Back to Journals » Neuropsychiatric Disease and Treatment » Volume 10
Frontotemporal dementia and primary progressive aphasia, a review
Authors Kirshner H
Received 28 November 2013
Accepted for publication 24 February 2014
Published 12 June 2014 Volume 2014:10 Pages 1045—1055
DOI https://doi.org/10.2147/NDT.S38821
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Howard S Kirshner
Department of Neurology, Vanderbilt University Medical Center, Nashville, TN, USA
Abstract: Frontotemporal dementias are neurodegenerative diseases in which symptoms of frontal and/or temporal lobe disease are the first signs of the illness, and as the diseases progress, they resemble a focal left hemisphere process such as stroke or traumatic brain injury, even more than a neurodegenerative disease. Over time, some patients develop a more generalized dementia. Four clinical subtypes characterize the predominant presentations of this illness: behavioral or frontal variant FTD, progressive nonfluent aphasia, semantic dementia, and logopenic primary progressive aphasia. These clinical variants correlate with regional patterns of atrophy on brain imaging studies such as MRI and PET scanning, as well as with biochemical and molecular genetic variants of the disorder. The treatment is as yet only symptomatic, but advances in molecular genetics promise new therapies.
Keywords: FTD, behavior variant or frontal variant FTD, pick's disease, PPA, progressive nonfluent aphasia
Introduction
Frontotemporal dementia (FTD) is a neurodegenerative disease, one of a series of disorders in which specific populations of neurons die off prematurely. Dementias are defined as syndromes of deterioration of cognitive function, sufficient to cause disability or affect activities of daily living. FTD refers to a family of disorders in which both the clinical syndromes and the regional pathology involve populations of neurons in the frontal and/or temporal lobes on one or both sides of the brain. The hallmark feature of FTD is the presentation with “focal” syndromes such as progressive language dysfunction, or aphasia, or behavioral changes characteristic of frontal lobe disorders. These syndromes mimic focal pathological states such as stroke, brain tumor, or traumatic brain injury. This family of focal, cortical disorders historically has been contrasted with the presentation of Alzheimer’s disease (AD), the most common dementing illness, and often considered to be a “generalized” dementia. AD most typically presents with a global short-term memory deficit, but this presentation is variable, and AD also affects specific populations of neurons in the cortical association areas, hippocampi, and medial temporal structures, and also the basal forebrain nuclei, such as the nucleus of Meynert. FTD is less common than AD, but it disproportionately affects younger patients, such that it is a major psychological and economic burden for patients and families still in the prime of life. In patients younger than 60 years, FTD is equal to or greater than AD in incidence.
Historical background
The concept of a “focal” dementia is not new. Arnold Pick, a German physician, published a case report in 1892, describing a focal disorder involving aphasia, as well as psychiatric manifestations.1 In Pick’s words, “simple progressive brain atrophy can lead to symptoms of local disturbance through local accentuation of the diffuse process”. In other words, a neuropathological disorder has to start somewhere in the brain, and then it may spread to other areas and develop into a recognizable dementing illness. This concept of the progression of a neurodegenerative disease from one region of the brain, or one neuron to another, remains central to current thinking on the evolution of dementing diseases in current neuroscience.
From the 1890s until the 1980s, the field advanced principally via case reports of unusual progressive aphasia and frontal neurobehavioral syndromes. In the 1980s, two patterns of publications appeared, describing neurodegenenerative disorders that presented with either aphasia or frontal lobe syndromes. In the US, the emphasis was on progressive language deterioration, or aphasia. Marsel Mesulam2 reported six patients with gradually progressive aphasia without a more generalized dementia. This disorder, called “primary progressive aphasia” (PPA), achieved syndrome status in the US and was supported by numerous other case reports and series. Mesulam’s group later defined PPA as a focal disease process, limited to progressive language deterioration and without general cognitive impairment or dementia, for at least 2 years.3–6 Over time, however, some patients with PPA evolve into a more generalized dementia.7,8 Cases of isolated right frontal or temporal degeneration have also been reported, although they have been less frequently recognized than those with either progressive aphasia or frontal lobe behavioral syndromes.9,10 For example, patients have been reported whose initial symptoms involved inability to recognize family members (prosopagnosia) or failure to remember common routes, others with traits such as religiosity, obsessive–compulsive tendencies, uninhibited behavior, and alterations in eating.9 In addition, some have increased artistic tendencies.9
In the UK and Europe, cases of “frontal lobe dementia” were described, with emphasis on progressive deterioration of frontal lobe functions. Neary and Snowden,11 Neary et al,12 and Snowden et al13 reported a series of disorders presenting with behavioral aberrations resembling a psychiatric illness. Examples of frontal lobe behavioral abnormalities included loss of inhibition, poor impulse control, or on the other hand, lack of initiative and “inertia” or abulia, inability to persist in a task, loss of social awareness, making comments to other people that would normally not be uttered in their presence, neglect of personal hygiene, rigidity of mental attitudes, stereotyped behavior, inappropriate sexual interests, and other “frontal lobe behaviors” such as imitation (imitating the examiner’s moves and gestures) and utilization behavior (the patient picks up and plays with any object in the environment, such as items on the examiner’s desk14). Frontal lobe behaviors also involve language, although the UK investigators emphasized reduced speech output, mutism, echolalia, and perseveration, rather than frank aphasia. Frontal lobe dementia was noted to have a very different neuropathology from the more common changes of AD. Even on gross inspection of the brain, there was atrophy of the frontal and/or temporal lobes unilaterally or bilaterally. On microscopic study, the affected areas of the brain had loss of neurons, gliosis, and vacuolation of the cortical neuropil, a pathology very distinct from the changes of AD, which typically involve neuritic plaques and neurofibrillary tangles.11,13
Investigators on both sides of the Atlantic have recognized that PPA and frontal lobe dementia are aspects of the same disorder, now usually called “frontotemporal dementia”. Some neuropathologists have preferred the term “frontotemporal lobar degeneration”. FTD is now divided into the following two main sub-syndromes: 1) behavioral variant FTD (sometimes called frontal variant FTD, bvFTD or fvFTD), and 2) PPA. Both disorders can be associated with motor neuron disease (MND) (FTD-MND).15 PPA has now been divided into three separate variants, which will be elaborated on later. The term “Pick’s disease” is increasingly used as a neuropathological term for cases in which the neuronal Pick inclusion bodies are found. Kertesz and Munoz16 proposed the term “Pick complex” to include all of the FTD and PPA variants, but this term has not been adopted in the literature.
Clinical subtypes of FTD
BvFTD/fvFTD
In bvFTD, or fvFTD, patients present with behavioral changes involving personality change and alterations in social conduct (Table 1). Patients with bvFTD may become uninhibited, saying inappropriate comments in front of other people, or delighting in a punning, silly type of humor called “witzelsucht”. Other patients have a general reduction in behavior, becoming abulic or apathetic, such that they just sit, speaking little and not interacting with other people or the environment. Patients become inattentive to personal hygiene and grooming, and they are unaware of the effects of their behaviors on others, a phenomenon referred to as loss of the “theory of mind”. Other frontal lobe behavioral changes include hyperorality, utilization and imitation behavior, and inappropriate sexuality. Again, the UK group did report changes in speech, but they emphasized reduced spontaneous speech or muteness, or perseveration, stereotyped utterances in answering questions, or a tendency to repeat vocalizations (echolalia).11–13 Formal diagnostic criteria have been proposed for bvFTD.17 Warren et al18 have suggested that in taking a history from family members, key questions include whether the patient has said or done anything to embarrass other people, whether they seem less affectionate to relatives and pets, whether their food preferences have changed or table manners declined, whether they seem overly concerned with the time, whether they have an altered sense of humor, and whether they have developed new hobbies or interests, especially those involving religious or spiritual investment, or those which are “pursued obsessively”. These changes in the basic personality of the patient are especially distressing for families.
 | Table 1 Variants of frontotemporal dementia and their clinical features |
Since Pick’s original report,1 and throughout the early literature on both frontal lobe dementias and PPA, most reported cases have had a neuropathology other than that of AD.11,13 The bvFTD cases and some of the PPA patients were later discovered to have a primary disturbance of tau proteins, referred to as a “tauopathy”.19 As we shall see, however, recent case studies have made it clear that there is an overlap between AD and FTD presentations. AD can begin with behavior disturbance or executive dysfunction, suggesting a frontal lobe syndrome.20,21 This presentation of AD is now being referred to as the frontal variant of AD.21
Another issue in FTD is the overlap with MND, with fasciculations, muscle atrophy, weakness, and increased tone and reflexes. Some patients develop bulbar symptoms, including dysarthria, dysphagia, and even respiratory failure.15 Another, rare variant of FTD is associated with inclusion body myopathy.22
PPA
The presenting symptoms in PPA involve changes in both expressive and receptive language functions. In Mesulam’s original description,2 other cognitive functions and behavior remain unaffected. Some PPA patients are able to continue working, provided that they do not depend on language skills for their living; they do not act forgetful, and they can travel and pursue hobbies independently. Later, some patients go on to develop symptoms of dementia, or they may acquire behavioral aberrations more typical of the frontal lobe dementia syndrome. Visual spatial functions and motor abilities tend to be the least involved, with the exception of apraxia in corticobasal degeneration, to be discussed later. Artistic expressions may not only continue but may even increase to new outlets; patients with PPA may develop new artistic expressions or hobbies,9,23,24 although Miller has pointed to the more simplified, stereotyped stylistic expressions.23
Patients with PPA most commonly have difficulty with naming or word-finding. Other common presenting symptoms include hesitancy or nonfluent speech production, motor speech abnormalities such as dysarthria, and impaired language comprehension and repetition. In recent years, the syndromes of PPA have been divided into three separate patterns: 1) progressive non-fluent aphasia (PNFA);3,5 2) semantic dementia;25–28 and 3) logopenic progressive aphasia (LPA), also called the logopenic/phonological variant of PPA.29–31 A recent review article describes in detail the language characteristics of the three syndromes.31 These language syndromes will be presented in briefer form here.
PNFA
PNFA may be thought of as a Broca-like syndrome; speech production is effortful and halting, with phoneme (speech sound) errors, and simplified or “agrammatic” productions. Word and simple sentence comprehension, as well as recognition of nouns (object knowledge), are preserved, although usually with some loss of comprehension for complicated syntactic constructions. Patients with PNFA, in recent series, rarely have underlying AD, fitting instead into the FTD/PPA spectrum. Most have a tauopathy, and this is especially true of patients who manifest both agrammatism and apraxia of speech.32 In a recent study, language testing in 24 patients with PNFA was categorized into four groups: those with apraxia of speech and agrammatism, those with speech apraxia but no agrammatism, those with no speech apraxia but with agrammatism, and those with neither speech apraxia nor agrammatism. The two groups with apraxia of speech appeared to have the classic PNFA, usually secondary to a tauopathy. The authors suggested that apraxia of speech develops earlier in the syndrome, whereas agrammatism comes later. The less common group with agrammatism but no apraxia of speech had progranulin mutations. These mutations will be discussed later. Finally, those with neither apraxia of speech nor agrammatism tend to have AD pathology or biomarkers, corresponding to the syndrome of logopenic/phonological progressive aphasia.32 Some patients with this language variant may appear relatively non-fluent, but they do not have the more specific features of agrammatism and apraxia of speech.
Semantic dementia
Semantic dementia is a syndrome involving fluent aphasia, with long strings of word production, but early deficits include anomia and impaired knowledge of word meanings, even for single words. For example, a patient with semantic dementia not only cannot name uncommon objects like “refrigerator” or “carburetor”, but may be unable to say what a refrigerator or carburetor is. These patients often have surface dyslexia (reading that is literally phonetic, such that patients cannot pronounce irregular nouns such as “colonel” or “yacht”). In addition, they can repeat without difficulty. They may also have impaired object and person knowledge. Semantic dementia was first described by Snowden et al,25 later expanded by Hodges et al,26 Hodges and Patterson,27 and Davies et al.28 The pathology can be variable, usually involving an FTD pathology, and genetically many have progranulin mutations, but a few have had the pathology of AD at postmortem.28
Logopenic PPA
The logopenic or logopenic/phonological variant of PPA involves impaired naming, or retrieval of single words. Along with this anomia, which can make speech sound relatively non-fluent, patients show inability to repeat phrases and sentences, and they sometimes make phonemic or speech sound errors, but they usually have no dysarthria or motor speech abnormalities, or even agrammatism. They also demonstrate normal comprehension of single words and simple sentences and preserved object knowledge.29–31 Logopenic PPA most often is a manifestation of AD, one of three “focal” presentations of AD, along with frontal variant AD referred to earlier, and the “posterior cortical atrophy” variant of AD, which presents with progressive visual complaints. The recognition of this overlap between PPA and AD has made the distinction between AD and FTD more complex. PPA is a syndrome, not a pathological diagnosis. Although the term initially implied a pathology other than AD, we now must consider that some cases may have a syndrome of PPA but a pathological diagnosis of AD. For example, in a series of 100 patients with “focal” presentations of dementing illness, 34 had autopsy-proved AD.33 These cases comprised 12 of 26, or 44.1%, of patients with PNFA. In comparison, only 2 of 28, or 7.1%, of the bvFTD patients and 2 of 20, or 10%, with semantic dementia had postmortem evidence of AD. Xiong et al34 reported 33 autopsy-studied cases of PNFA; 13 had AD pathology, and 20 had FTD pathology. Four clinical features were useful in predicting FTD pathology: age of onset before 60; “sweet tooth”, or preference for sweet foods; disinhibited behaviors; and the presence of “knife-edge” atrophy in the frontal and/or temporal lobes. These features were only weakly predictive individually, but a majority of the FTD cases had at least one of the features, whereas most AD cases had none. Features that did not distinguish between FTD and AD included progression to generalized dementia within 2 years (the defining feature in Mesulam’s original definition of PPA), or presence of memory deficits (the usual presenting symptom in AD), or impairment of activities of daily living. In addition, onset before age 65 did not distinguish between cases of FTD and AD in this series, although the series had disproportionately young patients because of the authors’ interest in early dementia. Hu et al35 described 19 patients with PNFA and 19 with LPA. Twelve of the 19 LPA patients and 6/19 PNFA patients had either autopsy confirmation or cerebrospinal fluid biomarkers suggestive of AD. Naming was more impaired in the AD patients, whereas letter-based fluency was more impaired in the non-AD cases.
Patterns of regional brain atrophy in the PPA syndromes
In a related manner, these somewhat distinct patterns of language presentation have shown correlations with differences in the patterns of regional brain atrophy, by voxel based magnetic resonance imaging (MRI) morphometry. Those with AD have tended to show more posterior–superior temporal atrophy, whereas those with FTD had perisylvian atrophy (LPA) or dorsolateral prefrontal and insular atrophy (PNFA).35 These regional atrophy patterns correspond well to our knowledge of the regional anatomy of the symptoms. Rohrer et al36 recently described 14 cases of LPA. All of the patients had MRI findings of cortical thinning in the left posterior and superior temporal, inferior parietal, medial temporal, and posterior cingulate regions. All had either an autopsy-confirmed or cerebrospinal fluid biomarker-confirmed diagnosis of AD. Both the clinical syndrome and the regional atrophy on MRI are distinct from those of PNFA, in which the atrophy is most marked in the dorsolateral frontal lobe and insula (Figure 1). These regional atrophy patterns are also supported by Deramecourt et al;37 frontal-temporal opercular and insular atrophy in PNFA, left frontotemporal atrophy with transactive response (TAR)-DNA mutations and PNFA, bilateral-anterior and lateral temporal cortex in FTD-TDP-43 semantic dementia, and left parietotemporal cortex in LPA associated with AD.
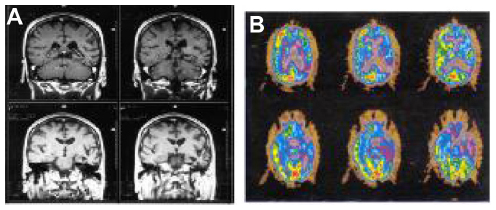 | Figure 1 Personal patient with progressive non-fluent aphasia. |
Course of PPA
PPA is almost always slowly progressive, involving slowly worsening language function. One area of controversy concerns whether all patients with PPA eventually develop a generalized dementia. Some cases of PPA have shown slowly progressive language dysfunction over courses as long as 10–12 years, without obvious deterioration of other cognitive functions or dementia. The incidence of dementia in patients with PPA is unknown, but by one estimate, it probably approaches 50% over several years.38
Other neurological symptoms and signs in PPA
Patients with FTD and PPA may develop other neurological abnormalities, in addition to language and cognitive deficits. Frontal release signs often develop, particularly in bvFTD, such as a positive glabellar (Myerson’s) sign, or a grasp, snout, or palmomental response. Movement disorders and extrapyramidal signs, such as rigidity or tremor, or even full-blown Parkinsonism, may develop in a minority of patients. As mentioned earlier, the syndrome of corticobasal degeneration is one of the underlying disorders in FTD and PPA. Corticobasal degeneration involves motor signs such as asymmetric rigidity and apraxia of the upper limbs, along with neurocognitive disorders, especially aphasia.39–41 As discussed earlier, some patients with FTD develop MND, with muscle atrophy, weakness, fasciculations, bulbar signs, and hyperreflexia.15 Muscle weakness is also seen in the rare variant with inclusion body myopathy.22
Immediate versus short-term memory in PPA and FTD
Grossman et al pointed to a double dissociation between immediate and short-term memory in a comparison study of four patients with PPA versus 25 patients with presumed AD.42 Immediate memory was more impaired in PPA patients, whereas short-term memory deficits characterized the deficits of patients with AD. The frontal cortex, especially on the left side, is thought to be the site of working or immediate memory, whereas the hippocampus and other medial temporal structures, often affected early in AD, represent the site of short-term memory. Other cognitive functions also showed differences between the two groups. Patients with PPA were more deficient than patients with AD on tasks of syntactic and speech fluency, correlating with the aphasia, and they had more severe impairment of attention (eg, digit span), another measure of immediate memory. Patients with PPA showed preserved memory and visuospatial functions, whereas those with AD were almost invariably impaired in these functions. As we have discussed, especially in the syndrome of logopenic PPA, however, atypical cases of AD can present with “focal” syndromes of aphasia7,8,33,34 or frontal lobe executive dysfunction.20,21 This issue of “focal” versus more generalized dementia, of course, harkens back to Pick’s original discussion1 of how neurodegenerative diseases progress. Recent evidence supports neuron to neuron transmission of pathology in both AD and FTD, whereby the abnormal misfolded proteins of tau or beta-amyloid can transmit from neuron to neuron in a way reminiscent of the “prion” diseases like Creutzfeldt-Jakob disease.43 A simplistic way of expressing this concept of progression of neurodegenerative disease is that what eventually becomes a “diffuse” disease has to start somewhere; it can start in one region of the brain and then spread, neuron to neuron, into unaffected areas.
Epidemiology
FTD is much less common than AD, but the incidence and prevalence may be equal to or greater than that of AD among early onset dementia patients, especially those presenting before the age of 60 years.18,34,38 Studies from a brain bank in Lund, Sweden, and Manchester, England have estimated that FTD accounts for 8%–10% of dementia patients.44 Stevens et al,45 in a study from Holland, found 74 cases of FTD out of a population of 15 million (about 5 cases per million). Among those aged 60–70 years, the prevalence was 28 cases per 100,000. In the United States, estimates have been lower, but there is some question about the recognition of these disorders, at least prior to 1990. A recent estimate placed FTD as occurring in 4–15 cases per 100,000, based on both US and European sources.46 In terms of prevalence, FTD ranks after AD, vascular dementia, and Lewy body dementia, although the percentages are likely higher in young-onset dementia cases.
Most patients with FTD dementia present in their 50s or 60s, or even 40s, clearly a younger age distribution, as compared with AD. Westbury and Bub,38 in a review of 112 cases published before 1997, found that the mean age of onset was 59 years. The series included 66% males and 34% females. Many cases of FTD have a positive family history, suggesting a genetic disorder in as many as one in four cases.18 In the Stevens et al45 study from Holland, 38% of the index cases of FTD had a first-degree relative with language or behavioral symptoms. A review44 reported autosomal dominant inheritance in 27% of cases. The genetic and molecular biology of FTD will be covered later in this current review.
Clinical diagnosis
The symptoms of patients with PPA and FTD are indicative of a focal lesion of the frontal lobes, in bvFTD, or the left hemisphere language cortex, in PPA, but brain imaging typically shows only focal brain atrophy. Laboratory tests of blood and spinal fluid are generally unrevealing, except for the genetic tests emerging in FTD. Brain imaging studies are helpful in ruling out focal lesions such as stroke, brain tumor, trauma, or infection. MRI is much more sensitive and accurate than computed tomography for delineating cerebral atrophy. Some patients have striking focal atrophy in the frontal or temporal lobe on one or both sides; coronal MRI makes it especially easy to detect temporal lobe atrophy, as well as to look at the medial temporal structures, which are more typically affected in AD. MRI, combined with voxel-based morphometry, provides quantitative mapping of localized cortical atrophy. As discussed earlier, the various subtypes of FTD have specific patterns of atrophy on MR imaging studies. Patients with bvFTD have striking atrophy of the frontal cortex, unilaterally or bilaterally. Patients with PNFA tend to have perisylvian, left hemisphere atrophy, involving the left frontal cortex. Those with semantic dementia have temporal lobe atrophy,24,25 often bilateral. Those with logopenic PPA have more posterior, temporal, and parietal atrophy, sometimes predominantly left-sided, but often bilateral. Whitwell et al47 reported that cases associated with MND have more paracentral atrophy by voxel-based MRI morphometry.47
Functional imaging techniques, particularly positron emission tomography (PET), make focal areas of hypometabolism easy to visualize. An early report by Tyrrell et al48 showed left temporal hypometabolism in all of six cases of PPA. More advanced cases also showed hypometabolism in the left frontal lobe and, in one case, the right temporal lobe. These regional patterns of cortical hypometabolism have been confirmed in subsequent studies.49,50 The pattern of frontal and/or temporal involvement is distinct from that of AD, in which the earliest detectable hypometabolism for glucose is typically evident in the parietal lobes. New ligands used to bind to amyloid protein deposits (eg, Pittsburgh compound B and florbetapir F18 [Amyvid™]) are helpful in the diagnosis of AD, showing abnormal deposits of amyloid in the brain. These new PET ligands are mainly helpful in the diagnosis of FTD by the exclusion of AD pathology. Interestingly, the amyloid ligand PET studies do not seem to show the regional involvement of specific brain regions that is the hallmark of fluorodeoxyglucose PET studies in the diagnosis of fvFTD and PPA; patients with logopenic PPA showed the typical topography of amyloid by Pittsburgh compound B imaging,51–53 and frontal variant AD patients also failed to show regional deposition of amyloid in the frontal lobes.54 In some instances, both types of PET scan might be needed to demonstrate both the regional areas of predilection and the underlying pathology in a neurodegenerative disorder. The cost of performing both studies, however, is prohibitive in most cases. Some evidence can also be found on lumbar puncture, in which elevated spinal fluid tau and reduced spinal fluid beta-amyloid can support the diagnosis of AD.
In general, a “probable” diagnosis of FTD or its variants requires a carefully studied clinical syndrome. A “definite” diagnosis requires either autopsy or a proved genetic mutation.17
Molecular genetics and pathology of FTD and PPA
The past 15 years have witnessed great progress in the molecular pathology and genetics of FTD. Until then, these disorders were defined by clinical syndromes and by light microscopic histopathology. Pick’s disease, the first pathologic disorder associated with the syndrome of PPA, has neuropathological features of focal atrophy of the frontal and/or temporal lobes of one or both hemispheres, prominent gliosis, swollen neurons and loss of neurons, microvacuolation of the neuropil, and silver-staining inclusions called Pick bodies. Several case reports of pathologically proved Pick’s disease have appeared.55–57 These patients presented with slowly progressive language deficits, especially involving naming. In these patients, memory and visuospatial functions were spared early on, distinguishing this disorder from typical AD. In the many autopsy verified cases of PPA, most do not have Pick bodies, but rather have the less specific findings of frontal and/or temporal atrophy, neuronal loss, gliosis, and microvacuolation. In one of Mesulam’s original cases,2 a brain biopsy yielded only nonspecific changes. Kirshner et al58 reported two autopsied cases of PPA, in which the microscopic pathology showed focal neuronal loss, gliosis, and microvacuolation of the cortex.58 At the time, these changes seemed new in PPA, but they are very similar to those reported in FTD,11–13 and also those termed “nonspecific dementia” or “dementia without specific histological features”.59 Based on the clinical and histopathological similarity between FTD and PPA, the term “frontotemporal dementia” for the syndromes and “frontotemporal lobar degeneration” for the pathology are now the preferred terms for both conditions.
Recent years have witnessed a new era of molecular genetic advances in the FTD syndromes. This started with the tau (MAPT) gene, described in 1997 by Heutink et al,60 and later by Hutton et al61 and Poorkaj et al,62 in families with FTD. Mutations in the MAPT gene, located on chromosome 17, were first reported in Dutch families, but “tau” mutations have now been reported in patients around the world. FTD is now not just a syndrome, but rather is associated with specific molecular genetic mutations. In the words of Wilhelmsen,63 this genetic linkage has put FTD “on the map” (ie, the gene map). Tau is the major component of Pick bodies and is seen in a number of other neurodegenerative diseases, including cases from two families with “hereditary dysphasic dementia” reported by Morris et al:64 progressive supranuclear palsy, corticobasal degeneration, and amyotrophic lateral sclerosis (ALS)-Parkinson dementia disease complex of Guam. These other disorders can also be associated with language changes resembling PPA and FTD. FTD is now one of many “tauopathies.” In particular, the tau mutations are seen most commonly in the bvFTD cases and the progressive non-fluent type of PPA. Seelaar et al65 reported that, in a series of 364 patients with FTD, 27% had positive family histories suggesting autosomal dominant inheritance. Of these, 11% had tauopathies secondary to mutations of the MAPT gene on chromosome 17.
The majority of cases of FTD, however, are not associated with tau pathology or mutations. Most of the remaining cases have been noted to have ubiquitin immunoreactive inclusions in the cytoplasm or nucleus, or ubiquitin immunoreactive neurites. This group of cases was initially designated “FTD-U.” A second gene, called the progranulin gene, also located on chromosome 17, was discovered to be associated with mutations in FTD-U by two groups of investigators in 2006.66,67 Patients with many different progranulin mutations have been reported. These progranulin mutations result in a premature termination codon, or haploinsufficiency, a loss of function; whereas tau mutations result in a toxic gain of function.68 In cases of the progranulin mutation, the TAR-DNA binding protein, also referred to as TDP-43 (transactive response DNA binding protein 43 kDa), aggregates in neurons and glia; this protein is a major component of the ubiquitinated inclusions in FTD-U cases. Progranulin mutations and TDP-43 positive inclusions have been reported in patients with clinical diagnoses of FTD, PPA, and corticobasal degeneration, including some with MND.69–73 Currently, FTD-U cases appear to outnumber tau mutation cases, although the terminology of FTD-U has been supplanted by specific terminology of the progranulin mutations or TDP-43 or FUS (fused in sarcoma) protein accumulation . Most cases of FTD with MND show ubiquitin staining (sometimes called FTD-MND).
Recently, the FTD-U group has been further subdivided. In one classification scheme, FTD-U type 1 is associated with semantic dementia, FTD-U type 2 is associated with FTD-MND and bvFTD, and FTD-U type 3 is associated with both bvFTD and PNFA. These FTD-U variants are inherited in autosomal dominant fashion, via the progranulin mutation.69 In the series by Seelaar et al,65 6% of the cases had progranulin mutations. They reported 10% with autosomal dominant inheritance patterns without a specific molecular genetic mutation, adding up to 27% with autosomal dominant inheritance. The remaining 73% of patients in their series did not have clear evidence of a genetic abnormality; the molecular genetic basis of these cases remains not understood.
Whereas most FTD patients are referred to by histopathology as “dementia lacking specific histological features”,59 most familial cases of FTD with autopsy information have either a tau or progranulin mutation, with a few other, rarer genetic disorders. At present, only rare cases are truly nonspecific.68 Other gene loci implicated in FTD include cases of inclusion body myopathy with Paget’s disease of the bone, caused by mutations involving the valosin-containing protein (VCP) gene on chromosome 9.74 Some patients with this mutation have TDP-43 protein accumulation. Different mutations on chromosome 9 have been associated with FTD-ALS.75,76 The chromosome 9 C9OEF72 mutation has been found to have hexanucleotide repeats and is associated with both familial ALS and FTD, as well as some seemingly sporadic cases.76–80 Most cases are associated with ubiquitin inclusions and TDP-43 accumulation. The hexanucleotide repeat phenomenon in this disorder is unique in both FTD and ALS.
Mutations of the chromatin-modifying protein 2B (CHMP2B) gene on chromosome 3 have been reported in a Danish family, not associated with TDP-43 protein accumulation.81 One last mutation involves cases with ubiquitin inclusions, but no TDP-43 accumulation, rather an accumulation of FUS proteins; reported cases have included both FTD and ALS.82,83
Patients with ALS and FTD, as mentioned earlier, have ubiquitin-immunoreactive inclusions in the frontal cortex, most associated with progranulin mutations. At least two genetic defects have been reported in patients with FTD and MND: one on chromosome 975,76 and one on chromosome 3.81 These cases of ALS have a strong association with bvFTD. Some patients with FTD develop MND as their illness progresses.68 Looked at another way, at least 10% of patients with ALS have significant behavioral and executive function abnormalities.
To summarize the breakdown of specific FTD syndromes with these molecular genetic disorders, most familial cases of PNFA have an MAPT mutation. Semantic dementia patients mostly have ubiquitin inclusions, associated with progranulin mutations. The logopenic variant of PPA usually has neither tau nor ubiquitin accumulation, but rather represents a focal presentation of AD. On the other hand, apolipoprotein E4, which is associated with an increased risk for late-onset, sporadic AD, does not appear to be of increased frequency in FTD or PPA patients.84–86
The new genotypes of FTD have been correlated with patterns of regional atrophy on MRI scans. Rohrer et al87 reported that MAPT mutations are associated with symmetric atrophy on MRI, whereas mutations in the progranulin gene are associated with more asymmetric atrophy. It seems more straightforward, however, to correlate the regional patterns of MRI atrophy with syndrome diagnoses. As mentioned earlier, bvFTD is associated with MAPT mutations and with bifrontal atrophy, PNFA is associated also with MAPT mutations and left frontal convexity atrophy, semantic dementia is associated with progranulin mutations and bitemporal atrophy, and LPA is associated with AD and temporal-parietal atrophy. Patients with the other tau-related disorders are also associated with regional predilection areas: patients with progressive supranuclear palsy have midbrain tegmentum and superior cerebellar peduncle atrophy, and those with corticobasal degeneration have frontoparietal atrophy.
Treatment of FTD and PPA
The treatment of FTD, at present, is largely symptomatic. Tremendous strides have been made in the diagnosis of FTD and delineating the molecular pathology and genetics of the specific variants. There is a hope that once the abnormal gene products are better worked out, specific medical treatments will become available. Social interventions, including counseling and speech/language/cognitive therapy, help patients and families adapt to the neurocognitive deficits. In AD, exercise and cognitive therapies have been associated in slowed progression of the disease, but this has not been proved as yet in FTD. Treatment of depression with a selective serotonin reuptake inhibitor, such as paroxetine, sertraline, or citalopram, is frequently helpful, although one study of paroxetine was positive,88 and another negative.89 Trazodone may be helpful for sleep, as well as for “frontal” behavioral aberrations.90 One study found a positive effect of oxytocin.91
Neurotransmitter-based treatments, analogous to the use of dopaminergic agents in Parkinson’s disease or anticholinesterase agents in AD, have not proved beneficial in FTD. Anticholinesterase agents such as donepezil, rivastigmine, and galantamine, are approved for AD. They have no clear rationale in FTD however, given the lack of a specific cholinergic deficiency; they may help memory in some patients, but they may worsen behavior in others.92 There are no large clinical trials on these agents in FTD patients. Memantine, a partial blocker of the NMDA glutamate receptor, has been anecdotally reported to be helpful in FTD, but two recent, small clinical trials have not confirmed its efficacy.93,94
Dopaminergic drugs have been tested in patients with transcortical motor aphasia secondary to strokes, but the evidence for efficacy is lacking. Personal and anecdotal experience with dopamine agonist agents such as bromocriptine or levodopa, moreover, has not been convincing. Stimulant drugs such as amphetamines and modafinil may benefit patients with frontal lobe syndromes. Large, randomized, double-blind studies have not been reported; all such therapies should be considered investigational and off-label.
Conclusion
FTDs are neurodegenerative diseases in which frontal and temporal lobe symptoms predominate, resembling a focal left hemisphere process such as a stroke or traumatic brain injury. Over time, the symptoms progress, and some patients develop a more generalized dementia. Four clinical subtypes characterize the predominant presentations of this illness: bvFTD, or fvFTD, PNFA, semantic dementia, and logopenic PPA. These clinical variants correlate with regional patterns of atrophy on brain imaging studies, and recently with biochemical and neurogenetic variants of the disease. Treatment is currently symptomatic, but advances in molecular genetics promise new therapies. The new field of proteomics promises that genetic mutations, clinical syndromes, imaging characteristics, and ultimately scientifically based treatments will emerge.95
Disclosure
The author reports no conflicts of interest in this work.
References
Pick A. On the relationship between aphasia and senile atrophy of the brain. In: Rottenberg DA, Hochberg FH, editors. Neurological Classics in Modern Translation. 39(5). Hafner Press; 1977. | |
Mesulam MM. Slowly progressive aphasia without generalized dementia. Ann Neurol. 1982;11(6):592–598. | |
Weintraub S, Rubin NP, Mesulam MM. Primary progressive aphasia. Longitudinal course, neuropsychological profile, and language features. Arch Neurol. 1990;47(12):1329–1335. | |
Mesulam MM. Primary progressive aphasia. Ann Neurol. 2001; 49(4):425–432. | |
Mesulam MM. Primary progressive aphasia – a language-based dementia. N Engl J Med. 2003;349(16):1535–1542. | |
Sonty SP, Mesulam MM, Thompson CK, et al. Primary progressive aphasia: PPA and the language network. Ann Neurol. 2003;53(1):35–49. | |
Kirshner HS, Webb WG, Kelly MP, et al. Language disturbance. An initial symptom of cortical degenerations and dementia. Arch Neurol. 1984;41(5):491–496. | |
Green J, Morris JC, Sandson J, et al. Progressive aphasia: a precursor of global dementia? Neurology. 1990;40(3 Pt 1):423–429. | |
Edwards-Lee T, Miller BL, Benson DF, et al. The temporal variant of frontotemporal dementia. Brain. 1997;120(Pt 6):1027–1040. | |
Josephs KA, Whitwell JL, Knopman DS, et al. Two distinct subtypes of right temporal variant frontotemporal dementia. Neurology. 2009;73:1443–1450. | |
Neary D, Snowden J. Fronto-temporal dementia: nosology, neuropsychology, and neuropathology. Brain Cogn. 1996;31(2):176–187. | |
Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546–1554. | |
Snowden J, Neary D, Mann D. Frontotemporal lobar degeneration: clinical and pathological relationships. Acta Neuropathol. 2007;114(1):31–38. | |
Lhermitte F, Pillon B, Serdaru M. Human autonomy and the frontal lobes. Part I: Imitation and utilization behavior: a neuropsychological study of 75 patients. Ann Neurol. 1986;19:326–334. | |
Neary D, Snowden JS, Mann DM, et al. Frontal lobe dementia and motor neuron disease. J Neurol Neurosurg Psychiatry. 1990;53(1):23–32. | |
Kertesz A, Munoz DG. Primary progressive aphasia and Pick complex. J Neurol Sci. 2003;206(1):97–107. | |
Roskovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134:2456–2477. | |
Warren JD, Rohrer JD, Rossor MN. Clinical review. Frontotemporal dementia. BMJ. 2013;347:f4827. | |
Bird TD. Genotypes, phenotypes, and frontotemporal dementia: take your pick. Neurology. 1998;50(6):1526–1527. | |
Woodward M, Jacova C, Black SE, et al. Differentiating the frontal variant of Alzheimer’s disease. Int J Geriatr Psychiatry. 2010;25:732–738. | |
Johnson JK, Head E, Kim R, et al. Clinical and pathological evidence for a frontal variant of Alzheimer’s disease. Arch Neurol. 1999;56:1233–1239. | |
Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36(4):377–381. | |
Miller BL, Cummings J, Mishkin F, et al. Emergence of artistic talent in frontotemporal dementia. Neurology. 1998;51:978–982. | |
Miller BL, Hou CE. Portraits of artists: emergence of visual creativity in dementia. Arch Neurol. 2004;61(6):842–844. | |
Snowden JS, Goulding PS, Neary D. Semantic dementia: a form of circumscribed cerebral atrophy. Behav Neurol. 1989;2:167–182. | |
Hodges JR, Patterson K, Oxbury S, et al. Semantic dementia. Progressive fluent aphasia with temporal lobe atrophy. Brain. 1992; 115(Pt 6):1783–1806. | |
Hodges JR, Patterson K. Semantic dementia: a unique clinicopathological syndrome. Lancet Neurol. 2007;6(11):1004–1014. | |
Davies RR, Hodges JR, Kril JJ, et al. The pathological basis of semantic dementia. Brain. 2005;128:1984–1995. | |
Gorno-Tempini ML, Brambati SM, Ginex V, et al. The logopenic/phonological variant of primary progressive aphasia. Neurology. 2008;71(16):1227–1234. | |
Gorno-Tempini ML, Dronkers NF, Rankin KP, et al. Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol. 2004;55(3):335–346. | |
Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76:1006–1014. | |
Rohrer JD, Rossor MN, Warren JD. Syndromes of nonfluent primary progressive aphasia: a clinical and neurolinguistic analysis. Neurology. 2010;75:603–610. | |
Alladi S, Xuereb J, Bak T, et al. Focal cortical presentations of Alzheimer’s disease. Brain. 2007;130:2636–2645. | |
Xiong L, Xuereb JH, Spillantini MG, et al. Clinical comparison of progressive aphasia associated with Alzheimer versus FTD-spectrum pathology. J Neurol Neurosurg Psychiatry. 2011;82:254–260. | |
Hu WT, McMillan C, Libon D, et al. Multimodal predictors for Alzheimer disease in nonfluent primary progressive aphasia. Neurology. 2010;75:595–602. | |
Rohrer JD, Ridgway GR, Modat M, Ourselin S, Mead S, Fox NC. Distinct profiles of brain atrophy in frontotemporal lobar degeneration caused by progranulin and tau mutations. Neuroimage. 2010;53:1070–1076. | |
Deramecourt V, Lebert F, Debachy B, Mackowiak-Cordoliani MA, Bombois S, Kerdraon O. Prediction of pathology in primary progressive language and speech disorders. Neurology. 2010;74:42–49. | |
Westbury C, Bub D. Primary progressive aphasia: a review of 112 cases. Brain Lang. 1997;60(3):381–406. | |
Lippa CF, Cohen R, Smith TW, et al. Primary progressive aphasia with focal neuronal achromasia. Neurology. 1991;41(6):882–886. | |
Kertesz A, Martinez-Lage P, Davidson W, et al. The corticobasal degeneration syndrome overlaps progressive aphasia and frontotemporal dementia. Neurology. 2000;55:1368–1375. | |
Graham NL, Bak T, Patterson K, Hodges JR. Language function and dysfunction in corticobasal degeneration. Neurology. 2003;61:493–499. | |
Grossman M, Mickanin J, Onishi K. Progressive nonfluent aphasia: language, cognitive, and PET measures contrasted with probable Alzheimer’s disease. J Cogn Neurosci. 1996;8:135–154. | |
Raj A, Kuceyeski A, Weiner M. A network diffusion model of disease progression in dementia. Neuron. 2012;73:1204–1215. | |
[No authors listed] Clinical and neuropathological criteria for frontotemporal dementia. The Lund and Manchester Groups. J Neurol Neurosurg Psychiatry. 1994;57(4):416–418. | |
Stevens M, van Duijn CM, Kamphorst W, et al. Familial aggregation in frontotemporal dementia. Neurology. 1998;50(6):1541–1545. | |
Rabinovici GD, Miller BL. Frontotemporal lobar degeneration: epidemiology, pathophysiology, diagnosis and management. CNS Drugs. 2010;24:375–398. | |
Whitwell JL, Jack CR Jr, Senjem ML, et al. Patterns of atrophy in pathologically confirmed FTLD with and without motor neuron degeneration. Neurology. 2006;66(1):102–104. | |
Tyrrell PJ, Warrington EK, Frackowiak RS, et al. Heterogeneity in progressive aphasia due to focal cortical atrophy. A clinical and PET study. Brain. 1990;113(Pt 5):1321–1336. | |
Abe K, Ukita H, Yanagihara T. Imaging in primary progressive aphasia. Neuroradiology. 1997;39(8):556–559. | |
Diehl J, Grimmer T, Drzezga A, et al. Cerebral metabolic patterns at early stages of frontotemporal dementia and semantic dementia. A PET study. Neurobiol Aging. 2004;25(8):1051–1056. | |
Leyton CE, Villemagne VL, Savage S, et al. Subtypes of progressive aphasia: application of the International Consensus Criteria and validation using beta-amyloid imaging. Brain. 2011;134:3030–3043. | |
Wolk DA, Price JC, Madeira C, et al. Amyloid imaging in dementias with atypical presentation. Alzheimers Dement. 2012;8:389–398. | |
Lehmann M, Ghosh PM, Madison C, et al. Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer’s disease. Brain. 2013;136:844–858. | |
Rabinovici GD, Furst AJ, O’Neil JP, et al. 11C-PIB PET imaging in Alzheimer disease and frontotemporal lobar degeneration. Neurology. 2007;68:1205–1212. | |
Holland AL, McBurney DH, Moossy J, et al. The dissolution of language in Pick’s disease with neurofibrillary tangles: a case study. Brain Lang. 1985;24(1):36–58. | |
Wechsler AF, Verity MA, Rosenschein S, et al. Pick’s disease. A clinical, computed tomographic, and histologic study with Golgi impregnation observations. Arch Neurol. 1982;39(5):287–290. | |
Graff-Radford NR, Damasio AR, Hyman BT, et al. Progressive aphasia in a patient with Pick’s disease: a neuropsychological, radiologic, and anatomic study. Neurology. 1990;40(4):620–626. | |
Kirshner HS, Tanridag O, Thurman L, et al. Progressive aphasia without dementia: two cases with focal spongiform degeneration. Ann Neurol. 1987;22(4):527–532. | |
Knopman DS, Mastri AR, Frey WH 2nd, et al. Dementia lacking distinctive histologic features: a common non-Alzheimer degenerative dementia. Neurology. 1990;40:251–256. | |
Heutink P, Stevens M, Rizzu P, et al. Hereditary frontotemporal dementia is linked to chromosome 17q21-q22: a genetic and clinicopathological study of three Dutch families. Ann Neurol. 1997;41(2):150–159. | |
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702–705. | |
Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43(6):815–825. | |
Wilhelmsen KC. Frontotemporal dementia is on the MAPtau. Ann Neurol. 1997;41(2):139–140. | |
Morris JC, Cole M, Banker BQ, et al. Hereditary dysphasic dementia and the Pick–Alzheimer spectrum. Ann Neurol. 1984;16(4):455–466. | |
Seelaar H, Kamphorst W, Rosso SM, et al. Distinct genetic forms of frontotemporal dementia. Neurology. 2008;71(16):1220–1226. | |
Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):916–919. | |
Cruts M, Gijselinck I, van den Zee J, et al. Null mutations in progranulin cause ubiquitin positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. | |
Bigio EH. Update on recent molecular and genetic advances in frontotemporal lobar degeneration. J Neuropathol Exp Neurol. 2008;67(7):635–648. | |
Yu CE, Bird TD, Bekris LM, Montine TJ, Leverenz JB, Steinbart E. The spectrum of mutations in progranulin: a collaborative study screening 545 cases of neurodegeneration. Arch Neurol. 2010;67(2):161–170. | |
Neumann M, Kwong LK, Sampathu DM, Trojanowski JQ, Lee VM. TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: protein misfolding diseases without amyloidosis. Arch Neurol. 2007;64(10):1388–1394. | |
Schymick JC, Yang Y, Andersen PM, Vonsattel JP, Greenway M, Momeni P. Progranulin mutations and amyotrophic lateral sclerosis or amyotrophic lateral sclerosis-frontotemporal dementia phenotypes. J Neurol Neurosurg Psychiatry. 2007;78(7):754–756. | |
Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol. 2007;171(1):227–240. | |
van Swieten JC, Heutink P. Mutations in progranulin (GRN) within the spectrum of clinical and pathological phenotypes of frontotemporal dementia. Lancet Neurol. 2008;7:965–974. | |
Forman MS, Mackenzie IR, Cairns NJ, et al. Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J Neuropathol Exp Neurol. 2006;65(6):571–581. | |
Hosler BA, Siddique T, Sapp PC, et al. Linkage of familial amyotrophic lateral sclerosis with frontotemporal dementia to chromosome 9q21-q22. JAMA. 2000;284:1664–1669. | |
Mahoney CJ, Beck J, Rohrer JD, et al. Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain. 2012;135:736–750. | |
Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. | |
Bigio EH. Motor neuron disease: the C9ORF72 hexanucleotide repeat expansion in FTD and ALS. Nat Rev Neurol. 2012;8:249–250. | |
Le Ber I, Camuzat A, Guillot-Noel L, et al. C9ORF72 repeat expansions in the frontotemporal dementias spectrum of diseases: a flow-chart for genetic testing. J Alzheimers Dis. 2013;34:485–499. | |
Liu Y, Yu JT, Sun FR, et al. The clinical and pathological phenotypes of frontotemporal dementia with C9ORF72 mutations. J Neurol Sci. 2013;335:26–35. | |
Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37(8):806–808. | |
Unwin H, Josephs KA, Rohrer JD, et al. FUS pathology defines the majority of tau- and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol. 2010;120:33–41. | |
Neumann M, Rademakers R, Roeber S, et al. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132:2922–2931. | |
Mesulam MM, Johnson N, Grujic Z, et al. Apolipoprotein E genotypes in primary progressive aphasia. Neurology. 1997;49(1):51–55. | |
Gustafson L, Abrahamson M, Grubb A, et al. Apolipoprotein-E genotyping in Alzheimer’s disease and frontotemporal dementia. Dement Geriatr Cogn Disord. 1997;8(4):240–243. | |
Geschwind D, Karrim J, Nelson SF, Miller B. The apolipoprotein E epsilon4 allele is not a significant risk factor for frontotemporal dementia. Ann Neurol. 1998;44:134–138. | |
Rohrer JD, Lashley T, Schott JM, et al. Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain. 2011;134:2565–2581. | |
Moretti R, Torre P, Antonello RM, et al. Frontotemporal dementia: paroxetine as a possible treatment of behavior symptoms. A randomized, controlled, open 14-month study. Eur Neurol. 2003;49(1):13–19. | |
Deakin JB, Rahman S, Nestor PJ, et al. Paroxetine does not improve symptoms and impairs cognition in frontotemporal dementia: a double-blind randomized controlled trial. Psychopharmacology (Berl). 2004;172:400–408. | |
Lebert F, Stekke W, Hasenbroekx C, et al. Frontotemporal dementia: a randomised, controlled trial with trazodone. Dement Geriatr Cogn Disord. 2004;17(4):355–359. | |
Jesso S, Morlog E, Ross S, et al. The effects of oxytocin on social cognition and behavior in frontotemporal dementia. Brain. 2011;134:2493–2501. | |
Huey ED, Putnam KT, Grafman J. A systematic review of neurotransmitter deficits and treatments in frontotemporal dementia. Neurology. 2006;66(1):17–22. | |
Boxer AL, Knopman DS, Kaufer DI, et al. Memantine in patients with frontotemporal lobar degeneration: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2013;12:149–156. | |
Vercelletto M, Boutoleau-Brettoniere C, Volteau C, et al. Memantine in behavioral variant frontotemporal dementia: negative results. J Alzheimers Dis. 2011;23:749–759. | |
Ghidoni R, Paterlini A, Benussi L. Translational proteomics in Alzheimer’s disease and related disorders. Clin Biochem. 2013;46:480–486. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.