. Curr Opin Infect Dis. 2011;24:230–234. doi:10.1097/QCO.0b013e328344b73e
259. Luo F, Sun X, Qu Z, Zhang X. Salmonella typhimurium-induced M1 macrophage polarization is dependent on the bacterial O antigen. World J Microbiol Biotechnol. 2016;32:1–7. doi:10.1007/s11274-015-1978-z
260. Quiding-Järbrink M, Raghavan S, Sundquist M. Enhanced M1 macrophage polarization in human helicobacter pylori-associated atrophic gastritis and in vaccinated mice. PLoS One. 2010;5:e15018. doi:10.1371/journal.pone.0015018
261. Fehlings M, Drobbe L, Moos V, et al. Comparative analysis of the interaction of Helicobacter pylori with human dendritic cells, macrophages, and monocytes. Infect Immun. 2012;80:2724–2734. doi:10.1128/IAI.06224-11
262. Gobert AP, Verriere T, Asim M, et al. Heme oxygenase-1 dysregulates macrophage polarization and the immune response to Helicobacter pylori. J Immunol. 2014;193:3013–3022. doi:10.4049/jimmunol.1401075
263. Moyat M, Velin D. Immune responses to Helicobacter pylori infection. World J Gastroenterol. 2014;20:5583–5593. doi:10.3748/wjg.v20.i19.5583
264. Borlace GN, Jones HF, Keep SJ, Butler RN, Brooks DA. Helicobacter pylori phagosome maturation in primary human macrophages. Gut Pathog. 2011;3:3–14. doi:10.1186/1757-4749-3-3
265. Huang Z, Luo Q, Guo Y, et al. Mycobacterium tuberculosis-induced polarization of human macrophage orchestrates the formation and development of tuberculous granulomas in vitro. PLoS One. 2015;10:e0129744. doi:10.1371/journal.pone.0129744
266. Guirado E, Schlesinger LS, Kaplan G. Macrophages in tuberculosis: friend or foe. Semin Immunopathol. 2013;35:563–583. doi:10.1007/s00281-013-0388-2
267. Roy S, Schmeier S, Kaczkowski B, et al. Transcriptional landscape of Mycobacterium tuberculosis infection in macrophages. Sci Rep. 2018;8:1–13. doi:10.1038/s41598-018-24509-6
268. Huang L, Nazarova EV, Tan S, Liu Y, Russell DG. Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J Exp Med. 2018;215:1135–1152. doi:10.1084/jem.20172020
269. Lerner TR, Borel S, Greenwood DJ, et al. Mycobacterium tuberculosis replicates within necrotic human macrophages. J Cell Biol. 2017;216:583–594. doi:10.1083/jcb.201603040
270. Fallows D, Peixoto B, Kaplan G, Manca C. Mycobacterium leprae alters classical activation of human monocytes in vitro. J Inflamm. 2016;13:4–8. doi:10.1186/s12950-016-0117-4
271. Yang D, Shui T, Miranda JW, et al. Mycobacterium leprae-infected macrophages preferentially primed regulatory T cell responses and was associated with lepromatous leprosy. PLoS Negl Trop Dis. 2016;10:1–13. doi:10.1371/journal.pntd.0004335
272. de Sousa JR, Sotto MN, Quaresma JAS. Leprosy as a complex infection: breakdown of the Th1 and Th2 immune paradigm in the immunopathogenesis of the disease. Front Immunol. 2017;8:18–21. doi:10.3389/fimmu.2017.01635
273. Lockwood DNJ, Suneetha L, De Sagili K, et al. Cytokine and protein markers of leprosy reactions in skin and nerves: baseline results for the north indian infir cohort. PLoS Negl Trop Dis. 2011;5:e1327. doi:10.1371/journal.pntd.0001370
274. Silva CAM, Belisle JT. Host lipid mediators in leprosy: the hypothesized contributions to pathogenesis. Front Immunol. 2018;9:134. doi:10.3389/fimmu.2018.00134
275. Aarão TL, de Sousa JR, Falcão ASC, Falcão LFM, Quaresma JAS. Nerve growth factor and pathogenesis of leprosy: review and update. Front Immunol. 2018;9:1–8. doi:10.3389/fimmu.2018.00939
276. Mukhopadhyay D, Mukherjee S, Roy S, et al. M2 polarization of monocytes-macrophages is a hallmark of Indian post kala-azar dermal leishmaniasis. PLoS Negl Trop Dis. 2015;9:1–19. doi:10.1371/journal.pntd.0004145
277. Lee SH, Charmoy M, Romano A, et al. Mannose receptor high, M2 dermal macrophages mediate nonhealing Leishmania major infection in a Th1 immune environment. J Exp Med. 2018;215:357–375. doi:10.1084/jem.20171389
278. Sousa JR, Dias F, Neto L, Sotto MN, Antonio J, Quaresma S. Immunohistochemical characterization of the M4 macrophage population in leprosy skin lesions. BMC Infect Dis. 2018;18:576. doi:10.1186/s12879-018-3109-6
279. Saha B, Szabo G. Innate immune cell networking in hepatitis C virus infection. J Leukoc Biol. 2014;96:757–766. doi:10.1189/jlb.4MR0314-141R
280. Revie D, Salahuddin SZ. Role of macrophages and monocytes in hepatitis C virus infections. World J Gastroenterol. 2014;20:2777–2784. doi:10.3748/wjg.v20.i11.2777
281. Cline TD, Beck D, Bianchini E. Influenza virus replication in macrophages: balancing protection and pathogenesis. J Gen Virol. 2017;98:2401–2412. doi:10.1099/jgv.0.000922
282. Duan M, Hibbs ML, Chen W. The contributions of lung macrophage and monocyte heterogeneity to influenza pathogenesis. Immunol Cell Biol. 2017;95:225–235. doi:10.1038/icb.2016.97
283. Newton AH, Cardani A, Braciale TJ. The host immune response in respiratory virus infection: balancing virus clearance and immunopathology. Semin Immunopathol. 2016;38:471–482. doi:10.1007/s00281-016-0558-0
284. Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. 2017;39:529–539. doi:10.1007/s00281-017-0629-x
285. Frieman M, Heise M, Baric R. SARS coronavirus and innate immunity. Virus Res. 2008;133:101–112. doi:10.1016/j.virusres.2007.03.015
286. Perlman S, Dandekar AA. Immunopathogenesis of coronavirus infections: implications for SARS. Nat Rev Immunol. 2005;5:917–927. doi:10.1038/nri1732
287. Pasquereau S, Al Moussawi F, Karam W, Diab Assaf M, Kumar A, Herbein G. Cytomegalovirus, macrophages and breast cancer. Open Virol J. 2017;11:15–27. doi:10.2174/1874357901711010015
288. Brinkmann MM, Dağ F, Hengel H, Messerle M, Kalinke U, Čičin-Šain L. Cytomegalovirus immune evasion of myeloid lineage cells. Med Microbiol Immunol. 2015;204:367–382. doi:10.1007/s00430-015-0403-4
289. Contreras A, Botero JE, Slots J. Biology and pathogenesis of cytomegalovirus in periodontal disease. Periodontol 2000. 2014;64:40–56. doi:10.1111/j.1600-0757.2012.00448.x
290. Bang B-R, Elmasry S, Saito T. Organ system view of the hepatic innate immunity in HCV infection. J Med Virol. 2016;88:2025–2037. doi:10.1002/jmv.24569
291. Horner SM, Gale M. Regulation of hepatic innate immunity by hepatitis C virus. Nat Med. 2013;19:879–888. doi:10.1038/nm.3253
292. Metzger DW, Sun K. Immune dysfunction and bacterial coinfections following influenza. J Immunol. 2013;191:2047–2052. doi:10.4049/jimmunol.1301152
293. Cauley LS, Vella AT. Why is coinfection with influenza virus and bacteria so difficult to control? Discov Med. 2015;19:33–40.
294. Mulcahy ME, McLoughlin RM. Staphylococcus aureus and influenza a virus: partners in coinfection. MBio. 2016;7:4–7. doi:10.1128/mBio.02068-16
295. Page C, Goicochea L, Matthews K, et al. Induction of alternatively activated macrophages enhances pathogenesis during severe acute respiratory syndrome coronavirus infection. J Virol. 2012;86:13334–13349. doi:10.1128/JVI.01689-12
296. Savarin C, Dutta R, Bergmann CC. Distinct gene profiles of bone marrow-derived macrophages and microglia during neurotropic coronavirus-induced demyelination. Front Immunol. 2018;9:1325. doi:10.3389/fimmu.2018.01325
297. Byrne AJ, Mathie SA, Gregory LG, Lloyd CM. Pulmonary macrophages: key players in the innate defence of the airways. Thorax. 2015;70:1189–1196. doi:10.1136/thoraxjnl-2015-207020
298. Bayer C, Varani S, Wang L, et al. Human cytomegalovirus infection of M1 and M2 macrophages triggers inflammation and autologous T-cell proliferation. J Virol. 2013;87:67–79. doi:10.1128/JVI.01585-12
299. Cousins SW, Espinosa-Heidmann DG, Miller DM, et al. Macrophage activation associated with chronic murine cytomegalovirus infection results in more severe experimental choroidal neovascularization. PLoS Pathog. 2012;8:e1002671. doi:10.1371/journal.ppat.1002671
300. Zheng S, Zhang P, Chen Y, Zheng S, Zheng L, Weng Z. Inhibition of Notch signaling attenuates schistosomiasis hepatic fibrosis via blocking macrophage M2 polarization. PLoS One. 2016;11:e0166808. doi:10.1371/journal.pone.0166808
301. Hussaarts L, García-Tardón N, van Beek L, et al. Chronic helminth infection and helminth-derived egg antigens promote adipose tissue M2 macrophages and improve insulin sensitivity in obese mice. Faseb J. 2015;29:3027–3039. doi:10.1096/fj.14-266239
302. Pinheiro RO, Schmitz V, de Andrade Silva BJ, et al. Innate immune responses in leprosy. Front Immunol. 2018;9:1–15. doi:10.3389/fimmu.2018.00518
303. Lopes MF, Costa-Da-Silva AC, Dosreis GA. Innate immunity to Leishmania infection: within phagocytes. Mediators Inflamm. 2014;2014:1–7. doi:10.1155/2014/754965
304. Kong L, Zhang Q, Chao J, et al. Polarization of macrophages induced by Toxoplasma gondii and its impact on abnormal pregnancy in rats. Acta Trop. 2015;143:1–7. doi:10.1016/j.actatropica.2014.12.001
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.