Back to Journals » Drug Design, Development and Therapy » Volume 18
Forsythoside B Mitigates Monocrotaline-Induced Pulmonary Arterial Hypertension via Blocking the NF-κB Signaling Pathway to Attenuate Vascular Remodeling
Authors Liu J, Fang G, Lan C, Qiu C, Yao L, Zhang Q, Hu J, Zhang Y, Yang Y, Zhang Y
Received 14 November 2023
Accepted for publication 24 February 2024
Published 11 March 2024 Volume 2024:18 Pages 767—780
DOI https://doi.org/10.2147/DDDT.S444605
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Jiying Liu,1– 3 Guangyao Fang,2,4 Cong Lan,2 Chenming Qiu,5 Li Yao,2 Qian Zhang,2 Jingtang Hu,2 Yaolei Zhang,6 Yongjian Yang,1,2 Yan Zhang1,2
1Department of Cardiology, The Affiliated Hospital of Southwest Medical University, Luzhou, Sichuan, 646000, People’s Republic of China; 2Department of Cardiology, General Hospital of Western Theater Command, Chengdu, Sichuan, 610083, People’s Republic of China; 3Department of Cardiology, The Third People’s Hospital of Yibin, Yibin, Sichuan, 644000, People’s Republic of China; 4College of Medicine, Southwest Jiaotong University, Chengdu, Sichuan, 610083, People’s Republic of China; 5Department of Burn and Plastic Surgery, General Hospital of Western Theater Command, Chengdu, Sichuan, 610083, People’s Republic of China; 6Basic Medical Laboratory, General Hospital of Western Theater Command, Chengdu, Sichuan, 610083, People’s Republic of China
Correspondence: Yongjian Yang, Department of Cardiology, General Hospital of Western Theater Command, Chengdu, Sichuan, 610083, People’s Republic of China, Tel +86-13880827421, Email [email protected] Yan Zhang, Department of Cardiology, General Hospital of Western Theater Command, Chengdu, Sichuan, 610083, People’s Republic of China, Tel +86-18980530960, Email [email protected]
Purpose: Pulmonary arterial hypertension (PAH) is a devastating disease with little effective treatment. The proliferation of pulmonary artery smooth muscle cells (PASMCs) induced by the nuclear factor-κB (NF-κB) signaling activation plays a pivotal role in the pathogenesis of PAH. Forsythoside B (FTS•B) possesses inhibitory effect on NF-κB signaling pathway. The present study aims to explore the effects and mechanisms of FTS•B in PAH.
Methods: Sprague–Dawley rats received monocrotaline (MCT) intraperitoneal injection to establish PAH model, and FTS•B was co-treated after MCT injection. Right ventricular hypertrophy and pulmonary artery pressure were measured by echocardiography and right heart catheterization, respectively. Histological alterations were detected by H&E staining and immunohistochemistry. FTS•B’s role in PASMC proliferation and migration were evaluated by CCK-8 and wound healing assay. To investigate the underlying mechanisms, Western blotting, immunofluorescence staining and ELISA were conducted. The NF-κB activator PMA was used to investigate the role of NF-κB in FTS•B’s protective effects against PAH.
Results: FTS•B markedly alleviated MCT-induced vascular remodeling and pulmonary artery pressure, and improved right ventricular hypertrophy and survival. FTS•B also reversed PDGF-BB-induced PASMC proliferation and migration, decreased PCNA and CyclinD1 expression in vitro. The elevated levels of IL-1β and IL-6 caused by MCT were decreased by FTS•B. Mechanistically, MCT-triggered phosphorylation of p65, IκBα, IKKα and IKKβ was blunted by FTS•B. FTS•B also reversed MCT-induced nuclear translocation of p65. However, all these protective effects were blocked by PMA-mediated NF-κB activation.
Conclusion: FTS•B effectively attenuates PAH by suppressing the NF-κB signaling pathway to attenuate vascular remodeling. FTS•B might be a promising drug candidate with clinical translational potential for the treatment of PAH.
Keywords: Forsythoside B, pulmonary arterial hypertension, NF-κB signaling pathway, pulmonary vascular remodeling, pulmonary artery smooth muscle cell
A Letter to the Editor has been published for this article.
Introduction
Pulmonary arterial hypertension (PAH) is defined hemodynamically by a mean pulmonary arterial pressure greater than 20 mmHg, a pulmonary artery wedge pressure lower than 15 mmHg, and a pulmonary vascular resistance of at least 3 wood units.1 PAH is known as a cardiovascular malignancy, and remains a key cause of death worldwide.2 Elevated pulmonary artery pressure in PAH leads to increased afterload of the right heart, resulting in decompensated hypertrophy of the right heart, ventricular dilation, and eventually right heart failure and fatality.3 Furthermore, in addition to posing a risk on its own, PAH can also complicate the identification and management of common illnesses such as chronic obstructive pulmonary disease and interstitial lung diseases, consequently exacerbating the progression of these conditions and increasing mortality rates.4 Currently, the treatment for PAH focuses on addressing the dysfunction of the endothelium by blocking the endothelin pathway and improving the pathways of prostacyclin (PGI2) and nitric oxide (NO).5 Despite significant advancements in the treatment strategy, achieving a full cure for PAH remains unattainable. The 3-year survival rate of PAH patients is only 68%,6 and the available medications are typically costly and accompanied by noticeable adverse reactions. Therefore, it is urgent to develop safe and effective medicines to fight against PAH.
Nuclear factor-κB (NF-κB) represents a family of transcription factors and plays an essential role in the regulation of inflammatory and immune responses.7 Many studies have revealed that NF-κB is involved in the pathogenesis of various cardiac diseases. For instance, activation of NF-κB signaling pathway promotes cardiac remodeling after myocardial infarction through unregulated inflammatory response;8 in streptozotocin-treated mice, NF-κB aggravates ferroptosis to promote the progress of diabetic cardiomyopathy.9 Recent years, it is widely accepted that inflammation is one of essential features of PAH, and both NF-κB and interlukin (IL)-1β have been identified to facilitate inflammation in human PAH and animal PAH models.10–12 Mitani Y et al reported that inhibition of NF-κB by pyrrolidine dithiocarbamate ameliorates pulmonary hypertension in MCT-induced PAH rats;13 Zinc finger protein A20 attenuates hypoxia-induced PAH by inhibiting NF-κB and PASMC proliferation.14 Hence, new strategies suppressing NF-κB activation and inflammation may provide opportunities to treat PAH.
Forsythoside B (FTS•B), derived from fructus forsythia, is a phenylethanoid glycoside with molecular formula of C34H44O19 (Figure 1) and is extensively used to treat inflammation, pyrexia, gonorrhea, carbuncles and erysipelas in China, Japan and Korea.15 By suppressing NF-κB signaling, FTS•B has the potential to effectively hinder the activation of internal inflammatory elements, positioning it as a promising pharmaceutical option for addressing inflammatory conditions.16,17 For example, FTS•B can attenuate memory impairment and neuroinflammation by inhibiting NF-κB signaling in Alzheimer’s disease.18 In addition, FTS•B was reported to inhibit the inflammatory response to improve cardiac function in ischemia‒reperfusion (I/R) injury.19 Nevertheless, the role of FTS•B in PAH is still unclear. Therefore, this study aims to investigate the effects and mechanisms of FTS•B on PAH through a series of in vivo and in vitro experiments.
 |
Figure 1 The chemical construction of forsythoside B. |
Materials and Methods
Drugs and Reagents
The following drugs and reagents were used monocrotaline (MCT, # C2401, Sigma‒Aldrich, St. Louis, MO, USA), phorbol-12-myristate-13-acetate (PMA, # P8139, Sigma‒Aldrich), Forsythoside B (FTS•B, # RFS-L01311807014, Chengdu Herbpurity CO., LTD, Chengdu, China), recombinant human platelet-derived growth factor-BB (PDGF-BB, # 100–14B, PeproTech, NJ, USA), anti-β-actin (# 4970S, Cell Signaling Technology, Boston, USA), anti-phospho-NF-κB p65 (p-p65, # 3033S, CST), anti-NF-κB p65 (p65, # 8242S, CST), anti-phospho-IκBα (p-IκBα, # 9246S, CST), anti-IκBα (# 4814, CST), anti-Cyclin D1 (# 55506S, CST), anti-rabbit IgG (HRP-linked antibody) (# 7074 CST), anti-mouse IgG (HRP-linked antibody) (# 5257, CST), anti-IKK alpha (IKKα, # R24674, ZENBIO, Chengdu, China), anti-IKK beta (IKKβ, # 381592, ZENBIO), anti-phospho-IKK alpha (p-IKKα, # 310201, ZENBIO), anti-phospho-IKK beta (p-IKKβ, # 347345, ZENBIO), goat anti-mouse IgG H&L (Alexa Fluor 488)(# 550036, ZENBIO), goat anti-rabbit IgG H&L (Alexa Fluor 594) (# 550043, ZENBIO), anti-CD68 (# R381310, ZENBIO), anti-PCNA (# 10205-2-AP, Proteintech, Wuhan, China), anti-Ki67 (# GB111499, Servicebio, Wuhan, China), anti-alpha smooth muscle actin (α-SMA, # AB7817, Abcam, Cambridge, UK), anti-CD31 (# AB182981, Abcam), an ECL chemiluminescent substrate kit (Hypersensitive) (# BL523B, Biosharp, Hefei, China), cell counting kit 8 (CCK-8, # BS350B, Biosharp), a hematoxylin and eosin staining kit (H&E, # BL700B, Biosharp), 4% paraformaldehyde (# P0099, Beyotime, Shanghai, China), 10% fetal bovine serum (FBS, # C0234, Beyotime), a whole cell lysis assay kit (# KGP250, KeyGEN BioTECH, Jiangsu, China), an enhanced BCA protein assay kit (# P0010S, Beyotime), an immunohistochemical kit (# SP-9000, ZSGB, Beijing, China), rat interleukin 1 beta (IL-1β) ELISA kit (# JL20884, Jianglai Biological, Shanghai, China), rat interleukin 6 (IL-6) ELISA kit (# JL20896, Jianglai Biological) and rat interleukin 6 (IL-10) ELISA kit (# JL20884, Jianglai Biological).
Animals
The animal experiments were approved by the Animal Research Ethics Committee of the Western Theater Command General Hospital (2022EC2-007). The guidelines of the National Research Council’s Guide for the Care and Use of Laboratory Animals were followed. Male Sprague–Dawley rats (6 weeks old, 200 ± 20 g) were obtained from Chengdu Dashuo Biotechnology Co., Ltd. All rats were raised at 22–24°C and 55–65% relative humidity with 12/12 h of light and dark; they had free access to water and food.
Rats were randomly assigned into four groups as follows: control group (n = 20), MCT group (n = 20), MCT + FTS•B group (n = 20) and MCT + FTS•B + PMA group (n = 20). PMA, a NF-κB stimulant, was used to further explore the underlying mechanisms. The MCT group received a single dose of MCT (60 mg/kg) on the first day and an equal volume of saline daily; the MCT + FTS•B group was administered MCT (60 mg/kg) on the first day and FTS•B (20 mg/kg) once a day;19,20 the MCT + FTS•B + PMA group was administered MCT (60 mg/kg) on the first day and FTS•B (20 mg/kg) plus PMA (5 mg/kg) once a day; the control group received an equal volume of saline daily; all of the drugs were intraperitoneally injected (i.p.) and the treatment lasted for 21 days. During this period, the death events in each group were recorded for survival analysis. Before being sacrificed, all the animals were subjected to echocardiographic analyses and right heart catheterization, then the blood, lungs and hearts were harvested.
Echocardiographic Analyses
A high-resolution ultrasound imaging system (Vevo 3100LT, Visual Sonics, Toronto, Ontario, Canada) was utilized for echocardiography. After the chest was shaved, all the rats were anesthetized by inhalation of isoflurane through a facemask. Pulse-wave Doppler examination was used to record pulmonary artery blood outflow at the annulus of the pulmonary valve in the parasternal short-axis view to measure the pulmonary arterial acceleration time (PAT) and pulmonary arterial ejection time (PET).21,22 The right ventricle free wall thickness (RVFWT) and right ventricle end-diastolic diameter (RVEDD) were calculated during end-diastole at the parasternal long-axis RV outflow tract level in M-mode.21 At least three consecutive beats were analyzed quantitatively using Vevo LAB analysis software (Vevo LAB 5.5.1).
Hemodynamic Measurements
Rats were anesthetized with phenobarbital, and a micromanometer-tipped pressure catheter (Millar Instruments, Houston, TX, USA) was inserted from the right external jugular vein into the right ventricle and then up to the pulmonary artery. Mean pulmonary artery pressure (mPAP) and right ventricular systolic pressure (RVSP) were recorded with PowerLab system (AD Instruments) and calculated using the LabChart software according to the previous experiments.23–25
Blood Tests and ELISA
Blood samples were collected from the abdominal aorta of rat and were tested using blood cell counter (BC-5000 Vet, Mindray company, Guangzhou, China) to observe blood cells. IL-1β, IL-6 and IL-10 levels in serum and lung tissue were measured by commercial ELISA kits according to the manufacturer’s instructions.
Histological Analyses
To determine the extent of right ventricular hypertrophy, the weight of the right ventricle was divided by the weight of the left ventricle and septum [RV/(LV + S)].26 The left lungs and hearts were immersed in 4% paraformaldehyde for over 48 h and then dehydrated, paraffin-embedded and cut into 4 μm slices. H&E staining was conducted by using a commercial kit. An optical scanning microscope (Olympus Optical Co., Ltd., Japan) was used to examine the tissue morphology. Pulmonary artery medial wall thickness (WT) was determined as follows: 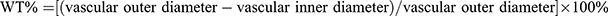 .27
.27
Immunohistochemistry
Lung tissue sections were dewaxed and rehydrated, followed by antigen retrieval. To block endogenous peroxidase, 3% H2O2 was used. After 10 min of incubation with goat serum, the sections were cultured with the specified primary antibodies (diluted 1:200) overnight at 4°C. Afterward, an immunohistochemical kit was used to identify protein localization and expression. The proportion of muscularization of more than 20 arterioles was determined in each section by α-SMA immunohistochemistry.28,29
Cell Culture and Cell Viability Assay
Rat pulmonary artery smooth muscle cells (PASMCs, # CL-048r, SAIOS, Wuhan, China) were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS and 1% penicillin‒streptomycin at 37°C in a 5% CO2 incubator. Passages between 3 and 8 were used for the experiments. All experiments were performed in triplicate. PASMCs were seeded in 96-well plates at a density of 4 × 103 cells per well and incubated with different concentrations of FTS•B (0, 2.5, 5, 10, 20 μmol/L, n = 5) in the presence or absence of PDGF-BB (20 ng/mL) at 37°C for 24 h.18 According to the manufacturer’s instructions, PASMCs were incubated with CCK-8 solution at 37°C for 2 h, and then the optical density was measured at 450 nm.
Wound Healing Assay
PASMCs were cultured in 6-well plates until they reached full confluence. To produce a pristine wound, a pipette tip with a volume of 200 µL was dragged across the center of the plate. Afterward, the cells were randomly separated into the control group (n = 5), PDGF-BB group (cultured with PDGF-BB 20 ng/mL, n = 5) and PDGF-BB + FTS•B group (cultured with PDGF-BB 20 ng/mL and FTS•B 10 μmol/L, n = 5) at 37°C for 24 h. A phase-contrast microscope was used to observe the wound area at both 0 and 24 h. Subsequently, the percentage of the migration area was measured with ImageJ software.15
Immunofluorescence Staining
PASMCs were fixed with 4% paraformaldehyde and lung tissue sections were subjected to dewaxing and antigen retrieval. The PASMCs and lung tissue sections were permeabilized with 0.5% Triton X-100 for 20 min and blocked in immunostaining blocking buffer containing 5% BSA at 37°C for 1 h. The cells and tissue sections were subsequently incubated with the indicated primary antibodies (1:100 dilution) at 4°C overnight and corresponding secondary antibodies (conjugated with Alexa Fluor 488 and 594, 1:1000 dilution) at 37°C for 1 h. Nuclei were counterstained with DAPI for 5 min. Immunofluorescence images were obtained under a laser confocal microscope (Nikon, Japan) or a fluorescence microscope (Nikon, Japan).
Western Blot Analyses
The KeyGEN whole cell lysis assay kit was used to extract the total protein, and an enhanced BCA kit was applied to detect concentrations of total protein. The protein samples were separated by sodium dodecyl sulfate‒polyacrylamide gel electrophoresis (SDS‒PAGE) and then transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% nonfat milk for 1 h and then exposed to specific primary antibodies overnight at 4°C. After washing, the membranes were incubated with secondary antibodies conjugated with horseradish peroxidase for 1 h. The ECL chemiluminescent substrate reagent was then added to the membranes. A chemiluminescent imager (A300, Azure, USA) detected the glowing indication. The protein bands were analyzed by ImageJ.
Statistical Analysis
The values were analyzed by SPSS software (version 25.0) and expressed as the mean ± SD. The Shapiro–Wilk test was used to test the normality of the data distribution. For normally distributed data, one-way ANOVA was used to determine the differences among groups. The survival rates were evaluated by Kaplan–Meier survival analysis and compared using the Log rank test. P < 0.05 was considered statistically significant.
Results
FTS•B Relieved MCT-Induced PAH and Vascular Remodeling in Rats
To test the effects of FTS•B on PAH, rats were first administered a single dose of MCT (60 mg/kg) to establish PAH models, and then subjected to echocardiography and right heart catheterization. Echocardiographic and hemodynamic analysis revealed that rats in the MCT group exhibited significantly decreased PAT and PAT/PET (Figure 2A) and elevated mPAP, RVSP, RVFWT and RVEDD (Figure 2B–D), indicating the successful establishment of PAH models. However, administration of FTS•B significantly inhibited the MCT-induced decreases of PAT and PAT/PET and increases of mPAP, RVSP, RVFWT and RVEDD (Figure 2A–D). FTS•B also markedly improved survival of MCT-treated rats (Figure 2E). Histological analysis and RV/(LV + S) further revealed obvious right ventricular hypertrophy in PAH model rats, while, FTS•B effectively blocked this alteration (Figure 2F). We also observed that FTS•B attenuated pulmonary vascular remodeling, indicated by ameliorated medial thickening and neomuscularization in pulmonary arterioles in MCT-treated rats (Figure 2G–H). In addition, PCNA immunohistochemical staining showed that FTS•B administration markedly attenuated PCNA-positive cells in the pulmonary artery in MCT rats (Figure 2I), suggesting that FTS•B markedly inhibited PASMC proliferation. It is reported that NF-κB-mediated inflammation promotes endothelial-to-mesenchymal transition (EndMT), subsequently the pathogenesis of PAH.30–32 In line with previous studies, we also observed MCT-induced EndMT, while, FTS•B suppressed this phenomenon (Supplemental Figure 1). Furthermore, FTS•B treatment significantly restricted MCT-triggered increases of blood cells, such as total white blood cell, neutrophils, lymphocytes and monocytes, suggesting the anti-inflammatory role of FTS•B in MCT-induced PAH (Supplemental Figure 2A–D). To explore the curative effect of FTS•B, rats were given intraperitoneal injection of FTS•B (20 mg/kg, daily for 21 days) from Day 14 after MCT stimulation. Echocardiographic and hemodynamic analysis were performed at Day 35 and the results showed that FTS•B improved MCT-induced adverse effects on mPAP, RVSP, PAT and PAT/PET, indicating the curative effect of FTS•B on PAH (Supplemental Figure 3A–C). Taken together, these data suggest that MCT administration effectively ameliorated MCT-induced PAH and vascular remodeling in rats.
 |
Figure 2 FTS•B relieved MCT-induced PAH and vascular remodeling in rats. Rats received intraperitoneal injection of MCT with/without cotreatment with FTS•B. (A) Echocardiographic PW Doppler tracing of pulmonary artery outflow. PAT, PET and quantitative PAT and PAT/PET data (n = 6). (B) RVFWT and RVEDD in M-mode echo and quantitative data of RVFWT and RVEDD (n = 6). Quantitative analysis of the mPAP (C) and RVSP (D) (n = 6). (E) Survival curves of the rats during the 21-day period (n = 20). (F) Representative images of H&E staining of heart sections (scale bar = 2 mm) and quantitative analysis of the RV/ (LV + S) (n = 6). (G) Representative images of H&E staining of lung sections (scale bar = 50 μm) and quantitative analysis of the WT (n = 6). (H) Representative images of α-SMA immunohistochemical staining of the pulmonary artery (scale bar = 50 μm). 10–20 pulmonary arteries were quantified per rats and quantitative assessments of muscularized distal arterioles (diameter 25–100 μm, n = 6). (I) Representative images of PCNA immunohistochemical staining of the pulmonary artery (scale bar = 50 μm) and quantitative analysis of PCNA-positive cells in the pulmonary artery (n = 6). The data are presented as the mean ± SD. *p < 0.05, **p < 0.01. Abbreviations: MCT, monocrotaline; FTS•B, forsythoside B; PAH, pulmonary arterial hypertension; mPAP, mean pulmonary arterial pressure; RVSP, right ventricular systolic pressure; PAT, pulmonary artery acceleration time; PCNA, proliferating cell nuclear antigen; PET, pulmonary arterial ejection time; RVFWT, right ventricular free wall thickness; RVEDD, right ventricular end-diastolic diameter; [RV/(LV + S)], weight ratio of the right ventricle divided by the sum of the left ventricle and septum; WT, medial wall thickness of pulmonary artery. |
FTS•B Alleviated the Proliferation and Migration of PASMCs Induced by PDGF-BB in vitro
Excessive proliferation and migration of PASMCs are vital factors in PAH.33 Thus, we examined the impacts of FTS•B on the proliferation and migration of PASMCs. The results of CCK-8 assay indicated that FTS•B exerted no impact on the viability of PASMCs under basal conditions (Figure 3A). Nonetheless, FTS•B mitigated the proliferation of PASMCs induced by PDGF-BB in a dose-dependent manner. When the concentration reached 10 µmol/L, FTS•B exerted the most effective effects in suppressing PASMC proliferation (Figure 3B); therefore, the dose of FTS•B was set at 10 µmol/L in subsequent experiments. Immunofluorescence staining further showed increased proliferation of PASMCs in the PDGF-BB group, as indicated by the increase in the percentage of Ki-67-positive nuclei, and FTS•B treatment ameliorated this change (Figure 3C). Furthermore, Western blot analysis indicated that FTS•B treatment blunted PDGF-BB-induced upregulation of PCNA and CyclinD1 (Figure 3D), implying the inhibitory effect of FTS•B on PASMCs proliferation. The wound healing experiments also demonstrated that PDGF-BB-induced migration of PASMCs was drastically blocked by FTS•B (Figure 3E). These findings suggest that FTS•B may protect against vascular remodeling by inhibiting PASMC proliferation and migration.
 |
Figure 3 FTS•B alleviated the proliferation and migration of PASMCs induced by PDGF-BB in vitro. PASMCs were subjected to PDGF-BB in the presence or absence of FTS•B for 24 h. (A) Cell viability of PASMCs subjected to different concentrations of FTS•B (n = 5). (B) Cell viability of PASMCs subjected to PDGF-BB and different concentrations of FTS•B (n = 5). (C) Representative images of Ki-67 immunofluorescence staining (scale bar = 50 μm, Ki-67, red; DAPI, blue) and quantitative data of Ki-67-positive cells (n = 5). (D) Representative Western blotting of PCNA and CyclinD1and quantitative analysis of PCNA/β-actin and CyclinD1/β-actin (n = 5). (E) Phase-contrast images from the wound healing assay, and quantitative analysis of the migration area (scale bar = 50 μm, n = 5). The data are presented as the mean ± SD. Ns, p ≥ 0.05, *p < 0.05, **p < 0.01. Abbreviations: PASMCs, pulmonary artery smooth muscle cells; FTS•B, forsythoside B; PDGF-BB, platelet-derived growth factor-BB. |
FTS•B Blocked the Activation of the NF-κB Signaling Pathway
Prior researches have shown that activation of the NF-κB signaling pathway promotes PASMC proliferation and the progression of PAH.34 NF-κB activation was also observed in PASMC proliferation under PDGF-BB exposure. Therefore, we investigated whether FTS•B affects the NF-κB signaling pathway in PDGF-BB-induced PASMC proliferation. Western blot analysis demonstrated that FTS•B downregulated PDGF-BB-induced phosphorylation of p65, IκBα, IKKα and IKKβ, indicating inhibitory effects on the NF-κB pathway (Figure 4A). In addition, immunofluorescence staining showed that p65 proteins were distributed in the cytoplasm in untreated PASMCs; however, they accumulated in the nucleus after PDGF-BB stimulation; when co-treated with FTS•B, the nuclear transfer of p65 was largely blunted (Figure 4B). Inhibitory effect of FTS•B on nuclear translocation of p65 in PASMCs was also observed in vivo (Figure 4C). These findings indicate that FTS•B blocks the activation of the NF-κB signaling pathway.
 |
Figure 4 FTS•B blocked the activation of the NF-κB signaling pathway. PASMCs were treated with PDGF-BB in the presence or absence of FTS•B for 24 h. (A) Representative Western blotting of p-p65, p65, p-IκBα, IκBα, p-IKKα, IKKα, p-IKKβ and IKKβ, and quantitative analysis of p-p65/p65, p-IκBα/IκBα, p-IKKα/IKKα and p-IKKβ/IKKβ (n = 5). (B) Immunofluorescence staining of p65 in PASMCs (p65, red; DAPI, blue; scale bar = 50 μm) and quantitative data of nuclear p65 immunofluorescence intensity (n = 6). (C) Representative immunofluorescence staining of p65 in pulmonary artery (p65, red; DAPI, blue; scale bar = 50 μm) and quantitative data of nuclear p65 immunofluorescence intensity (n = 6). The data are presented as the mean ± SD. *p < 0.05,**p < 0.01. Abbreviations: PASMCs, pulmonary artery smooth muscle cells; NF-κB, nuclear factor-κB; PDGF-BB, platelet-derived growth factor-BB; FTS•B, forsythoside B. |
Activation of the NF-κB Signaling Pathway Blocked the Protective Effects of FTS•B in vivo
To further investigate whether the NF-κB signaling pathway plays a crucial role in FTS•B-mediated protection against PAH, MCT + FTS•B rats were co-treated with NF-κB activator PMA (5 mg/kg, i.p. daily, for 21 days). Western blotting revealed that the inhibitory effect of FTS•B on NF-κB pathway was reversed by PMA (Figure 5A). Previous studies have shown that the activation of NF-κB promotes the perivascular macrophage infiltration and inflammation, eventually leading to PASMC proliferation and the occurrence of PAH.12,35 Our current study also observed macrophage infiltration, upregulated levels of IL-1β and IL-6 in MCT-induced PAH rats, while FTS•B treatment suppressed these proinflammatory factors (Supplemental Figure 4A–C and E–F). On the other hand, FTS•B treatment also induced increase of anti-inflammatory cytokine IL-10 (Supplemental Figure 4D and G). However, the anti-inflammatory effect was blunted by PMA-mediated activation of NF-κB (Supplemental Figure 4A–G). As expected, FTS•B-induced decreases of pulmonary artery pressure and right ventricular sizes in PAH rats were also offset by PMA (Figure 5B–E). Furthermore, PMA also reversed FTS•B’s beneficial effects in right ventricular hypertrophy (Figure 5F), pulmonary arteriole medial thickness (Figure 5G) and neomuscularization (Figure 5H). In addition, PMA nearly diminished the inhibitory effect of FTS•B on proliferation of PASMCs (Figure 5I). Collectively, these data suggest that FTS•B alleviates pulmonary hypertension and vascular remodeling by blocking activation of the NF-κB signaling pathway.
 |
Figure 5 NF-κB activation blocked the protective effect of FTS•B against PAH and vascular remodeling. Rats were subjected to MCT and FTS•B with/without cotreatment with PMA. (A) Representative Western blotting of p-p65, p65, p-IκBα, IκBα, p-IKKα, IKKα, p-IKKβ and IKKβ in lung lysates, and quantitative data of p-p65/p65, p-IκBα/IκBα, p-IKKα/IKKα and p-IKKβ/IKKβ (n = 6). (B) Echocardiographic PW Doppler tracing of pulmonary artery outflow and quantitative analysis of PAT and PAT/PET (n = 6). (C) RVFWT and RVEDD in M-mode echo and relative quantification (n = 6). (D) Quantitative analysis of the mPAP (n = 6). (E) Quantitative analysis of the RVSP (n = 6). (F) Representative images of H&E staining of heart sections (scale bar = 2 mm) and quantitative analysis of the RV/ (LV + S) (n = 6). (G). Representative images of H&E staining of lung sections (scale bar = 50 μm) and quantitative analysis of the WT (n = 6). (H) Representative images of α-SMA immunohistochemical staining in the pulmonary artery (scale bar = 50 μm) and quantitative assessments of muscularized pulmonary arteries (n = 6). (I) Representative images of PCNA immunohistochemical staining in the pulmonary artery (scale bar = 50 μm) and quantitative data of PCNA-positive cells (n = 6). The data are presented as the mean ± SD. *p < 0.05, **p < 0.01. Abbreviations: MCT, monocrotaline; FTS•B, forsythoside B; NF-κB, nuclear factor-κB; PMA, phorbol-12-myristate-13-acetate; mPAP, mean pulmonary arterial pressure; RVSP, right ventricular systolic pressure; PAT, pulmonary artery acceleration time; PET, pulmonary arterial ejection time; RVFWT, right ventricular free wall thickness; RVEDD, right ventricular end-diastolic diameter; RV/(LV + S), weight ratio of the right ventricle divided by the sum of the left ventricle and septum; WT, medial wall thickness of pulmonary artery. |
Discussion
Current therapies for PAH target 3 pathways: stimulating the nitric oxide-cyclic guanosine monophosphate biological pathway, increasing prostacyclin effects on receptors, and antagonizing the endothelin pathway.1 However, many patients still suffer from clinical worsening in response to combined targeted therapy, which is considered much better than monotherapy for PAH.36,37 Furthermore, the available medications are costly and come with evident adverse reactions, and the current therapeutic approaches have not significantly improved the long-term outlook for PAH.14,38 Hence, there is an urgent requirement for the creation of novel, efficient and secure medications to combat PAH. The present study discovered that FTS•B treatment significantly inhibited the activation of NF-κB pathway, subsequently the nuclear translocation of p65, leading to the suppression of macrophage infiltration and the expression of IL-1β and IL-6 in MCT-induced PAH. FTS•B also suppressed PASMC proliferation and pulmonary arteriole neomuscularization, decreased mPAP, RVSP and RV/(LV + S), ultimately improved cardiac function and survival rate in MCT-induced PAH. These results indicate that FTS•B holds promise as a potential medication for addressing PAH.
The use of natural compounds derived from plants to treat diseases has a long history, such as that of the drug aspirin.39,40 In recent years, there has been an increased focus on the functions of plant-derived natural compounds in different illnesses. Paclitaxel suppresses FoxO1-mediated autophagy to inhibit pulmonary vascular remodeling in the treatment of MCT-induced PAH.41 The use of perillyl alcohol demonstrates significant protective and therapeutic properties against MCT-induced PAH.42 As one of the natural compounds, FTS•B has been reported to possess anti-inflammatory, anti-oxidant and anti-bacterial activities and has been widely studied for the treatment of inflammatory diseases.43–45 Liu et al, revealed that FTS•B suppresses inflammation in lipopolysaccharide-induced acute lung injury by attenuating the TLR4/NF-κB pathway.17 FTS•B also improves rat cardiac function after ischemia-reperfusion injury by its anti-inflammatory effects.19 Our study also showed that FTS•B was effective in alleviating pulmonary artery pressure and vascular remodeling, thus highlighting it as a potential anti-PAH drug.
PAH is a persistent and serious heart and lung condition resulted from the excessive growth and fibrosis of pulmonary arterioles, which leads to the progression of pulmonary vascular resistance.46 Complex pathological cascade reactions lead to the excessive growth of smooth muscle and endothelial cells in the lungs, inflammation and the formation of in situ thrombi, which results in narrowing and remodeling of the pulmonary artery. Although there are different etiologies, initiating factors and pathophysiological processes, vascular remodeling is a key step.47 Hence, addressing vascular restructuring is a crucial objective in managing pulmonary hypertension. For example, strategies via enhancing BMPR-II signaling to treat PAH include a new generation of drugs that specifically target known defective molecular regulators of pulmonary vascular remodeling.47,48 Hypoxia-inducible factor (HIF)-2α-selective inhibitors can reverse PAH by targeting vascular remodeling.49 Consistent with these studies, our study revealed that FTS•B alleviated pulmonary vascular remodeling, suggesting that blocking vascular remodeling is the key mechanism by which this compound protects against PAH.
PAH has a complex molecular mechanism with involvement of numerous signaling pathways. The data obtained from in vivo and in vitro experiments confirmed that HIF signaling plays a crucial and central role in the development of PAH.50 Abnormalities in K+ channels and Ca2+ channels have been associated with narrowing and remodeling in the pulmonary artery.51 The development of PAH in humans is linked to the Notch signaling pathway.51,52 Hong J performed single-cell RNA sequencing on PAH model rats and strong NF-κB pathway activation was observed across cell types.53 It is well established that NF-κB has involved in PAH pathogenesis. As a key transcription factor, NF-κB regulates a great deal of proinflammatory genes including TNF-α, IL-1β and IL-6, which mediate perivascular inflammation, PASMC proliferation and ultimately PAH.7,54–56 Previous studies have reported that IMD-0354 ameliorates PAH via NF-κB inhibition.57 In line with these studies, our present study also revealed that FTS•B suppressed NF-κB activation, downregulated IL-1β and IL-6 expression and finally attenuated PASMC proliferation. Therefore, we speculated that FTS•B attenuates PAH via blocking the NF-κB signaling pathway to attenuate vascular remodeling.
In summary, the current study demonstrated that FTS•B alleviated MCT-induced PAH and significantly inhibited vascular remodeling by blocking the NF-κB signaling pathway. Still, there have some limitations: 1) only adult male SD rats were tested in this study, thus the impact of gender on the beneficial effect of FTS•B remains unknown; 2) genetic tools were not applied to explore the underlying mechanisms; 3) the curative effects of FTS•B were only partially surveyed in rodents, more experiments in primates are needed to determine the safety and optimal drug dose.
Data Sharing Statement
The corresponding author can provide the datasets upon reasonable request.
Acknowledgment
The research was funded by the Sichuan Provincial Administration of Traditional Chinese Medicine (NO.2023MS197), the Natural Science Foundation of Sichuan Province (NO.2022NSFSC0822), and Hospital Management Research of Western Theater General Hospital (NO.2021XZYGC16).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ruopp NF, Cockrill BA. Diagnosis and treatment of pulmonary arterial hypertension: a review. JAMA. 2022;327(14):1379–1391. doi:10.1001/jama.2022.4402
2. Jiang Y, Song S, Liu J, et al. Epigenetic regulation of programmed cell death in hypoxia-induced pulmonary arterial hypertension. Front Immunol. 2023;14:1206452. doi:10.3389/fimmu.2023.1206452
3. Naeije R, Richter MJ, Rubin LJ. The physiological basis of pulmonary arterial hypertension. Eur Respir J. 2022;59(6):2102334. doi:10.1183/13993003.02334-2021
4. Olsson KM, Corte TJ, Kamp JC, et al. Pulmonary hypertension associated with lung disease: new insights into pathomechanisms, diagnosis, and management. Lancet Respir Med. 2023;11(9):820–835. doi:10.1016/s2213-2600(23)00259-x
5. Hassoun PM. Pulmonary Arterial Hypertension. N Engl J Med. 2021;385(25):2361–2376. doi:10.1056/NEJMra2000348
6. Hurdman J, Condliffe R, Elliot CA, et al. ASPIRE registry: assessing the spectrum of pulmonary hypertension identified at a REferral centre. Eur Respir J. 2012;39(4):945–955. doi:10.1183/09031936.00078411
7. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2(17023):17023. doi:10.1038/sigtrans.2017.23
8. Gong Y, Kong B, Shuai W, Chen T, Zhang JJ, Huang H. USP38 regulates inflammatory cardiac remodeling after myocardial infarction. Clin Sci. 2023;137(21):1665–1681. doi:10.1042/cs20230728
9. Wu S, Zhou Y, Liang J, et al. Upregulation of NF-κB by USP24 aggravates ferroptosis in diabetic cardiomyopathy. Free Radic Biol Med. 2024;210:352–366. doi:10.1016/j.freeradbiomed.2023.11.032
10. Parpaleix A, Amsellem V, Houssaini A, et al. Role of interleukin-1 receptor 1/MyD88 signalling in the development and progression of pulmonary hypertension. Eur Respir J. 2016;48(2):470–483. doi:10.1183/13993003.01448-2015
11. Krzyżewska A, Baranowska-Kuczko M, Jastrząb A, Kasacka I, Kozłowska H. Cannabidiol improves antioxidant capacity and reduces inflammation in the lungs of rats with monocrotaline-induced pulmonary hypertension. Molecules. 2022;27(10):3327. doi:10.3390/molecules27103327
12. Hu Y, Chi L, Kuebler WM, Goldenberg NM. Perivascular inflammation in pulmonary arterial hypertension. Cells. 2020;9(11):2338. doi:10.3390/cells9112338
13. Sawada H, Mitani Y, Maruyama J, et al. A nuclear factor-kappaB inhibitor pyrrolidine dithiocarbamate ameliorates pulmonary hypertension in rats. Chest. 2007;132(4):1265–1274. doi:10.1378/chest.06-2243
14. Chen M, Ding Z, Zhang F, et al. A20 attenuates hypoxia-induced pulmonary arterial hypertension by inhibiting NF-κB activation and pulmonary artery smooth muscle cell proliferation. Exp Cell Res. 2020;390(2):111982. doi:10.1016/j.yexcr.2020.111982
15. Wang Z, Xia Q, Liu X, et al. Phytochemistry, pharmacology, quality control and future research of Forsythia suspensa (Thunb.) Vahl: a review. J Ethnopharmacol. 2018;210:318–339. doi:10.1016/j.jep.2017.08.040
16. Jiang WL, Yong X, Zhang SP, Zhu HB, Jian H. Forsythoside B protects against experimental sepsis by modulating inflammatory factors. Phytother Res. 2012;26(7):981–987. doi:10.1002/ptr.3668
17. Liu JX, Li X, Yan FG, et al. Protective effect of forsythoside B against lipopolysaccharide-induced acute lung injury by attenuating the TLR4/NF-κB pathway. Int Immunopharmacol. 2019;66:336–346. doi:10.1016/j.intimp.2018.11.033
18. Kong F, Jiang X, Wang R, Zhai S, Zhang Y, Wang D. Forsythoside B attenuates memory impairment and neuroinflammation via inhibition on NF-κB signaling in Alzheimer’s disease. J Neuroinflammation. 2020;17(1):305. doi:10.1186/s12974-020-01967-2
19. Jiang WL, Fu FH, Xu BM, Tian JW, Zhu HB, Jian H. Cardioprotection with forsythoside B in rat myocardial ischemia-reperfusion injury: relation to inflammation response. Phytomedicine. 2010;17(8–9):635–639. doi:10.1016/j.phymed.2009.10.017
20. Yu M, Wu X, Peng L, et al. Inhibition of Bruton’s tyrosine kinase alleviates monocrotaline-induced pulmonary arterial hypertension by modulating macrophage polarization. Oxid Med Cell Longev. 2022;2022:6526036. doi:10.1155/2022/6526036
21. Zhu Z, Godana D, Li A, et al. Echocardiographic assessment of right ventricular function in experimental pulmonary hypertension. Pulm Circ. 2019;9(2):2045894019841987. doi:10.1177/2045894019841987
22. Yared K, Noseworthy P, Weyman AE, McCabe E, Picard MH, Baggish AL. Pulmonary artery acceleration time provides an accurate estimate of systolic pulmonary arterial pressure during transthoracic echocardiography. J Am Soc Echocardiogr. 2011;24(6):687–692. doi:10.1016/j.echo.2011.03.008
23. Lan C, Fang G, Qiu C, Li X, Yang F, Yang Y. Inhibition of DYRK1A attenuates vascular remodeling in pulmonary arterial hypertension via suppressing STAT3/Pim-1/NFAT pathway. Clin Exp Hypertens. 2024;46(1):2297642. doi:10.1080/10641963.2023.2297642
24. Guo J, Yang ZC, Liu Y. Attenuating pulmonary hypertension by protecting the integrity of glycocalyx in rats model of pulmonary artery hypertension. Inflammation. 2019;42(6):1951–1956. doi:10.1007/s10753-019-01055-5
25. Gao L, Fan Y, Hao Y, et al. Cysteine-rich 61 (Cyr61) upregulated in pulmonary arterial hypertension promotes the proliferation of pulmonary artery smooth muscle cells. Int J Med Sci. 2017;14(9):820–828. doi:10.7150/ijms.19282
26. Mendes-Ferreira P, Maia-Rocha C, Adão R, et al. Neuregulin-1 improves right ventricular function and attenuates experimental pulmonary arterial hypertension. Cardiovasc Res. 2016;109(1):44–54. doi:10.1093/cvr/cvv244
27. Yang JM, Zhou R, Zhang M, Tan HR, Yu JQ. Betaine attenuates monocrotaline-induced pulmonary arterial hypertension in rats via inhibiting inflammatory response. Molecules. 2018;23(6). doi:10.3390/molecules23061274
28. Sheikh AQ, Lighthouse JK, Greif DM. Recapitulation of developing artery muscularization in pulmonary hypertension. Cell Rep. 2014;6(5):809–817. doi:10.1016/j.celrep.2014.01.042
29. Tang L, Cai Q, Wang X, et al. Canagliflozin ameliorates hypobaric hypoxia-induced pulmonary arterial hypertension by inhibiting pulmonary arterial smooth muscle cell proliferation. Clin Exp Hypertens. 2023;45(1):2278205. doi:10.1080/10641963.2023.2278205
30. Tang BL, Liu Y, Zhang JL, Lu ML, Wang HX. Ginsenoside Rg1 ameliorates hypoxia-induced pulmonary arterial hypertension by inhibiting endothelial-to-mesenchymal transition and inflammation by regulating CCN1. Biomed Pharmacother. 2023;164:114920. doi:10.1016/j.biopha.2023.114920
31. Dougherty EJ, Chen LY, Awad KS, et al. Inflammation and DKK1-induced AKT activation contribute to endothelial dysfunction following NR2F2 loss. Am J Physiol Lung Cell Mol Physiol. 2023;324(6):L783–l798. doi:10.1152/ajplung.00171.2022
32. Mahler GJ, Farrar EJ, Butcher JT. Inflammatory cytokines promote mesenchymal transformation in embryonic and adult valve endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33(1):121–130. doi:10.1161/atvbaha.112.300504
33. Li Y, Ren W, Wang X, et al. MicroRNA-150 relieves vascular remodeling and fibrosis in hypoxia-induced pulmonary hypertension. Biomed Pharmacother. 2019;109:1740–1749. doi:10.1016/j.biopha.2018.11.058
34. Hinz M, Krappmann D, Eichten A, Heder A, Scheidereit C, Strauss M. NF-kappaB function in growth control: regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol Cell Biol. 1999;19(4):2690–2698. doi:10.1128/mcb.19.4.2690
35. Chen S, Yan D, Qiu A. The role of macrophages in pulmonary hypertension: pathogenesis and targeting. Int Immunopharmacol. 2020;88:106934. doi:10.1016/j.intimp.2020.106934
36. Lajoie AC, Lauzière G, Lega JC, et al. Combination therapy versus monotherapy for pulmonary arterial hypertension: a meta-analysis. Lancet Respir Med. 2016;4(4):291–305. doi:10.1016/s2213-2600(16)00027-8
37. Chin KM, Sitbon O, Doelberg M, et al. Three- versus two-drug therapy for patients with newly diagnosed pulmonary arterial hypertension. J Am Coll Cardiol. 2021;78(14):1393–1403. doi:10.1016/j.jacc.2021.07.057
38. Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369(9):809–818. doi:10.1056/NEJMoa1213917
39. Mirhadi E, Roufogalis BD, Banach M, Barati M, Sahebkar A. Resveratrol: mechanistic and therapeutic perspectives in pulmonary arterial hypertension. Pharmacol Res. 2021;163:105287. doi:10.1016/j.phrs.2020.105287
40. Davidson KW, Barry MJ, Mangione CM, et al. Aspirin use to prevent cardiovascular disease: us preventive services task force recommendation statement. JAMA. 2022;327(16):1577–1584. doi:10.1001/jama.2022.4983
41. Feng W, Wang J, Yan X, et al. Paclitaxel alleviates monocrotaline-induced pulmonary arterial hypertension via inhibition of FoxO1-mediated autophagy. Naunyn Schmiedebergs Arch Pharmacol. 2019;392(5):605–613. doi:10.1007/s00210-019-01615-4
42. Beik A, Najafipour H, Joukar S, Rajabi S, Iranpour M, Kordestani Z. Perillyl alcohol suppresses monocrotaline-induced pulmonary arterial hypertension in rats via anti-remodeling, anti-oxidant, and anti-inflammatory effects. Clin Exp Hypertens. 2021;43(3):270–280. doi:10.1080/10641963.2020.1860080
43. Nazemiyeh H, Rahman MM, Gibbons S, et al. Assessment of the antibacterial activity of phenylethanoid glycosides from Phlomis lanceolata against multiple-drug-resistant strains of Staphylococcus aureus. J Nat Med. 2008;62(1):91–95. doi:10.1007/s11418-007-0194-z
44. Li Y, Yang Y, Kang X, et al. Study on the anti-inflammatory effects of Callicarpa nudiflora based on the spectrum-effect relationship. Front Pharmacol. 2021;12:806808. doi:10.3389/fphar.2021.806808
45. Yoo TK, Jeong WT, Kim JG, et al. UPLC-ESI-Q-TOF-MS-based metabolite profiling, antioxidant and anti-inflammatory properties of different organ extracts of Abeliophyllum distichum. Antioxidants. 2021;10(1):70. doi:10.3390/antiox10010070
46. Lau EMT, Giannoulatou E, Celermajer DS, Humbert M. Epidemiology and treatment of pulmonary arterial hypertension. Nat Rev Cardiol. 2017;14(10):603–614. doi:10.1038/nrcardio.2017.84
47. Thompson AAR, Lawrie A. Targeting vascular remodeling to treat pulmonary arterial hypertension. Trends Mol Med. 2017;23(1):31–45. doi:10.1016/j.molmed.2016.11.005
48. Smith ER, Wang JQ, Yang DH, Xu XX. Paclitaxel resistance related to nuclear envelope structural sturdiness. Drug Resist Updat. 2022;65:100881. doi:10.1016/j.drup.2022.100881
49. Dai Z, Zhu MM, Peng Y, et al. Therapeutic targeting of vascular remodeling and right heart failure in pulmonary arterial hypertension with a HIF-2α inhibitor. Am J Respir Crit Care Med. 2018;198(11):1423–1434. doi:10.1164/rccm.201710-2079OC
50. Pullamsetti SS, Mamazhakypov A, Weissmann N, Seeger W, Savai R. Hypoxia-inducible factor signaling in pulmonary hypertension. J Clin Invest. 2020;130(11):5638–5651. doi:10.1172/jci137558
51. Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol. 2011;8(8):443–455. doi:10.1038/nrcardio.2011.87
52. Li X, Zhang X, Leathers R, et al. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat Med. 2009;15(11):1289–1297. doi:10.1038/nm.2021
53. Hong J, Arneson D, Umar S, et al. Single-cell study of two rat models of pulmonary arterial hypertension reveals connections to human pathobiology and drug repositioning. Am J Respir Crit Care Med. 2021;203(8):1006–1022. doi:10.1164/rccm.202006-2169OC
54. Soon E, Holmes AM, Treacy CM, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 2010;122(9):920–927. doi:10.1161/circulationaha.109.933762
55. Savai R, Pullamsetti SS, Kolbe J, et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186(9):897–908. doi:10.1164/rccm.201202-0335OC
56. He Y, Zuo C, Jia D, et al. Loss of DP1 aggravates vascular remodeling in pulmonary arterial hypertension via mTORC1 signaling. Am J Respir Crit Care Med. 2020;201(10):1263–1276. doi:10.1164/rccm.201911-2137OC
57. Hosokawa S, Haraguchi G, Sasaki A, et al. Pathophysiological roles of nuclear factor kappaB (NF-kB) in pulmonary arterial hypertension: effects of synthetic selective NF-kB inhibitor IMD-0354. Cardiovasc Res. 2013;99(1):35–43. doi:10.1093/cvr/cvt105
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.