Back to Journals » OncoTargets and Therapy » Volume 13
FOXC2 Promotes Oxaliplatin Resistance by Inducing Epithelial-Mesenchymal Transition via MAPK/ERK Signaling in Colorectal Cancer
Authors Chen Y, Deng G, Fu Y, Han Y, Guo C, Yin L, Cai C, Shen H ![]() , Wu S, Zeng S
, Wu S, Zeng S
Received 7 December 2019
Accepted for publication 7 February 2020
Published 21 February 2020 Volume 2020:13 Pages 1625—1635
DOI https://doi.org/10.2147/OTT.S241367
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Yihong Chen,1 Ganlu Deng,2 Yaojie Fu,1,3 Ying Han,1,3 Cao Guo,1,3 Ling Yin,1 Changjing Cai,1,3 Hong Shen,1,3 Shaobin Wu,4 Shan Zeng1,5
1Department of Oncology, Xiangya Hospital, Central South University, Changsha, Hunan 410008, People’s Republic of China; 2Department of Oncology, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi 530021, People’s Republic of China; 3Key Laboratory for Molecular Radiation Oncology of Hunan Province, Xiangya Hospital, Central South University, Changsha, Hunan 410008, People’s Republic of China; 4Department of Gastrointestinal Surgery, Xiangya Hospital, Central South University, Changsha, Hunan 410008, People’s Republic of China; 5National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, Hunan 410008, People’s Republic of China
Correspondence: Shan Zeng
Department of Oncology, Xiangya Hospital, Central South University, Changsha, Hunan 410008, People’s Republic of China
Tel +86 731-8975-3034
Email [email protected]
Shaobin Wu
Department of Gastrointestinal Surgery, Xiangya Hospital, Central South University, Changsha, Hunan 410008, People’s Republic of China
Tel +86 731-8975-3007
Email [email protected]
Background: Chemoresistance is a major obstacle to improving the survival rate of colorectal cancer (CRC) patients. Forkhead box protein C2 (FOXC2), a member of the forkhead box (Fox) transcription factor family, is reported to be an important regulator of epithelial-to-mesenchymal transition (EMT) and plays a key role in tumor progression. However, little is known about the effects of FOXC2 on oxaliplatin (OXA) resistance in CRC.
Methods: OXA-resistant cells were generated from HCT116 cells. CCK-8, colony formation, flow cytometry and Transwell assays were used to compare the characteristics of OXA-resistant HCT116/OXA cells and the corresponding parental HCT116 cells. The expression of FOXC2 was confirmed by qRT-PCR and Western blotting in HCT116/OXA and HCT116 cells. Gain- and loss-of-function assays were performed to evaluate the effects of FOXC2 on OXA sensitivity and EMT in HCT116/OXA and HCT116 cells both in vitro and in vivo, and the possible molecular mechanisms were investigated.
Results: The relative expression of FOXC2 was significantly increased in HCT116/OXA cells compared with the parental HCT116 cells. Upregulation of FOXC2 in HCT116 cells reduced OXA sensitivity and promoted EMT. However, knockdown of FOXC2 in HCT116/OXA cells markedly increased the in vitro and in vivo sensitivity of HCT116/OXA cells to OXA by regulating EMT progression. Furthermore, FOXC2 activated MAPK/ERK signaling, and blockade of ERK attenuated FOXC2-induced EMT and FOXC2-enhanced OXA resistance.
Conclusion: FOXC2 induced EMT to promote oxaliplatin resistance by activating the MAPK/ERK signaling pathway. FOXC2 may be a potential therapeutic target for overcoming OXA resistance in human CRC.
Keywords: forkhead box protein C2, epithelial-mesenchymal transition, colorectal cancer, oxaliplatin resistance, MAPK/ERK
Introduction
Colorectal cancer (CRC) is a leading cause of cancer-related death worldwide, and its incidence is increasing.1,2 Therapeutic strategies for CRC, including surgical resection, radiotherapy and systemic chemotherapy, have been used to improve the survival of CRC patients in past decades. However, the five-year survival rate of CRC patients still dismal.3 Drug resistance is a major obstacle to effective treatment and results in poor prognosis in CRC patients.4,5 Oxaliplatin (OXA), a third-generation platinum drug, has been used as a first-line chemotherapeutic drug for systemic treatment of advanced CRC patients.6 Therefore, clarifying the underlying mechanism of OXA resistance is imperative for the success of CRC therapy.
Epithelial-mesenchymal transition (EMT) is a cellular process in which epithelial cells lose their typical characteristics and acquire mesenchymal traits. Previous studies have indicated that EMT plays a crucial role in the invasion and metastasis of cancer.7,8 Cells undergoing EMT develop downregulation of epithelial markers (such as E-cadherin) and upregulation of mesenchymal markers (for example, Vimentin) and EMT-associated transcription factors (Snail, Twist, et al), subsequently acquiring the capacity for motility and invasion.9 In recent years, EMT has been implicated as an indispensable mediator of drug resistance.10–12 Exploring the relationship between EMT and OXA resistance can be a promising approach for the development of therapeutic regimens to treat OXA-resistant patients.
Forkhead box protein C2 (FOXC2) is a member of the Fox transcription factor family that was originally identified in the process of embryogenesis. Ectopic expression of FOXC2 has been found to participate in tumorigenesis and the development of human malignances.13 A high expression level of FOXC2 has been found to be a prognostic factor in gliomas,14 nasopharyngeal cancer,15 and gastric cancer.16 In addition, FOXC2 has been reported to regulate chemosensitivity in some human cancers.17–20 In colorectal cancer, FOXC2 has been shown to promote proliferation and metastasis.21 However, whether FOXC2 plays a role in OXA resistance in CRC is unknown. In the present study, we investigated the role of FOXC2 in mediating OXA resistance in colorectal cancer cell lines and explored the underlying mechanism involved in FOXC2-regulated OXA resistance both in vitro and in vivo.
Materials and Methods
Chemicals
Oxaliplatin and the MAPK/ERK inhibitor SCH772984 were purchased from Selleck Chemicals (Houston, TX) and diluted according to the manufacturer’s protocol. Primary antibodies against FOXC2, Ki67, E-cadherin, Vimentin and Snail were purchased from Abcam (Cambridge, MA, USA), and those against ERK1/2, phospho-ERK1/2 and GAPDH were purchased from Cell Signaling Technology (Cambridge, MA, USA).
Cell Lines and Culture Conditions
The human colorectal cancer (CRC) cell line HCT116 was obtained from the Institutes of Biomedical Sciences (IBS, Shanghai, China) and cultured in RPMI 1640 medium (Gibco, USA) containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Invitrogen, USA) at 37°C in 5% CO2. The oxaliplatin-resistant cell line HCT116/OXA was established by exposing parental HCT116 cells to gradually increasing concentrations of oxaliplatin and was maintained in RPMI 1640 medium supplemented with 7.5μM oxaliplatin (Selleck Chemicals, Houston, TX).
Chemosensitivity Assays
Cells were seeded in a 96-well plate at a density of 3.0×103 cells/well overnight and were then exposed to various concentrations of OXA for 48 hr. Cell viability was assessed with a Cell Counting Kit-8 (CCK-8, Dojindo Molecular Technologies, Tokyo, Japan). Briefly, 10μL of CCK-8 solution was added to each well and incubated for 1 hr at 37°C. Then, the absorbance was measured at a wavelength of 450 nm. The 50% inhibitory concentration (IC50) was calculated from the survival curves. Each assay was performed in triplicate.
Transwell Invasion and Migration Assays
The invasion and migration abilities of HCT116 and HCT116/OXA cells were assessed by using Transwell chambers containing polycarbonate filters with an 8-μm pore size (BD Biosciences) and precoated with or without Matrigel. Culture medium containing 10% FBS was placed in the lower chambers to act as a chemoattractant. Cells were suspended at a density of 4×104 cells/well in 200μL of serum-free DMEM and were then transferred into the upper chambers. After incubation at 37°C for 48 hr, the invaded cells were fixed and stained with 0.1% crystal violet hydrate solution for 30 min. Images were acquired using an inverted microscope (Leica DMI-4000B), and the numbers of invaded cells in each group were counted and compared.
Quantitative Real-Time Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
Total RNA was isolated using Trizol Reagent (Invitrogen, Waltham, MA), and cDNA was synthetized using a PrimeScript™ Kit (TaKaRa Bio Inc., Otsu, Japan) following the manufacturer’s instructions. qRT-PCR was performed in triplicate using a SYBR Green fluorescence-based assay (TaKaRa Bio Inc.) on a ViiATM7 RT-PCR system (Applied Biosystems, Carlsbad, CA). The primers used for real-time PCR were as follows: FOXC2 Forward: 5ʹ- CCTACCTGAGCGAGCAGAAT −3ʹ, Reverse: 5ʹ- ACCTTGACGAAGCACTCGTT −3ʹ; E-cadherin Forward: 5ʹ- TACGCCTGGGACTCCACCTA −3ʹ, Reverse: 5ʹ- CCAGAAACGGAGGCCTGAT −3ʹ; Vimentin Forward: 5ʹ- TGTGGATGT TTCCAAGCCTGAC −3ʹ, Reverse: 5ʹ- GAGTGGGTATCAACCAGAGGGAG −3ʹ; and GAPDH Forward: 5ʹ- CGGAGTCAACGGATTTGGTCGTAT −3ʹ, Reverse: 5ʹ- AGCCTTCTCCATGGTGGTGAAGAC −3ʹ. The relative mRNA expression values was normalized to GAPDH expression values and were calculated based on the Ct value according to the equation 2−ΔΔCt [ΔCt=Ct (targeting gene)-Ct (GAPDH)].
Western Blot Analysis
Cells were harvested and lysed in RIPA buffer. Protein extraction and Western blotting were performed as described previously.22 Primary antibodies against the following proteins were used: FOXC2 (1:500, Abcam, US), E-cadherin (1:1000, Abcam, US), Vimentin (1:1000, Abcam, US), ERK1/2 (1:1000, Cell Signaling Technology, Beverly, MA), p-ERK1/2 (1:1000, Cell Signaling Technology, Beverly, MA), Snail (1:500, Abcam, US) and GAPDH (1:10,000, Cell Signaling Technology, Beverly, MA).
Plasmid Construction and Cell Transfection
The human FOXC2 construct was generated by cloning PCR-amplified full-length FOXC2 cDNA into pBabe (Addgene, Inc., Cambridge, MA, USA). To knock down FOXC2, an shRNA sequence specifically targeting FOXC2 (5′-CCACACGTTTGCAACCCAA-3′) was cloned into pSuper-retro.neo (Oligo-Engine, Seattle, WA, USA) to generate the FOXC2-shRNA construct. Transfection was performed using LipofectamineTM 2000 (Invitrogen, USA) according to the protocol supplied by the manufacturer.
Xenograft Model
To investigate the effects of FOXC2 on OXA sensitivity in vivo, a xenograft animal model was established in male BALB/c mice (4 weeks old). Briefly, 5×106 sh-control or sh-FOXC2 HCT116/OXA cells were suspended in 200 μL of serum-free DMEM and injected subcutaneously into the left flank regions. Ten days after implantation, mice received an intraperitoneal injection of OXA (0.8 mg/kg/w) for 20 days. After one month, the mice were sacrificed, and the xenograft tumors were harvested. The tumor weights and tumor volumes were measured. The tumor volumes were calculated using the following formula: volume (cm3) = (length × width2) × 0.5. All animal studies were conducted at the Animal Institute of Central South University according to the protocols approved by the Medical Experimental Animal Care Commission of the university.
Immunohistochemistry (IHC)
CRC tissues from xenograft tumors were fixed with 10% formalin, dehydrated, and embedded in paraffin. Four-micron-thick sections were stained with antibodies against FOXC2 (1:200, Abcam), Ki67 (1:200, Abcam), E-cadherin (1:300, Abcam) and Vimentin (1:500, Abcam).
Statistical Analysis
Statistical analysis was performed using SPSS software (Version 19.0, SPSS Inc., Chicago, IL). All data were presented as the means ± SDs and were analyzed by Student’s t-test or one-way ANOVA. p < 0.05 was considered statistically significant.
Results
Characterization of Oxaliplatin-Resistant Cells
To investigate the mechanism underlying the resistance of CRC to oxaliplatin (OXA) treatment, we established an OXA-resistant CRC cell line, HCT116/OXA, by continuously exposing HCT116 cells to OXA, and we compared the characteristics of parental HCT116 and OXA-resistant HCT116/OXA cells. As shown in Figure 1A, the resistant HCT116/OXA cells exhibited higher resistance to various concentrations of OXA than the parental HCT116 cells, as determined by CCK-8 assays (Figure 1A). According to the CCK-8 assay results, the IC50 value in HCT116/OXA cells was significantly higher than that in HCT116 cells, confirming the acquisition of OXA resistance by HCT116/OXA cells (Figure 1B, p < 0.001). The IC50 values in HCT116 cells and HCT116/OXA cells were 7.53 ± 0.63 μM and 145.5 ± 3.52 μM, respectively. We further compared the difference in the proliferation ability between the two cell lines and found that HCT116/OXA cells had a higher proliferation capacity than HCT116 cells (Figure 1C, p < 0.05). Flow cytometric analyses showed that HCT116/OXA cells exhibited a significantly lower apoptosis rate than HCT116 cells when treated with 7.5 μM OXA (Figure 1D, p < 0.01). A clonogenic assay was performed to measure the sensitivity of these cells to OXA treatment. Consistent with the results of the CCK-8 assay, treatment with 7.5 μM OXA decreased the colony numbers of HCT116 cells compared with those of HCT116/OXA cells (Figure 1E, p < 0.01). The results of Transwell migration and invasion assays revealed that HCT116/OXA cells demonstrated greater migration and invasion capacities than HCT116 cells (Figure 1F and G, p < 0.01).
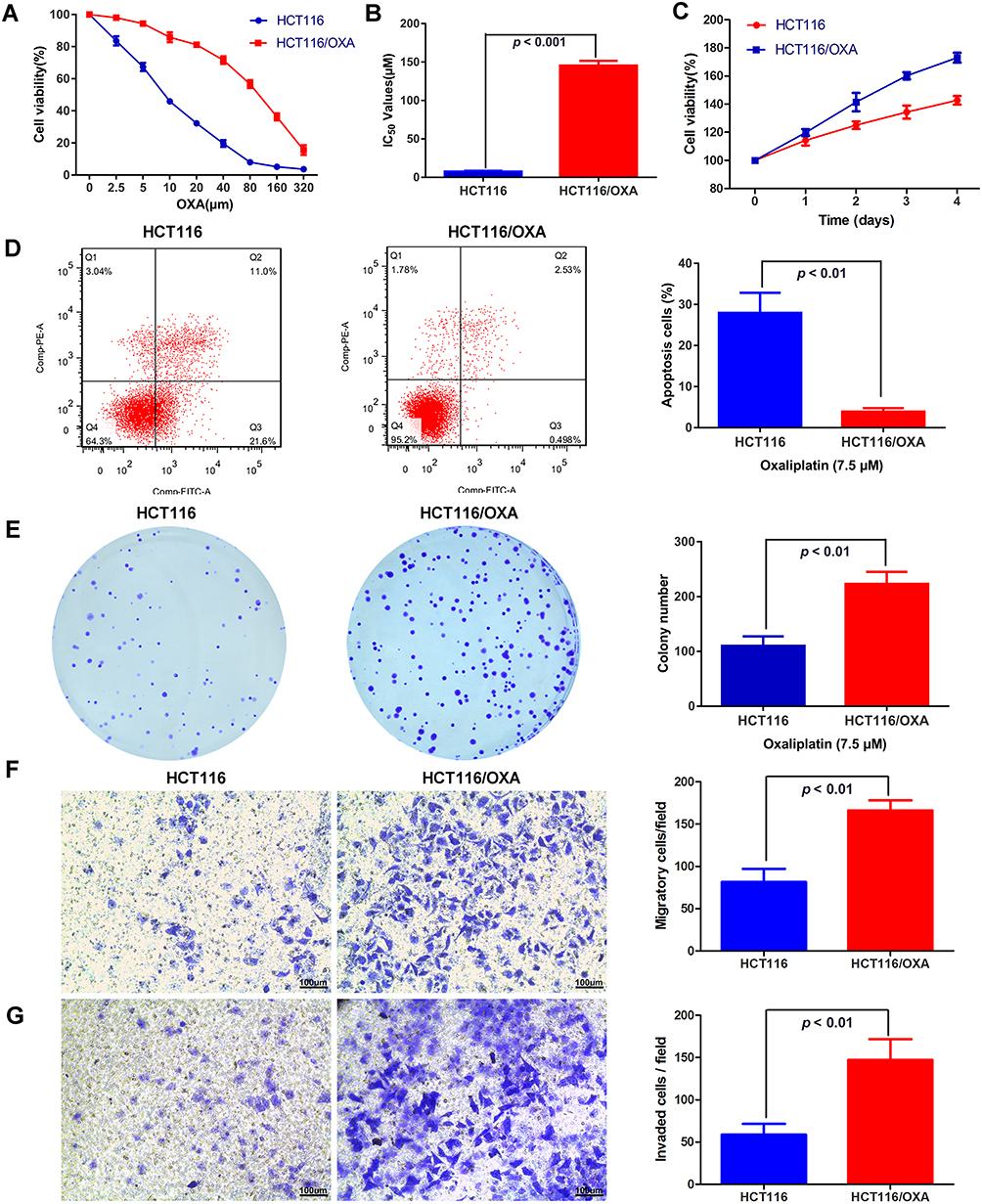 |
Figure 1 Cellular characterization of oxaliplatin-resistant HCT116/OXA cells: (A) The viability of HCT116 and HCT116/OXA cells treated with different concentrations of OXA for 48 hrs was determined by a CCK-8 assay. (B) The IC50 values of OXA in HCT116 and HCT116/OXA cells were measured by a CCK-8 assay. IC50 values in HCT116: 7.53 ± 0.63μM, in HCT116/OXA: 145.5 ± 3.52μM. (C) The proliferation ability of HCT116/OXA cells was higher than that of HCT116 cells (p < 0.05). (D) Flow cytometric detection of apoptosis in HCT116 and HCT116/OXA cells with Annexin V/PI staining. (E) The colony formation assay results indicated that the colony numbers of HCT116 cells were significantly lower than those of HCT116/OXA cells. (p < 0.01). (F & G) Transwell migration and invasion assays were used to evaluate the migration and invasion capacities of HCT116 and HCT116/OXA cells (scale bars =100μm). All experiments are repeated in triplicate, and the data are shown as the means ± SDs. |
The Expression of FOXC2 Was Significantly Upregulated in OXA-Resistant CRC Cells
We first examined the expression of FOXC2 in the HCT116 and HCT116/OXA cell lines by qRT-PCR and Western blotting. Compared to parental HCT116 cells, HCT116/OXA cells exhibited significant upregulation of FOXC2 at both the mRNA (p < 0.001) and protein levels (p < 0.05, Figure 2A).
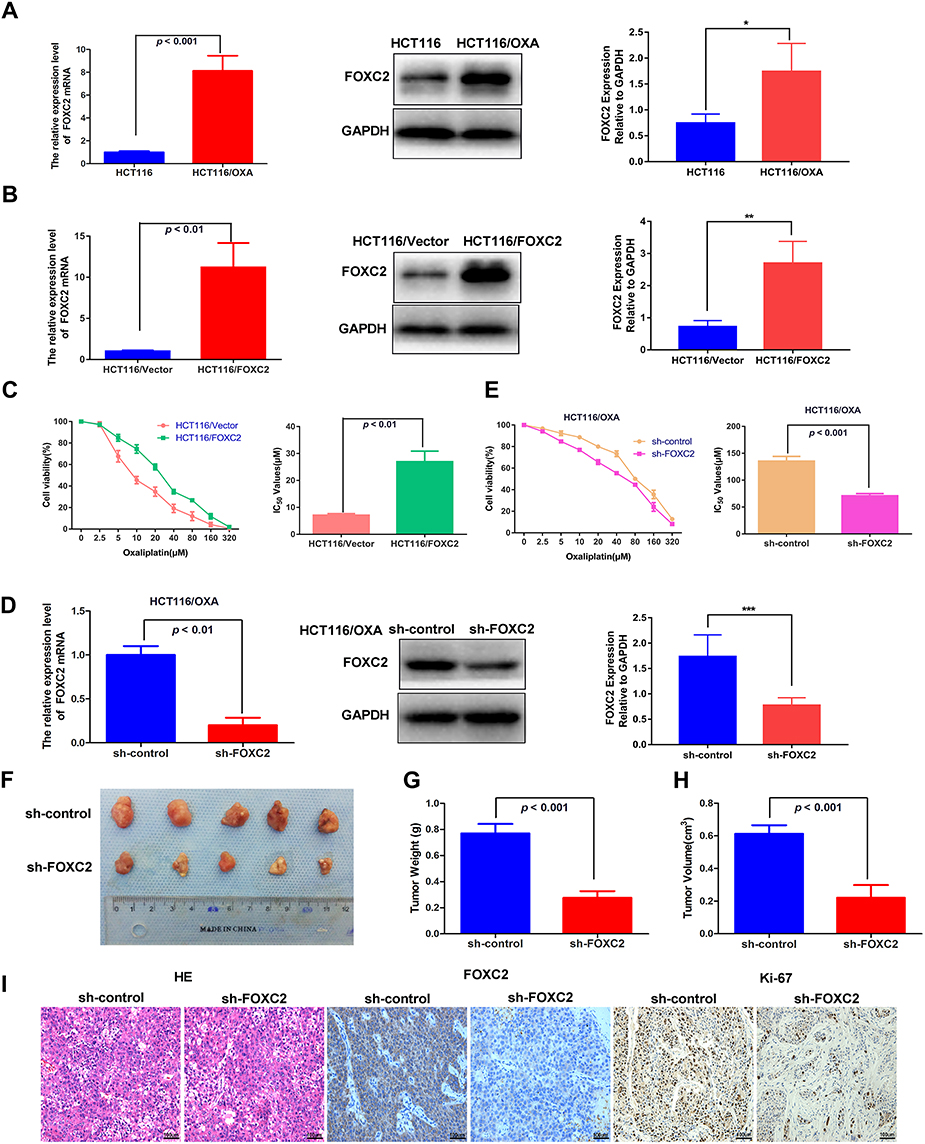 |
Figure 2 The effect of FOXC2 dysregulation on the sensitivity of HCT116 and HCT116/OXA cells to OXA. (A) The mRNA and protein levels of FOXC2 in HCT116 and HCT116/OXA cell lines were measured by qRT-PCR and Western blotting. GAPDH was used as the internal control. The differences in FOXC2 expression levels among different groups were tested by one-way ANOVA. All values represent the average of three independent experiments (mean ± SD). (B) mRNA and protein levels of FOXC2 in HCT116-FOXC2 cells. (C) CCK-8 assays were used to analyze the effects of FOXC2 overexpression on the IC50 values of HCT116. (D) qRT-PCR and Western blot detection of FOXC2 mRNA and protein expression after FOXC2 knockdown in HCT116/OXA cells. (E) The influence of FOXC2 knockdown on the sensitivity of HCT116/OXA cells to OXA was assessed by CCK-8 assays. (F) HCT116/OXA sh-control cells or sh-FOXC2 cells were subcutaneously injected into the left flank regions of nude mice (n = 5 in each group). Four weeks after subcutaneous implantation and OXA treatment, the tumors were removed from the mice. (G) The tumor weights were calculated using a precision electronic balance (p < 0.001). (H) The average tumor volumes in each group were measured by the following formula: V (cm3) = (L ×W2) ×0.5 (L: tumor length, W: tumor width) (p < 0.001). The data are shown as the mean tumor volumes ± SDs. (I) HE and immunohistochemical staining of FOXC2 and Ki67 expression in subcutaneously implanted tumors (scale bars = 100μm). The data are from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001. |
Overexpression of FOXC2 Reduced the Sensitivity of HCT116 Cells to OXA
To assess whether FOXC2 plays a role in drug resistance in CRC, we first established a stable cell line overexpressing FOXC2 in chemosensitive HCT116 cells (HCT116/FOXC2 cells). The results of qRT-PCR and Western blotting confirmed the efficiency of FOXC2 overexpression (Figure 2B). Compared with control vector treatment, stable overexpression of FOXC2 inhibited the sensitivity of HCT116 cells to various concentrations of OXA and significantly increased the IC50 of OXA in HCT116 cells (Figure 2C).
Downregulation of FOXC2 Increased the Sensitivity of HCT116/OXA Cells to OXA Both in vitro and in vivo
To further confirm the effects of FOXC2 on OXA sensitivity, we knocked down FOXC2 expression in HCT116/OXA cells and established a stable FOXC2 knockdown cell line. The results of qRT-PCR and Western blotting confirmed the success of the knockdown assay (Figure 2D). Regarding OXA sensitivity, the CCK-8 assay results demonstrated that knockdown of FOXC2 enhanced the sensitivity of HCT116/OXA cells to OXA, which significantly reduced the IC50 of OXA in HCT116/OXA cells (Figure 2E).
To determine whether knockdown of FOXC2 promotes tumor sensitivity to OXA in vivo, we then established a CRC xenograft mouse model using HCT116/OXA cells (sh-control) and FOXC2 knockdown HCT116/OXA cells (sh-FOXC2) for further experiments. HCT116/OXA sh-control cells or sh-FOXC2 cells were subcutaneously implanted into the left flank regions of nude mice (n = 5 in each group). Four weeks after HCT116/OXA cell implantation, mice receiving OXA treatment in the sh-control group exhibited a larger average tumor size than mice in the sh-FOXC2 group (Figure 2F). Compared to sh-control treatment, FOXC2 knockdown resulted in a significant decrease in tumor weight and a marked decrease in tumor volume (p < 0.001, respectively; Figure 2G and H). As shown in Figure 2I, HE staining confirmed the successful establishment of the xenograft tumor model. Immunohistochemical assays on the subcutaneous tumors confirmed the knockdown efficiency of FOXC2 in vivo (Figure 2I). FOXC2 knockdown in HCT116/OXA cells led to downregulated expression of the proliferation index Ki-67. Taken together, these results identified FOXC2 as an important factor mediating the OXA sensitivity of CRC cells.
OXA-Resistant Cells Acquired an EMT Phenotype
We observed an obvious difference between the parental cells and OXA-resistant cells. As shown in Figure 3A, OXA-resistant HCT116/OXA cells displayed a dispersed, elongated spindle-like mesenchymal morphology, while HCT116 cells presented an epithelial cobblestone morphology (Figure 3A). This morphological change implied that CRC cells with OXA resistance might have undergone an EMT process. Previous studies have demonstrated that OXA-resistant CRC cells acquire an EMT phenotype.11
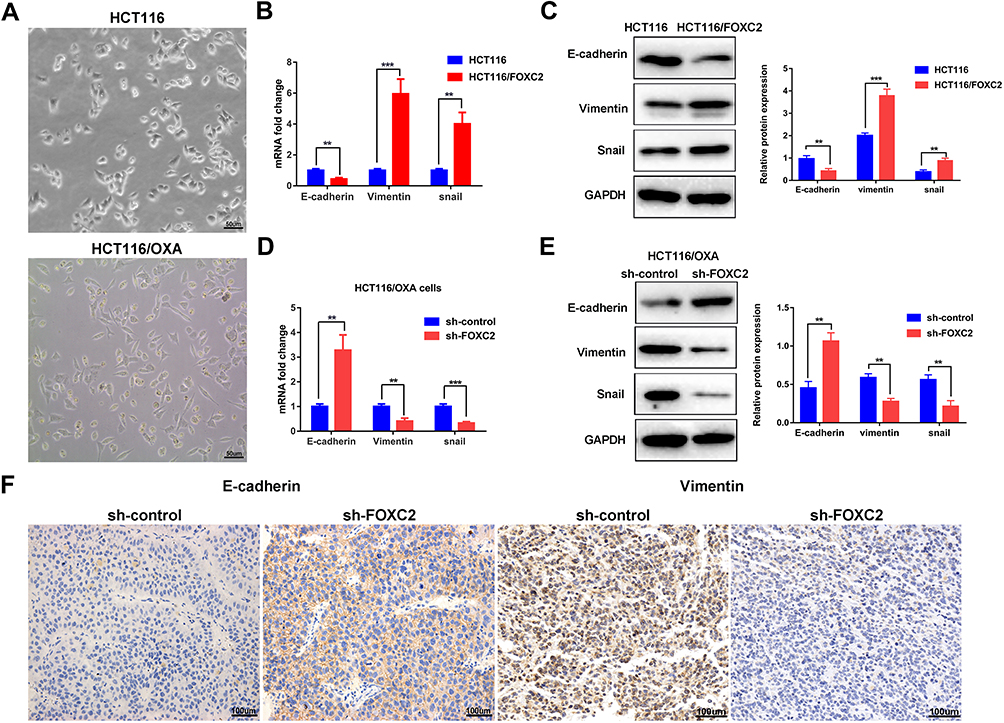 |
Figure 3 FOXC2 affected the oxaliplatin resistance of human colorectal cancer cells via the induction of EMT progression. (A) Morphological changes in OXA-resistant HCT116/OXA cells: elongated spindle-like mesenchymal morphology (scale bars = 50μm). (B) The mRNA expression of E-cadherin, Vimentin and Snail in HCT116-FOXC2 cells and control cells were determined by qRT-PCR. The data are presented as the means ± SDs. (C) Western blot analysis was used to detect the protein expression levels of E-cadherin, Vimentin and Snail in HCT116 and HCT116-FOXC2 cells. (D & E) Knockdown of FOXC2 in HCT116/OXA cells led to downregulation of Vimentin and Snail, while E-cadherin was upregulated both at the mRNA and protein levels. (F) Vimentin and E-cadherin expression in sh-FOXC2 tumors and sh-control tumors was visualized using immunohistochemical staining (scale bars =100μm). The data are presented as the means ± SDs of three independent experiments. **p < 0.01, ***p < 0.001. |
EMT Played an Important Role in FOXC2-Regulated CRC Drug Resistance
We then assessed whether FOXC2 plays a role in the acquisition of the EMT phenotype by OXA-resistant cells. The qRT-PCR results showed that compared with control vector treatment, FOXC2 overexpression suppressed the mRNA expression of E-cadherin, accompanied by elevated mRNA expression levels of Vimentin and Snail (Figure 3B). Western blotting confirmed the results of qRT-PCR at the protein level (Figure 3C). Furthermore, knockdown of FOXC2 in OXA-resistant HCT116/OXA cells by shRNA resulted in downregulation of the mesenchymal markers Vimentin and Snail but upregulation of E-cadherin both at the mRNA and protein levels (Figure 3D and E). Immunohistochemical staining for tumors derived from sh-FOXC2 cells exhibited downregulation of Vimentin but upregulation of E-cadherin compared with the corresponding levels in sh-control cells in vivo (Figure 3F). These data indicated that FOXC2 affects EMT progression in CRC drug resistance both in vitro and in vivo.
Activation of the MAPK/ERK Signaling Pathway Was Required for FOXC2-Regulated OXA Resistance
Mitogen-activated protein kinase (MAPK)/ERK signaling has been implicated in drug resistance. We further investigated whether the MAPK/ERK pathway is involved in FOXC2-regulated OXA resistance. As shown in Figure 4A, we found that knockdown of FOXC2 significantly decreased the level of phospho- ERK1/2 in OXA-resistant HCT116/OXA cells, while the level of total ERK1/2 showed no obvious difference. The overexpression of FOXC2 increased the phosphorylation of ERK1/2 in HCT116 cells (Figure 4B). To further confirm the role of the MAPK/ERK signaling pathway in FOXC2-regulated OXA sensitivity, we utilized the MAPK/ERK kinase inhibitor SCH772984 in HCT116/FOXC2 cells. Treatment with SCH772984 reversed the changes in the E-cadherin, Vimentin and phospho-ERK1/2 proteins in HCT116/FOXC2 cells (Figure 4B). Importantly, the results of CCK-8 assays revealed that the MAPK/ERK kinase inhibitor could sensitize HCT116/FOXC2 cells to OXA treatment by decreasing the IC50 of OXA in HCT116/FOXC2 cells (Figure 4C). These data suggested that FOXC2 induces EMT and thus contributes to OXA resistance in colorectal cancer cells via the activation of the MAPK/ERK signaling pathway.
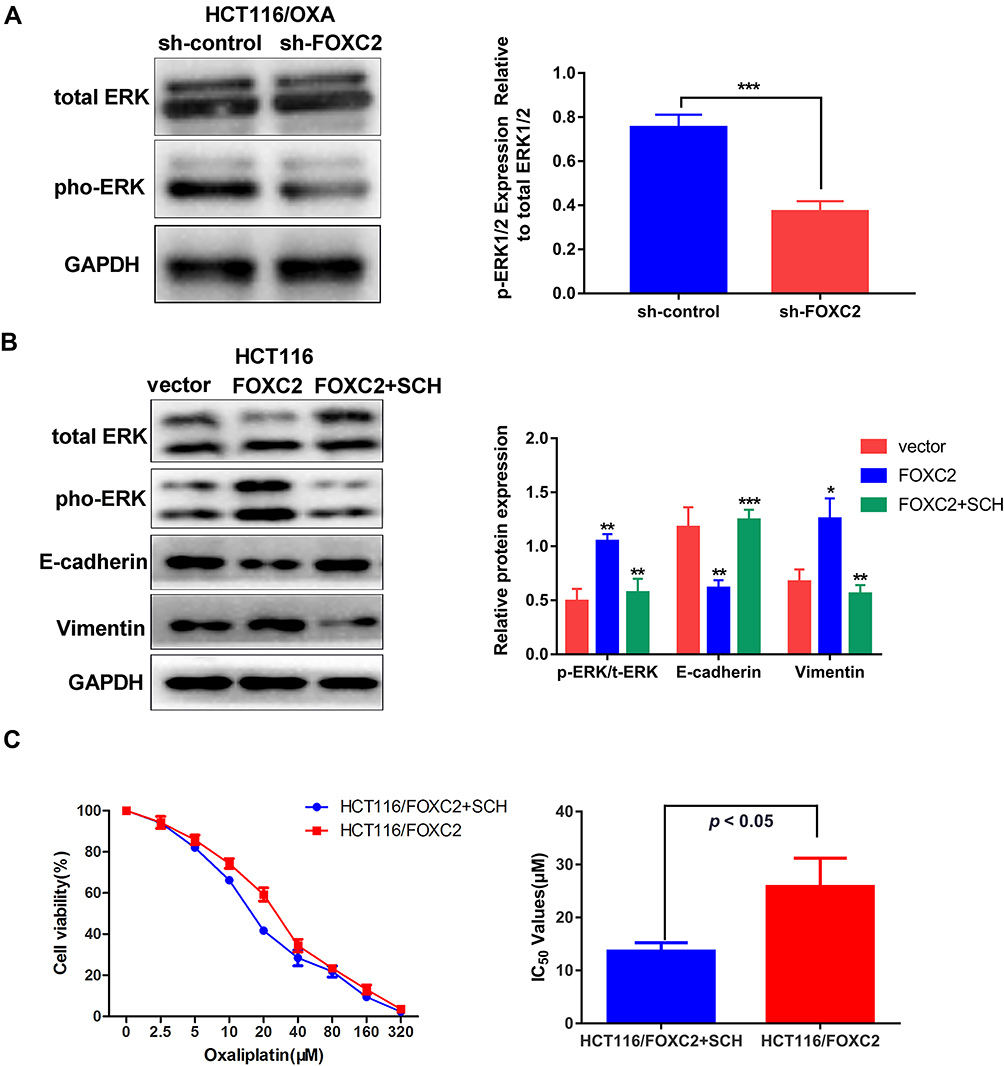 |
Figure 4 Activation of the MAPK/ERK signaling pathway was involved in FOXC2-regulated oxaliplatin resistance. (A) The ERK1/2 and phospho-ERK1/2 levels after FOXC2 silencing in OXA-resistant HCT116/OXA cells were determined by Western blotting. (B) Western blotting detection of E-cadherin, Vimentin, ERK1/2 and phospho-ERK1/2 proteins in HCT116/FOXC2 cells after MAPK/ERK kinase inhibitor SCH772984 treatment. GAPDH served as an internal control. (C) MAPK/ERK kinase inhibitor SCH772984 could reduce the IC50 of OXA in HCT116/FOXC2 cells (p < 0.05). IC50 values of HCT116/FOXC2 group: 13.65 ± 0.92μM, HCT116/FOXC2+ SCH772984 group: 25.85 ± 3.09μM. All values stand for the average of three independent experiments (means ± SDs). *p < 0.05, **p < 0.01, ***p < 0.001. |
Discussion
In recent decades, researchers have made a great effort to advance the treatment of CRC, including the development of radio- and chemotherapy combination treatments. However, patient prognosis is still poor due to the lack of therapeutic success. Chemotherapeutic resistance is a crucial cause of recurrence and leads to treatment cessation. New therapeutic strategies that overcome chemoresistance safely provide a promising approach for effective CRC treatment. In the present study, we first investigated the mRNA and protein expression of FOXC2 and found that FOXC2 was significantly upregulated in OXA-resistant colorectal cancer cells. In the parental HCT116 CRC cells, overexpression of FOXC2 decreased OXA sensitivity, while knockdown of FOXC2 sensitized HCT116/OXA cells to OXA treatment both in vitro and in vivo. OXA-resistant HCT116/OXA cells acquired a mesenchymal phenotype, but manipulating the expression of FOXC2 impaired EMT progression. Moreover, the signaling pathway that participated in FOXC2-regulated OXA resistance was the MAPK/ERK pathway, and an inhibitor of MAPK/ERK reversed FOXC2-induced OXA resistance and EMT in HCT116/OXA cells. These data indicate that specific inhibition of FOXC2 could be a novel strategy for the treatment of CRC patients.
A variety of mechanisms, such as ER stress, DNA damage, autophagy and EMT, have been reported to contribute to drug resistance in CRC.23–26 In general, EMT is traditionally thought to contribute to tumor invasion and metastasis. In recent years, accumulated evidence has indicated that EMT plays an essential role in the regulation of drug sensitivity in some human malignant cancers.27–29 Tumor cells that undergo EMT are resistant to chemotherapy. In addition, EMT progression can interact with other mechanisms to induce chemoresistance. For example, autophagy stimulation downregulates Snail and Slug, thus reversing EMT progression in glioblastoma.30 Alizadeh et al found that TGF-β1 can induce both autophagy and EMT in non-small cell lung cancer cells and that autophagy inhibition suppresses EMT progression.31 In addition, EMT is correlated with stem cell-like characteristics of cancer cells, an important contributor to chemoresistance progression.32 EMT can induce the stem cell-like phenotype, and Snail can modulate cancer stem cell properties to promote tumor progression.33,34 In our study, we found that OXA-resistant cells exhibit an EMT phenotype and are regulated by FOXC2, indicating that FOXC2-induced OXA resistance is modulated via EMT.
The MAPK/ERK signaling pathway is acknowledged to play a critical role in multiple cellular processes, including proliferation, differentiation and migration.35 A previous study reported that FOXC2 promotes colorectal cancer growth through the activation of the MAPK and AKT signaling pathways.21 Downregulation of FOXC2 has been shown to promote apoptosis induced by 5-fluorouracil through the activation of the MAPK and AKT pathways in colorectal cancer.36 Similarly, Li et al found that FOXC2 can promote EMT in CDDP-resistant ovarian cancer cells via the activation of the ERK or AKT/GSK-3β signaling pathways.37 Moreover, many studies have reported that the activation of AKT or ERK signaling can upregulate the nuclear expression and transcriptional activity of Snail, thus triggering the EMT process. Therefore, we hypothesize that FOXC2 regulates EMT to promote OXA resistance through the MAPK/ERK pathway in CRC. In our study, we demonstrated that FOXC2 modulates the expression of EMT-related proteins (E-cadherin, Vimentin and Snail) and that knockdown of FOXC2 in HCT116/OXA cells reverses sensitivity to OXA both in vitro and in vivo. As we assumed, MAPK/ERK signaling is activated in OXA-resistant HCT116/OXA cells, and inhibition of MAPK/ERK can sensitize HCT116/OXA cells to OXA treatment and reverse EMT in HCT116/OXA cells, indicating that FOXC2 regulates OXA resistance mediated by EMT activation via MAPK/ERK. In conclusion, this study demonstrates that the molecular mechanism of FOXC2-activated EMT in FOXC2-regulated OXA resistance in CRC operates by activating the MAPK/ERK signaling pathway. Specifically, targeting FOXC2 may be a potential strategy for the effective treatment of human colorectal cancer.
Ethics Approval
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and the protocols for treating animals were approved by the Animal Use Committee of Central South University (20170232).
Acknowledgments
This study was supported by grants from National Natural Science Foundation of China (Nos. 81070362, 81172470, 81372629, 81772627, 81874073, and 81974384). National Key R & D Program of China (No. 2018YFC1313303), two key projects from the Natural Science Foundation of Hunan Province (Nos. 2015JC3021 and 2016JC2037), and a project from China Cancer Elite Team Innovative Grant (No. 2016JY0006).
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.v69.1
2. Sjo OH, Berg M, Merok MA, et al. Peritoneal carcinomatosis of colon cancer origin: highest incidence in women and in patients with right-sided tumors. J Surg Oncol. 2011;104(7):792–797. doi:10.1002/jso.21959
3. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. doi:10.1002/ijc.29210
4. Fuertes MA, Castilla J, Alonso C, Pérez JM. Novel concepts in the development of platinum antitumor drugs. Curr Med Chem Anticancer Agents. 2002;2(4):539–551. doi:10.2174/1568011023353958
5. Li J, Chen Y, Zhao J, Kong F, Zhang Y. miR-203 reverses chemoresistance in p53-mutated colon cancer cells through downregulation of Akt2 expression. Cancer Lett. 2011;304(1):52–59. doi:10.1016/j.canlet.2011.02.003
6. Lee DW, Han SW, Bae JM, et al. Tumor mutation burden and prognosis in colorectal cancer patients treated with adjuvant fluoropyrimidine and oxaliplatin. Clin Cancer Res. 2019. doi:10.1158/1078-0432.CCR-19-1105
7. Tao YM, Huang JL, Zeng S, et al. BTB/POZ domain-containing protein 7: epithelial-mesenchymal transition promoter and prognostic biomarker of hepatocellular carcinoma. Hepatology. 2013;57(6):2326–2337. doi:10.1002/hep.26268
8. Zeng S, Zhang Y, Ma J, et al. BMP4 promotes metastasis of hepatocellular carcinoma by an induction of epithelial-mesenchymal transition via upregulating ID2. Cancer Lett. 2017;390:67–76. doi:10.1016/j.canlet.2016.12.042
9. Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15(6):740–746. doi:10.1016/j.ceb.2003.10.006
10. Deng G, Zeng S, Ma J, et al. The anti-tumor activities of Neferine on cell invasion and oxaliplatin sensitivity regulated by EMT via Snail signaling in hepatocellular carcinoma. Sci Rep. 2017;7:41616. doi:10.1038/srep41616
11. Guo C, Ma J, Deng G, et al. ZEB1 promotes oxaliplatin resistance through the induction of epithelial - mesenchymal transition in colon cancer cells. J Cancer. 2017;8(17):3555–3566. doi:10.7150/jca.20952
12. Ma J, Zeng S, Zhang Y, et al. BMP4 promotes oxaliplatin resistance by an induction of epithelial-mesenchymal transition via MEK1/ERK/ELK1 signaling in hepatocellular carcinoma. Cancer Lett. 2017;411:117–129. doi:10.1016/j.canlet.2017.09.041
13. Bollong MJ, Pietilä M, Pearson AD, et al. A vimentin binding small molecule leads to mitotic disruption in mesenchymal cancers. Proc Natl Acad Sci U S A. 2017;114(46):E9903–E9912. doi:10.1073/pnas.1716009114
14. Wang YW, Yin CL, Zhang HY, et al. High expression of forkhead box protein C2 is related to poor prognosis in human gliomas. Asian Pac J Cancer Prev. 2014;15(24):10621–10625. doi:10.7314/APJCP.2014.15.24.10621
15. Nishida N, Mimori K, Yokobori T, et al. FOXC2 is a novel prognostic factor in human esophageal squamous cell carcinoma. Ann Surg Oncol. 2011;18(2):535–542. doi:10.1245/s10434-010-1274-y
16. Zhu JL, Song YX, Wang ZN, et al. The clinical significance of mesenchyme forkhead 1 (FoxC2) in gastric carcinoma. Histopathology. 2013;62(7):1038–1048. doi:10.1111/his.12132
17. Cai J, Tian AX, Wang QS, et al. FOXF2 suppresses the FOXC2-mediated epithelial-mesenchymal transition and multidrug resistance of basal-like breast cancer. Cancer Lett. 2015;367(2):129–137. doi:10.1016/j.canlet.2015.07.001
18. Paranjape AN, Soundararajan R, Werden SJ, et al. Inhibition of FOXC2 restores epithelial phenotype and drug sensitivity in prostate cancer cells with stem-cell properties. Oncogene. 2016;35(46):5963–5976. doi:10.1038/onc.2015.498
19. Pietilä M, Vijay GV, Soundararajan R, et al. FOXC2 regulates the G2/M transition of stem cell-rich breast cancer cells and sensitizes them to PLK1 inhibition. Sci Rep. 2016;6:23070. doi:10.1038/srep23070
20. Zhou Z, Zhang L, Xie B, et al. FOXC2 promotes chemoresistance in nasopharyngeal carcinomas via induction of epithelial mesenchymal transition. Cancer Lett. 2015;363(2):137–145. doi:10.1016/j.canlet.2015.04.008
21. Cui YM, Jiang D, Zhang SH, et al. FOXC2 promotes colorectal cancer proliferation through inhibition of FOXO3a and activation of MAPK and AKT signaling pathways. Cancer Lett. 2014;353(1):87–94. doi:10.1016/j.canlet.2014.07.008
22. Du Y, Li D, Li N, et al. POFUT1 promotes colorectal cancer development through the activation of Notch1 signaling. Cell Death Dis. 2018;9(10):995. doi:10.1038/s41419-018-1055-2
23. Ceballos MP, Rigalli JP, Ceré LI, Semeniuk M, Catania VA, Ruiz ML. ABC transporters: regulation and association with multidrug resistance in hepatocellular carcinoma and colorectal carcinoma. Curr Med Chem. 2019;26(7):1224–1250.
24. Hsu HH, Chen MC, Baskaran R, et al. Oxaliplatin resistance in colorectal cancer cells is mediated via activation of ABCG2 to alleviate ER stress induced apoptosis. J Cell Physiol. 2018;233(7):5458–5467. doi:10.1002/jcp.26406
25. Sun L, Ke J, He Z, et al. HES1 promotes colorectal cancer cell resistance to 5-Fu by inducing of EMT and ABC transporter proteins. J Cancer. 2017;8(14):2802–2808. doi:10.7150/jca.19142
26. Wang J, Xie S, Yang J, et al. The long noncoding RNA H19 promotes tamoxifen resistance in breast cancer via autophagy. J Hematol Oncol. 2019;12(1):81. doi:10.1186/s13045-019-0747-0
27. Fischer KR, Durrans A, Lee S, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527(7579):472–476. doi:10.1038/nature15748
28. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14(10):611–629. doi:10.1038/nrclinonc.2017.44
29. Zheng X, Carstens JL, Kim J, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527(7579):525–530. doi:10.1038/nature16064
30. Catalano M, D’Alessandro G, Lepore F, et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol. 2015;9(8):1612–1625. doi:10.1016/j.molonc.2015.04.016
31. Alizadeh J, Glogowska A, Thliveris J, et al. Autophagy modulates transforming growth factor beta 1 induced epithelial to mesenchymal transition in non-small cell lung cancer cells. Biochim Biophys Acta Mol Cell Res. 2018;1865(5):749–768. doi:10.1016/j.bbamcr.2018.02.007
32. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9(4):265–273. doi:10.1038/nrc2620
33. Matsumoto T, Yokoi A, Hashimura M, Oguri Y, Akiya M, Saegusa M. TGF-β-mediated LEFTY/Akt/GSK-3β/Snail axis modulates epithelial-mesenchymal transition and cancer stem cell properties in ovarian clear cell carcinomas. Mol Carcinog. 2018;57(8):957–967. doi:10.1002/mc.v57.8
34. Singla M, Kumar A, Bal A, Sarkar S, Bhattacharyya S. Epithelial to mesenchymal transition induces stem cell like phenotype in renal cell carcinoma cells. Cancer Cell Int. 2018;18:57. doi:10.1186/s12935-018-0555-6
35. Buscà R, Pouysségur J, Lenormand P. ERK1 and ERK2 map kinases: specific roles or functional redundancy? Front Cell Dev Biol. 2016;4:53. doi:10.3389/fcell.2016.00053
36. Yang C, Cui X, Dai X, Liao W. Downregulation of Foxc2 enhances apoptosis induced by 5-fluorouracil through activation of MAPK and AKT pathways in colorectal cancer. Oncol Lett. 2016;11(2):1549–1554. doi:10.3892/ol.2016.4097
37. Li C, Ding H, Tian J, et al. Forkhead box protein C2 (FOXC2) promotes the resistance of human ovarian cancer cells to cisplatin in vitro and in vivo. Cel Physiol Biochem. 2016;39(1):242–252. doi:10.1159/000445620
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.