Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Frequency and risk factor analysis of cognitive and anxiety-depressive disorders in patients with amyotrophic lateral sclerosis/motor neuron disease
Authors Cui F, Zhu W ![]() , Zhou Z, Ren Y, Li Y, Li M, Huo Y, Huang X
, Zhou Z, Ren Y, Li Y, Li M, Huo Y, Huang X
Received 15 June 2015
Accepted for publication 16 September 2015
Published 3 November 2015 Volume 2015:11 Pages 2847—2854
DOI https://doi.org/10.2147/NDT.S90520
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Wai Kwong Tang
Fang Cui, Wenjia Zhu, Zhibin Zhou, Yuting Ren, Yifan Li, Mao Li, Yunyun Huo, Xusheng Huang
Department of Neurology, Chinese PLA General Hospital, Beijing, People’s Republic of China
Objectives: To examine the frequency and risk factors of cognitive and anxiety-depressive disorders in patients with amyotrophic lateral sclerosis/motor neuron disease (ALS/MND).
Methods: This was an observational study of 100 ALS/MND patients treated at our hospital outpatient and inpatient departments between January 2009 and April 2010 and 100 matched healthy controls. Subjects were surveyed using Mini Mental State Examination (MMSE), Zung Self-Rating Anxiety Scale (SAS), and Zung Self-Rating Depression Scale (SDS). Patient neurological status was graded by the ALS Functional Rating Scale (ALSFRS). Multivariate linear regression was used to identify factors associated with the MMSE, SAS, SDS, and ALSFRS scores.
Results: Patients had significantly lower MMSE scores than controls (P<0.05). MMSE score did not differ by sex or age (<50/≥50 years) (P>0.05). Patients with higher educational level (college and above), shorter disease course (<2 years), and lower ALSFRS score (<20) had significantly higher MMSE scores (all P<0.05). Multivariate analysis revealed that higher education, shorter disease course, and lower ALSFRS score were independent predictors of better cognitive function (higher MMSE score). Patients had significantly higher mean SAS and SDS total scores than controls (both P<0.05), indicating higher subjective anxiety and depression. Female patients, patients with higher education, and those with higher ALSFRS scores had significantly higher SAS and SDS scores (all P<0.05). Age, occupation, diagnostic classification, disease duration, and disease awareness did not influence SAS or SDS scores. Multivariate analysis indicated that lower education and lower ALSFRS were protective factors against anxiety and depression.
Conclusion: The frequency of anxiety-depressive disorders was high among patients with ALS/MND. High educational level, short course of disease, and lower ALSFRS were associated with preserved cognitive function. Female sex, higher education, and lower ALSFRS score conferred a greater risk of anxiety and depression. Tailored pharmacotherapy and psychological interventions may help in reducing anxiety and depression in these patients.
Keywords: motor neuron disease, amyotrophic lateral sclerosis, cross-sectional study, anxiety, depression
Background
The motor neuron diseases (MNDs) are a group of neurodegenerative disorders of unknown etiology that selectively destroy motor neurons in spinal cord, brain stem, and cerebral cortex, with considerable heterogeneity among MND types.1 Loss of motor neurons results in progressive amyotrophy, amyasthenia, and pyramidal signs, finally evolving to fatal dysphagia and respiratory muscle weakness. Amyotrophic lateral sclerosis (ALS; or classical MND) is often used interchangeably with MND. MND/ALS tends to progress more rapidly than other neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease. While the incidence is only approximately 3.9/100,000, 80%–90% of MND/ALS patients die within 3–5 years of disease onset.2 According to Chancellor et al,3,4 mean survival was only 2.5 years in half of MND patients, and 5-year overall survival was only 28%. Respiratory failure induced by respiratory muscle paralysis is the major cause of death.5
In 1896, Charcot reported the first case of MND, but the etiology still remains unclear more than 100 years later. Most recent studies have focused on the contributions of excitotoxicity,6 gene mutations,7 oxidative stress, inflammation, and/or hypermetabolism,8 but no theory of MND etiology has yielded effective therapies for substantially slowing disease progression.9 Investigated strategies include antiexcitotoxic agents like riluzole, neurotrophic agents, antioxidants, free radical scavengers,10 and gene therapy.7 In addition, supportive and symptomatic treatments have also been examined.11 Still, riluzole is the only drug approved by the USA and European Union for MND treatment, and it merely prolongs survival time by about half a year.12
Functional neuroimaging and neuropathology of MND have demonstrated structural and functional impairments beyond motor regions.13 Epidemiological studies have also found cognitive changes in a subgroup of patients, and MND patients generally demonstrate attenuations of attention, alertness, psychomotor speed, and memory compared to education- and age-matched controls. In many cases, MND coexists with other neurodegenerative diseases, particularly frontotemporal dementia (FTD).14 Cognitive impairment is now regarded as a common component of MND progression, ranging from mild to marked FTD.1,15 Many studies have proposed a strong association between classical MND, MND associated with dementia, and MND associated with FTD. For instance, over 100 cases of MND associated with FTD have been reported in Japan since 1964,16 and another study has shown that approximately 15% of FTD patients showed MND symptoms.17 Thus, MND is now recognized as a multisystem degenerative disease characterized by motor system degeneration and often associated with functional impairment induced by frontotemporal lobar degeneration.18 The most frequent early behavioral/cognitive changes are apathy and reduced executive function. Insight and executive function deficits may help in the diagnosis of FTD in patients with ALS. These patients are negatively affected when they know that their cognitive functions are affected, and they are often seeking help.19 Therefore, cognitive function in MND and MND symptoms in FTD should be carefully evaluated for improved treatment and to better understand the clinical and etiological relationship between MND and dementia.
Study of patients with ALS and their caregivers found that anxiety is common at the diagnostic stage.20 Recent studies have revealed that depressive symptoms in patients with ALS will negatively affect their quality of life21 and that higher depressive scores are associated with faster disease progression.13 In addition, depression has been shown to influence the cognitive functions in a number of diseases.22,23 Therefore, it is necessary for physicians to evaluate and treat patient anxiety when diagnosing MND/ALS. Surveyed caregivers of ALS patients using the Cambridge Behavior Scale found that cognitive and emotional disorders were frequently reported. Moreover, many lacked awareness of disease-related problems such as amyasthenia and dysphrasia. Once diagnosed, most MND/ALS patients present anxiety-depressive disorders, even suicidal ideation, because ALS/MND is a rapidly progressive disease with a low survival rate and short survival time. Moreover, motor dysfunction impairs socialization, which may further compound psychiatric effects.24 Rabkin et al25 reported that 44%–75% of ALS patients suffer from depression and approximately 10% reach the criteria for major depressive disorder. Nevertheless, in Western studies, the association between ALS and depression and the risk factors for depression vary between studies and remain controversial.26–28 A South Korean study revealed that decreased physical functioning was associated with depressive symptoms.29
Until now, no large-sample studies of anxiety-depressive disorders in ALS/MND have been conducted in the People’s Republic of China. To this end, we assessed the frequency and characteristics of cognitive impairments and anxiety-depressive symptoms in 100 patients with MND/ALS, and performed multiple linear regression analysis to identify clinical and demographic factors associated with low cognitive and psychiatric function. These results might help in tailoring interventions that are specific to these patients.
Methods
Study population
One hundred ALS/MND Chinese Han patients diagnosed at our outpatient or inpatient departments between January 2009 and April 2010 met the following inclusion criteria: 1) consistent with the El Escorial criteria revised by the World Federation of Neurology in 1998;30,31 2) no history of sedatives, psychotropic drugs, or agents affecting the central nervous system; and 3) not complicated with other diseases that may hamper the emotional expression of anxiety-depressive disorders such as severe dementia other than ALS-related FTD, aphasia, or deafness.
One hundred healthy Chinese Han volunteers matched for sex ratio, age, and educational level were enrolled as the control group. All controls were neurologically normal as shown by examination and without past history of neuropsychiatric diseases. Normal cognitive function and the absence of anxiety or depressive symptoms were confirmed by designated physicians.
Data collection
Clinical and demographic data including age, sex, occupation, educational level, site of neurodegeneration, course of disease, diagnostic classification, anxiety score, and depression score were entered into a medical records database. The study was approved by the ethics committee of Chinese PLA General Hospital, Beijing 100853, China. All procedures performed in studies involving human participants were in accordance with the ethical standards of Chinese PLA General Hospital and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study.
The Mini Mental State Examination (MMSE)32 was used to evaluate orientation to time, orientation to place, registration, attention, calculation, short-term memory, object naming, repetition, reading comprehension, language comprehension, language expression, and figure drawing ability. Total score ranges from 0 to 30 points. Cutoff values separating normal and abnormal correlate with educational levels: 17 points for the illiterate subgroup (no education), 20 points for the primary school subgroup (6 years of education), and 24 points for the middle school/higher subgroup (>6 years of education). Scores below the cutoff are indicative of cognitive dysfunction, while scores above the cutoff are indicative of normal cognitive status.
The ALS Functional Rating Scale (ALSFRS)33 evaluates neurological impairment of ALS from the following measures: speech, salivation, swallowing, handwriting, cutting food and handling utensils, dressing and hygiene, turning in bed and adjusting bed clothes, walking, climbing stairs, and breathing. For each measure, normal is scored as 4, while total loss is scored as 0. The total score is 40, and lower score indicates more severe neurological dysfunction.
The Self-Rating Anxiety Scale (SAS)34 is composed of 20 items (with choices of A to D) including both positive items scored from 1 (A) to 4 (D) and negative items (questions no 5, 9, 13, 17, and 19) scored from 4 (A) to 1 (D). A gross score is obtained by adding the scores of the 20 questions, and the standardized score is obtained by the integral part of 1.25-fold of the gross score. Subjective anxiety was defined as a standard score ≥50. The Self-Rating Depression Scale (SDS)35 includes 20 items, each graded from 1 to 4, for a maximum total score of 80. Subjective depression was defined as a total score ≥50.
Statistical methods
Quantitative data are expressed as mean ± standard deviation, and categorical data are expressed as frequency/ratio. The t-test was used to compare quantitative means between two groups, one-way analysis of variance to compare multiple quantitative means, and the chi-square test to compare two ratios. Multiple linear regression analysis was performed to evaluate candidate predictive factors for cognitive impairment and anxiety-depressive disorders. SPSS 17.0 (SPSS Inc., Chicago, IL, USA) was applied for statistical analysis with the significance level defined as P<0.05.
Results
Baseline group characteristics
Fifty-two men and 48 women (male-to-female ratio 1.08:1) with age of onset between 16 and 74 years (mean 54.7±10.2 years) and symptom-to-diagnosis time from 3 to 48 months (mean 13.2±10.4 months) were enrolled in the MND/ALS group (Table 1). One hundred neurologically healthy volunteers matched for mean age, sex ratio, and education level were enrolled as the control group.
 | Table 1 Clinical and demographic characteristics of the patient group |
MMSE evaluation of ALS/MND patients
The MMSE total score and subscores for recall, calculation, and figure drawing were significantly lower in the patient group compared to the control group (Table 2, all P<0.001).
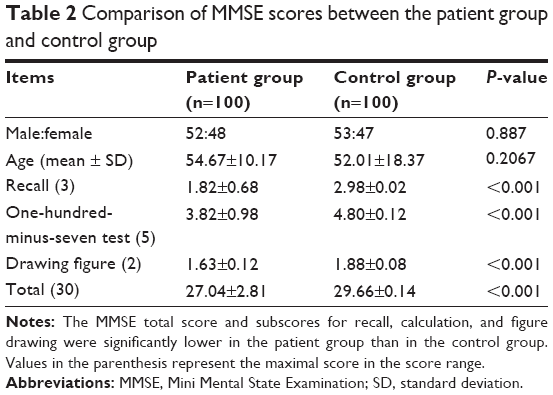 | Table 2 Comparison of MMSE scores between the patient group and control group |
Among patients, univariate analysis indicated that the MMSE score did not differ significantly between male and female patients or between those <50 years and those ≥50 years (both P>0.05). In contrast, patients with higher educational level (college and higher), shorter course of disease (<2 years), and lower ALSFRS score (<20) had higher MMSE scores than those with lower education, longer course of disease, and higher ALSFRS score, respectively (all P<0.05, Table 3).
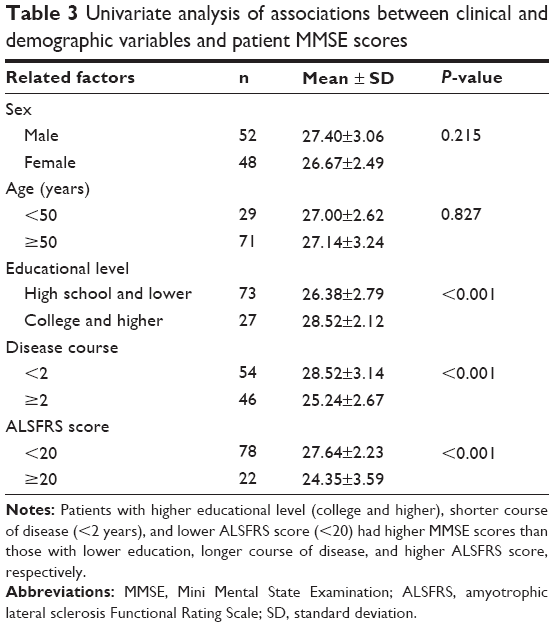 | Table 3 Univariate analysis of associations between clinical and demographic variables and patient MMSE scores |
Among patients, multiple linear regression analysis was performed with MMSE total score as the dependent variable and sex, age, educational level, disease course, and ALSFRS score as independent variables. Educational level (β=0.335, P<0.001), course of disease (β=0.252, P=0.001), and ALSFRS score (β=0.568, P<0.001) significantly affected MMSE total score. Specifically, higher educational level, short course of disease, and lower ALSFRS score were predictive of higher MMSE scores and thus greater preservation of cognitive function (Table 4).
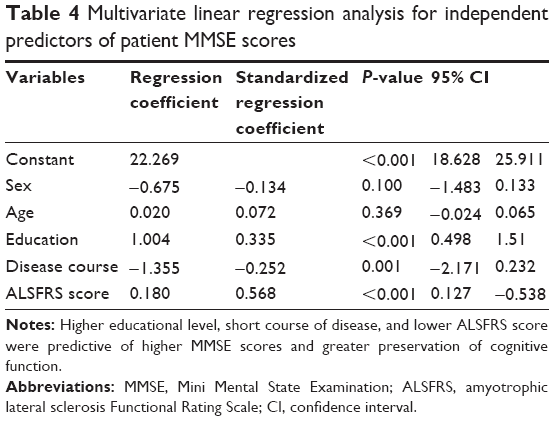 | Table 4 Multivariate linear regression analysis for independent predictors of patient MMSE scores |
Anxiety-depressive disorders in ALS/MND patients
The patient group also had significantly higher mean SAS standard score, mean SDS standard score, and frequencies of both anxiety and depression than the control group (all P<0.05, Table 5).
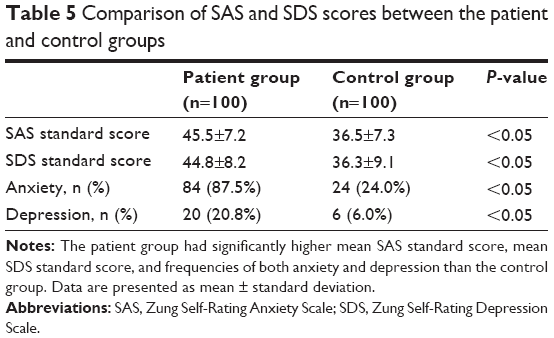 | Table 5 Comparison of SAS and SDS scores between the patient and control groups |
In the patient group, univariate analyses of candidate factors associated with anxiety and depression revealed higher SAS and SDS scores in females, those with higher education, and those with lower ALSFRS scores (<20) compared to males, those with lower educational level, and higher ALSFRS scores (≥20), respectively (Table 6). In contrast, age (<50 vs ≥50 years old), occupation (mental vs physical labor), ALS diagnosis (clinically confirmed vs clinically suspected), course of disease (<2 vs ≥2 years), and disease awareness had no significant influence on SAS and SDS scores.
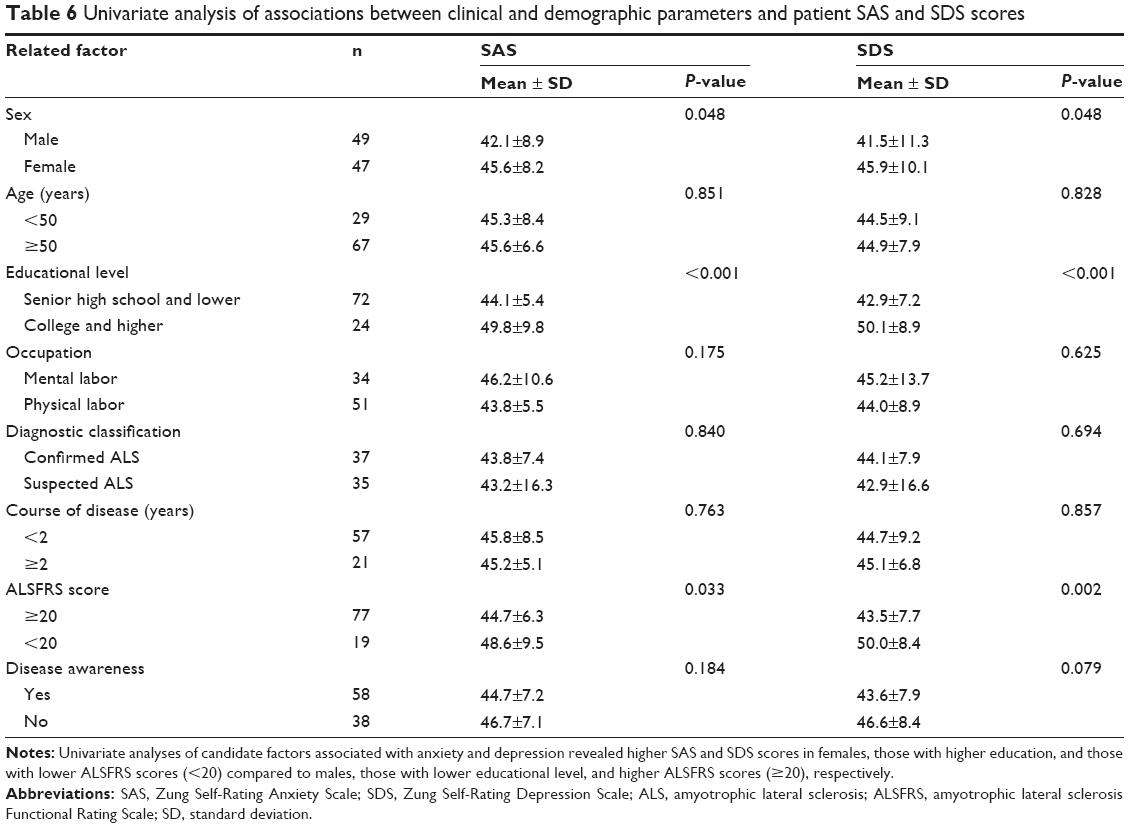 | Table 6 Univariate analysis of associations between clinical and demographic parameters and patient SAS and SDS scores |
Multivariate linear regression analysis revealed that educational level and ALSFRS score were independently associated with anxiety (β=0.410, P<0.001 and β=0.268, P=0.013, respectively) and depression (β=0.441, P<0.001 and β=0.385, P<0.001, respectively). Specifically, lower educational level and higher ALSFRS scores were predictive of lower SAS and SDS scores. Thus, lower education and higher ALSFRS scores were protective factors against anxiety and depressive symptoms (Tables 7 and 8).
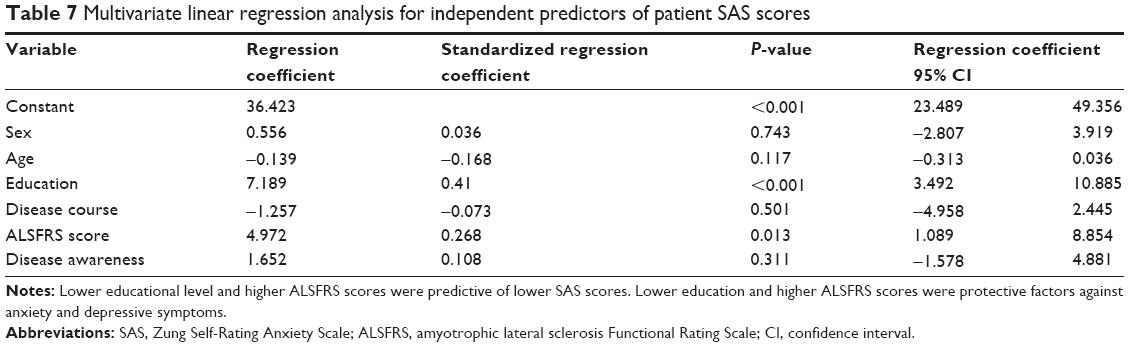 | Table 7 Multivariate linear regression analysis for independent predictors of patient SAS scores |
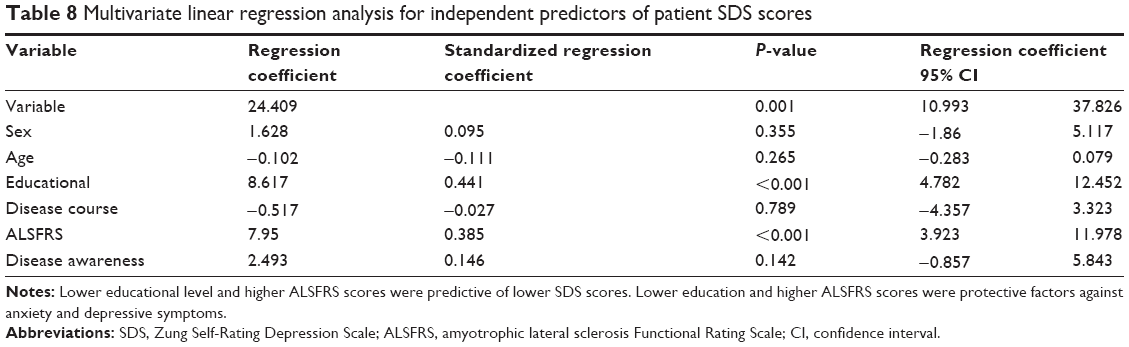 | Table 8 Multivariate linear regression analysis for independent predictors of patient SDS scores |
Discussion
The patient group had significantly lower MMSE scores for recall and calculation (the one-hundred-minus-seven test) compared to controls matched for age and education. The patient group also showed deficits in figure drawing, although this could be due to motor deficits (suggesting that the drawing item might be of little value for intellectual evaluation in this patient group). Multivariate analysis revealed that high educational level, short disease course, and lower ALSFRS scores were independently associated with higher MMSE scores, indicative of preserved cognitive function.
The prevalence of depression in patients with ALS compared to the general population is controversial. Indeed, Atassi et al28 reported a lower frequency of depression in patients with ALS. Nevertheless, in Western studies, the association between ALS and depression and the risk factors for depression vary between studies and remain controversial.26–28 A South Korean study revealed that decreased physical functioning was associated with depressive symptoms.29 Most of these studies report on small groups of patients, and it is thus difficult to compare the frequency of depression with the general population.
Cognitive function, operationally defined as skillful utilization of knowledge, depends on multiple neural processes such as acquisition, recall, working memory, attention, calculation, and abstraction/generalization. Cognitive functioning in MND is traditionally considered normal, but clinical observations have revealed progressive cognitive decline and even dementia symptoms in MND patients.36 Recent studies conducted in the People’s Republic of China and elsewhere have demonstrated cortical damage associated with speech comprehension and fluency impairments, memory deficits, and decline in calculation, as well as subcortical damage associated with impairments in abstraction.37 In addition, MND patients exhibited local reductions in cerebral blood flow and metabolic activity in frontal lobe, insular lobe, parahippocampal gyrus, and thalamic nuclei, and these reductions correlated with cognitive deficits.38 Such cognitive impairment may be mild at early stages and may be possibly neglected by patients, caregivers, and clinicians. However, cognitive decline may be more difficult to detect at a later stage due to exacerbation of dyskinesia and bulbar paralysis-induced speech disorder. Accurate evaluation of cognitive function at the early stages may enhance treatment efficacy, thereby enhancing patient quality of life.
Higher anxiety and depressive scores were reported by females, patients of higher educational level, and those with higher ALSFRS scores. Female sex was independently associated with higher anxiety-depressive symptoms (SDS and SAS scores). Recent studies have implicated hormonal changes in female anxiety.39 Moreover, as women play multiple roles in the family, they may worry about the family’s future due to their illness. The fact that patients with college and higher educational levels also exhibited more severe symptoms of anxiety than those with senior high school and lower educational levels may be explained by greater awareness of future outcome. ALSFRS for neurological deficit evaluation shows that severe neurological impairment and poor daily living ability can induce anxiety-depressive status, which is supported by a previous study.13 In contrast, age, occupation, disease duration, diagnostic classifications, and degree of disease awareness had no significant effects on SAS or SDS scores. The lack of effect of disease duration may reflect patient adjustment or acceptance of condition despite more severe symptoms.
In a previous report, approximately 25% of ALS patients were on antidepressants.40 In the present cross-sectional study, only a minority of patients were receiving psychotherapy or antidepressants, underscoring a lack of attention to anxiety and depression in MND patients. No randomized, double-blind, controlled trials on drug therapy for anxiety and depression associated with MND/ALS have been conducted so far.41 Clinically, the most commonly used agents are selective serotonin reuptake inhibitors and tricyclic antidepressants, of which the latter are also effective for anxious symptoms and useful for salivation, pseudobulbar palsy, and insomnia. We suggest that antianxiety and antidepressive treatments should be considered upon diagnosis.
The present study is not without limitations. The sample size was small, but considering the rarity of the disease and compared to previous studies, it was quite large. Nevertheless, the patients were from a single institution in the People’s Republic of China, and future multicenter studies should increase the sample size and validate the findings of this study. ALS/MND is influenced by genes such as C9ORF72, SOD1, TDP-43, and FUS;42 however, no genetic data are available for the patients included in this study. Nutrition is also a factor that could affect ALS progression, quality of life, and depressive symptoms;43 however, nutritional status of the patients was not available. Finally, hospitalized patients and outpatients may suffer differently from depression, but these data were not assessed in the present study. Future studies should include the assessment of polymorphisms in these genes and nutritional data as they might influence the psychological outcomes.
Conclusion
The frequency of symptoms of anxiety and depression in Chinese patients with ALS/MND was high. High educational level, short course of disease, and lower ALSFRS were independently associated with preserved cognitive function. Female sex, higher education, and lower ALSFRS score conferred a greater risk of anxiety and depression. Tailored pharmacotherapy and psychological interventions may help reduce anxiety and depression in these patients.
Author contributions
FC carried out the study design, data collection, and analysis, and wrote the manuscript. WJZ, ZBZ, YR, YFL, ML, and YYH participated in data collection, statistical analysis, and provided the critical revision. XSH conceived of the study, participated in its design and coordination, and provided the critical revision. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work. All authors approved the final version of the paper.
Disclosure
The authors report no conflicts of interest in this work.
References
Strong MJ, Lomen-Hoerth C, Caselli RJ, Bigio EH, Yang W. Cognitive impairment, frontotemporal dementia, and the motor neuron diseases. Ann Neurol. 2003;54(Suppl 5):S20–S23. | ||
Mehta P, Antao V, Kaye W, et al. Prevalence of amyotrophic lateral sclerosis – United States, 2010–2011. MMWR Surveil Summ. 2014;63(Suppl 7):1–14. | ||
Chancellor AM, Warlow CP. Adult onset motor neuron disease: worldwide mortality, incidence and distribution since 1950. J Neurol Neurosurg Psychiatry. 1992;55(12):1106–1115. | ||
Chancellor AM, Warlow CP, Carstairs V, Elton RA, Swingler RJ. Affluence, age, and motor neuron disease. J Epidemiol Community Health. 1992;46(2):172–173. | ||
Ringel SP, Murphy JR, Alderson MK, et al. The natural history of amyotrophic lateral sclerosis. Neurology. 1993;43(7):1316–1322. | ||
Blasco H, Mavel S, Corcia P, Gordon PH. The glutamate hypothesis in ALS: pathophysiology and drug development. Curr Med Chem. 2014;21(31):3551–3575. | ||
Scarrott JM, Herranz-Martin S, Alrafiah AR, Shaw PJ, Azzouz M. Current developments in gene therapy for amyotrophic lateral sclerosis. Expert Opin Biol Ther. 2015:1–13. | ||
Hooten KG, Beers DR, Zhao W, Appel SH. Protective and toxic neuroinflammation in amyotrophic lateral sclerosis. Neurotherapeutics. 2015;12(2):364–375. | ||
Bucchia M, Ramirez A, Parente V, et al. Therapeutic development in amyotrophic lateral sclerosis. Clin Ther. 2015;37(3):668–680. | ||
Graf M, Ecker D, Horowski R, et al. High dose vitamin E therapy in amyotrophic lateral sclerosis as add-on therapy to riluzole: results of a placebo-controlled double-blind study. J Neural Transm. 2005;112(5):649–660. | ||
Yacila G, Sari Y. Potential therapeutic drugs and methods for the treatment of amyotrophic lateral sclerosis. Curr Med Chem. 2014;21(31):3583–3593. | ||
Lee CT, Chiu YW, Wang KC, et al. Riluzole and prognostic factors in amyotrophic lateral sclerosis long-term and short-term survival: a population-based study of 1149 cases in Taiwan. J Epidemiol. 2013;23(1):35–40. | ||
Jelsone-Swain L, Persad C, Votruba KL, et al. The relationship between depressive symptoms, disease state, and cognition in amyotrophic lateral sclerosis. Front Psychol. 2012;3:542. | ||
Devenney E, Vucic S, Hodges JR, Kiernan MC. Motor neuron disease-frontotemporal dementia: a clinical continuum. Exp Rev Neurother. 2015;15(5):509–522. | ||
Abe K, Fujimura H, Toyooka K, Sakoda S, Yorifuji S, Yanagihara T. Cognitive function in amyotrophic lateral sclerosis. J Neurol Sci. 1997;148(1):95–100. | ||
Mitsuyama Y. Dementia with motor neuron disease. Neuropathology. 2000;20(Suppl):S79–S81. | ||
Snowden JS, Neary D, Mann DM. Frontotemporal dementia. Br J Psychiatry. 2002;180:140–143. | ||
Strong MJ, Grace GM, Freedman M, et al. Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009;10(3):131–146. | ||
Girardi A, Macpherson SE, Abrahams S. Deficits in emotional and social cognition in amyotrophic lateral sclerosis. Neuropsychology. 2011;25(1):53–65. | ||
Vignola A, Guzzo A, Calvo A, et al. Anxiety undermines quality of life in ALS patients and caregivers. Eur J Neurol. 2008;15(11):1231–1236. | ||
Tramonti F, Bongioanni P, Di Bernardo C, Davitti S, Rossi B. Quality of life of patients with amyotrophic lateral sclerosis. Psychol Health Med. 2012;17(5):621–628. | ||
Diamond BJ, Johnson SK, Kaufman M, Graves L. Relationships between information processing, depression, fatigue and cognition in multiple sclerosis. Arch Clin Neuropsychol. 2008;23(2):189–199. | ||
Sassoon SA, Rosenbloom MJ, Fama R, Sullivan EV, Pfefferbaum A. Selective neurocognitive deficits and poor life functioning are associated with significant depressive symptoms in alcoholism-HIV infection comorbidity. Psychiatry Res. 2012;199(2):102–110. | ||
Simmons Z, Bremer BA, Robbins RA, Walsh SM, Fischer S. Quality of life in ALS depends on factors other than strength and physical function. Neurology. 2000;55(3):388–392. | ||
Rabkin JG, Albert SM, Del Bene ML, et al. Prevalence of depressive disorders and change over time in late-stage ALS. Neurology. 2005;65(1):62–67. | ||
McLeod JE, Clarke DM. A review of psychosocial aspects of motor neurone disease. J Neurol Sci. 2007;258(1–2):4–10. | ||
McDonald ER, Wiedenfeld SA, Hillel A, Carpenter CL, Walter RA. Survival in amyotrophic lateral sclerosis. The role of psychological factors. Arch Neurol. 1994;51(1):17–23. | ||
Atassi N, Cook A, Pineda CM, Yerramilli-Rao P, Pulley D, Cudkowicz M. Depression in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2011;12(2):109–112. | ||
Oh H, Sin MK, Schepp KG, Choi-Kwon S. Depressive symptoms and functional impairment among amyotrophic lateral sclerosis patients in South Korea. Rehabil Nurs. 2012;37(3):136–144. | ||
Brooks BR, Miller RG, Swash M, Munsat TL. World Federation of Neurology Research Group on Motor Neuron D. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293–299. | ||
Ikemoto A, Hirano A, Akiguchi I. Neuropathology of amyotrophic lateral sclerosis with extra-motor system degeneration: characteristics and differences in the molecular pathology between ALS with dementia and Guamanian ALS. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(2):97–104. | ||
Molloy DW, Standish TI. A guide to the standardized Mini-Mental State Examination. Int Psychogeriatr. 1997;9(Suppl 1):87–94; discussion 143–150. | ||
The Amyotrophic Lateral Sclerosis Functional Rating Scale. Assessment of activities of daily living in patients with amyotrophic lateral sclerosis. The ALS CNTF treatment study (ACTS) phase I-II Study Group. Arch Neurol. 1996;53(2):141–147. | ||
Zung WW. A rating instrument for anxiety disorders. Psychosomatics. 1971;12(6):371–379. | ||
Zung WW. A Self-Rating Depression Scale. Arch Gen Psychiatry. 1965;12:63–70. | ||
Strong MJ, Grace GM, Orange JB, Leeper HA. Cognition, language, and speech in amyotrophic lateral sclerosis: a review. J Clin Exp Neuropsychol. 1996;18(2):291–303. | ||
Gallassi R, Montagna P, Ciardulli C, Lorusso S, Mussuto V, Stracciari A. Cognitive impairment in motor neuron disease. Acta Neurol Scand. 1985;71(6):480–484. | ||
Kiernan MC. Identification of cognitive deficits in amyotrophic lateral sclerosis. Clin Neurophysiol. 2009;120(4):645–646. | ||
Seeman MV. Psychopathology in women and men: focus on female hormones. Am J Psychiatry. 1997;154(12):1641–1647. | ||
Wicks P, Abrahams S, Masi D, Hejda-Forde S, Leigh PN, Goldstein LH. Prevalence of depression in a 12-month consecutive sample of patients with ALS. Eur J Neurol. 2007;14(9):993–1001. | ||
Kurt A, Nijboer F, Matuz T, Kubler A. Depression and anxiety in individuals with amyotrophic lateral sclerosis: epidemiology and management. CNS Drugs. 2007;21(4):279–291. | ||
Millecamps S, Boillee S, Le Ber I, et al. Phenotype difference between ALS patients with expanded repeats in C9ORF72 and patients with mutations in other ALS-related genes. J Med Genet. 2012;49(4):258–263. | ||
Van den Berg JP, Kalmijn S, Lindeman E, et al. Multidisciplinary ALS care improves quality of life in patients with ALS. Neurology. 2005;65(8):1264–1267. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.