Back to Journals » Drug Design, Development and Therapy » Volume 11
Formulation, release characteristics, and bioavailability study of gastroretentive floating matrix tablet and floating raft system of Mebeverine HCl
Authors El Nabarawi MA, Teaima MH ![]() , Abd El-Monem RA
, Abd El-Monem RA ![]() , El Nabarawy NA
, El Nabarawy NA ![]() , Gaber DA
, Gaber DA
Received 8 January 2017
Accepted for publication 18 February 2017
Published 3 April 2017 Volume 2017:11 Pages 1081—1093
DOI https://doi.org/10.2147/DDDT.S131936
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Georgios Panos
Mohamed A El Nabarawi,1 Mahmoud H Teaima,1 Rehab A Abd El-Monem,2 Nagla A El Nabarawy,3 Dalia A Gaber4
1Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Cairo University, Cairo, Egypt; 2Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Misr University for Science and Technology, 6th of October, Egypt; 3National Egyptian Center of Environment and Toxicological Research (NECTER), Faculty of Medicine, Cairo University, Cairo, Egypt; 4Department of Quality Control, Holding Company for Biological Products and Vaccines, Cairo, Egypt
Abstract: To prolong the residence time of dosage forms within the gastrointestinal tract until all drug is released at the desired rate is one of the real challenges for oral controlled-release drug delivery systems. This study was designed to develop a controlled-release floating matrix tablet and floating raft system of Mebeverine HCl (MbH) and evaluate different excipients for their floating behavior and in vitro controlled-release profiles. Oral pharmacokinetics of the optimum matrix tablet, raft system formula, and marketed Duspatalin® 200 mg retard as reference were studied in beagle dogs. The optimized tablet formula (FT-10) and raft system formula (FRS-11) were found to float within 34±5 sec and 15±7 sec, respectively, and both remain buoyant over a period of 12 h in simulated gastric fluid. FT-10 (Compritol/HPMC K,100M 1:1) showed the slowest drug release among all prepared tablet formulations, releasing about 80.2% of MbH over 8 h. In contrast, FRS-11 (Sodium alginate 3%/HPMC K,100M 1%/Precirol 2%) had the greatest retardation, providing sustained release of 82.1% within 8 h. Compared with the marketed MbH product, the Cmax of FT-10 was almost the same, while FRS-11 maximum concentration was higher. The tmax was 3.33, 2.167, and 3.0 h for marketed MbH product, FT-10, and FRS-11, respectively. In addition, the oral bioavailability experiment showed that the relative bioavailability of the MbH was 104.76 and 116.01% after oral administration of FT-10 and FRS-11, respectively, compared to marketed product. These results demonstrated that both controlled-released floating matrix tablet and raft system would be promising gastroretentive delivery systems for prolonging drug action.
Keywords: GRDDS, Precirol®, Compritol®, HPMC, Na alginate
Introduction
Oral formulations have earned a significant place among the various dosage forms due to the ease of administration, patient compliance, and flexibility in formulation. In most of the cases, the conventional oral delivery systems show limited bioavailability because of fast gastric emptying time among many other reasons involved.1,2 However, the recent technological development has resulted in too many novel pharmaceutical products, mainly the controlled release drug delivery systems to help overcome this problem. Controlled-release systems aim to maintain the steady plasma level of the drug over a prolonged time period, reduce the adverse side effects, and improve patient convenience and compliance. Gastroretentive drug delivery system (GRDDS) is one such example where attributes like gastric retention time coupled with the drug release for extended time have significantly improved patient compliance.3,4
GRDDS may be achieved by the mechanisms of mucoadhesion, flotation, sedimentation, expansion modified shape systems,5–7 or by the simultaneous administration of pharmacological agents that delay gastric emptying.8 Floating drug delivery systems are those systems having a bulk density less than that of the gastric fluids, and thus these systems remain buoyant for a prolonged period of time in the stomach without being affected by the gastric emptying rate. The drug is released slowly at the desired rate from the system, and after release of the drug the residual system is emptied from the stomach.9 Several floating dosage forms have been launched in the market.10–12
Floating matrix systems appear to be a very attractive approach in controlled-release system. Floating matrix-type formulations are prepared from either swellable hydrophilic polymers and/or nonswellable lipophilic excipients, like waxes and lipids, with carbonate or bicarbonate as the gas-generating agent.13,14
In situ gelling technique (also known as raft forming system) in combination with carbon dioxide bubble entrapment was also reported as another patient compliance design for gastroretention. This type of delivery system, initially as a solution form, contains sodium alginate as the in situ gel forming polymer along with carbonates or bicarbonates as effervescent agents. When they come in contact with the gastric fluid, they swell and generate a viscous cohesive gel that contains entrapped carbon dioxide bubbles, causing reduction in the density of the systems and contributes to its flotation above the gastric content.15 These gels can remain in the stomach for longer periods of time in comparison to a meal, and this was reported by others.7,16 Hydrophilic polymers are becoming very popular in formulating oral sustained-release formulations, such as xanthan gum, cellulose derivatives, alginate sodium, or carbopol.17 Hydroxypropyl methylcellulose (HPMC) is the most commonly and successfully used hydrophilic material for sustained drug delivery.18 It possesses some important characteristics including nontoxicity, pH independence, and high water swellability, which contribute to a desirable drug sustained release profile. In this investigation, HPMC was used as a release retardant carrier in the design of sustained release matrix tablets and raft system for Mebeverine HCl (MbH).19
The use of lipid and wax polymers seems to have a particular advantage in the preparation of controlled release dosage forms due to their chemical inertness against other materials, better characterization of lipid excipients and formulation versatility, and the choice of different drug delivery systems.20 Recently, much attention has been focused on the use of gelucires as carriers in drug delivery systems. The gelucires containing only glyceride are used in preparation of controlled-release formulations. In particular, Compritol 888 ATO5 (Cr), Precirol ATO5 (Pr), and Glycerol monostearate (GMS) were used as glyceride base for the preparation of controlled-release dosage forms.4
MbH is a musculotropic antispasmodic drug without atropic side effect, whose major therapeutic role is in the treatment of irritable bowel syndrome. It has a short biological half-life of 2.5 h, plasma protein binding of 75%, and is rapidly absorbed after oral administration from the upper part of gastrointestinal tract with peak plasma concentration occurring in 1–3 h. Hence, MbH has been selected as a model drug as it fulfills the required pharmacokinetic and physicochemical properties for controlled delivery.21
However, to our knowledge, no published data have been developed for a floating dosage form of MbH and nor has its pharmacokinetics been monitored in beagle dogs. Therefore, the present study was undertaken to investigate the efficacy of MbH floating matrix tablet (FT) and floating raft system (FRS) as a drug delivery system for better control of MbH release using both hydrophilic and lipid polymers in different ratios. The formulations, with the optimal buoyancy properties and in vitro drug release, would be selected for a pharmacokinetic study to investigate the in vivo superiority over the marketed product “Duspatalin® retard” in beagle dogs.
Materials and method
Materials
MbH (EIPICO, Mansoura, Egypt), Duspatalin® retard 200 mg capsule, (Reference) Batch no T4171, (Abbott Healthcare SAS, Rungis Cedex, France), HPMC with different grades – HPMC K100M and HPMC K15M (El Kahera Pharmaceuticals, Cairo, Egypt), Compritol® 888ATO (glyceryl behenate NF; Gattefossé s.a., Lyon, France), Precirol® ATO5 (glyceryl palmitostearate; Gattefossé s.a.), GMS (Loba Chemie Pvt. Ltd., Mumbai, India), sodium alginate LF R5/60 (Sisco Research Laboratories Pvt. Ltd., Mumbai, India), sodium citrate, sodium bicarbonate, and calcium carbonate (Adwic Pharmaceuticals, Cairo, Egypt), Acetonitrile high-performance liquid chromatography (HPLC) grade (Scharlau Chemie SA, Barcelona, Spain), sodium dihydrogen phosphate (Koch-Light Laboratories, Colnbrook Bucks, UK), and talc and magnesium stearate were all of pharmaceutical grade and used as received.
Method
Preparation of floating dosage forms of MbH
Preparation of FT of MbH
Twelve floating tablets formulations, each containing 200 mg of MbH, were prepared by a direct compression method. Briefly, 200 mg of the drug, polymer, and other additives as mentioned in Table 1 were mixed in incremental amounts, and then the mixture was screened through a 40-mesh sieve (425 μm). Magnesium stearate and purified talc were then added. Powder mixtures were compressed into tablets using a single-punch tablet compression machine (Cadmach, Ahmedabad, India) fit with 10 mm concave-faced punches. Compression was controlled to produce a 5-kg/cm2 tablet-crushing strength.22
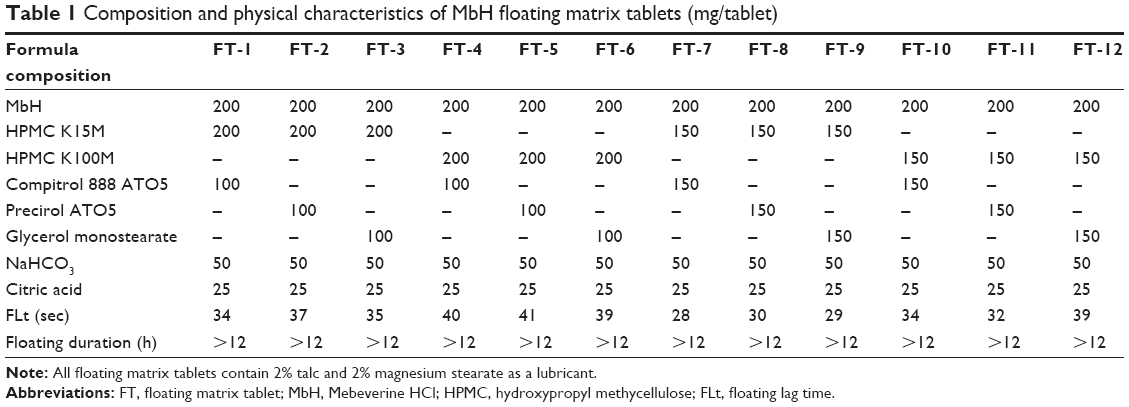 | Table 1 Composition and physical characteristics of MbH floating matrix tablets (mg/tablet) |
Preparation of FRS of MbH
Eleven liquid formulations with in situ gelling and floating properties were prepared. The formulations were liquid sols of alginate containing calcium carbonate as an effervescent agent. Different concentrations of HPMC K100M, Cr, and Pr were incorporated into alginate-based formulations to retard the drug release rate. Composition of the prepared FRS is shown in Table 2. Calculated amounts of sodium alginate and HPMC were individually dispersed in deionized water, and then sodium citrate 0.25% w/v was added to sodium alginate solution and heated to 90°C with stirring till a homogeneous viscous liquid was obtained; then MbH was dispersed (200 mg/10 mL formulation) in the viscous liquid. Carbonate dispersion providing 1% w/v concentration was then added to the prepared emulsion and mixed well with a homogenizer (Homogenizer T-25; IKA, Staufen, Germany) at a speed of 2,000 rpm for a duration of 10 min until a homogeneous stable emulsion was obtained. For lipid-containing formulations, Cr and Pr were melted in a water bath adjusted to a 90°C temperature and then MbH was dispersed in the molten lipid (200 mg/10 mL formulation). A calculated volume of the in situ gelling sol, containing sodium citrate 0.25% w/v and HPMC solution, was heated to the same temperature of molten lipid and then added to MbH lipid dispersion. Carbonate was then added (1% w/v) to the prepared emulsion and mixed well (as mentioned earlier).23,24
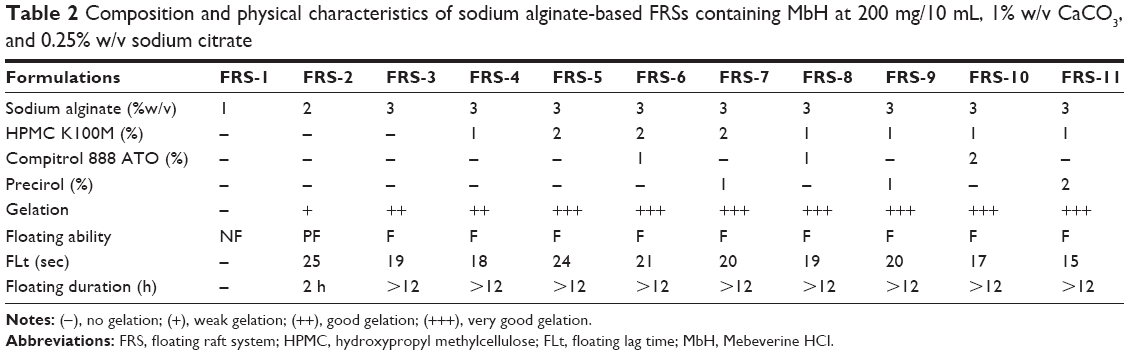 | Table 2 Composition and physical characteristics of sodium alginate-based FRSs containing MbH at 200 mg/10 mL, 1% w/v CaCO3, and 0.25% w/v sodium citrate |
In vitro evaluation of physicochemical characteristics of MbH FT and FRS
Evaluation of the precompression and postcompression parameters of FT of MbH
Angle of repose, compressibility index, and Hausner’s ratio of powder mixture were measured.25 Compressed tablets were characterized for weight variation, crushing strength, diameter, thickness, friability, and uniformity of drug content.26,27
In vitro gelation study of FRS of MbH
Gelation capacity was determined by placing 1 mL of the formulation in a test tube containing 5 mL of gelation medium (simulated gastric fluid pH 1.2). All were freshly prepared and equilibrated at 37°C and were added slowly. Gelation was observed by visual examination.2
In vitro floating study
The in vitro buoyancy was determined by investigation of floating lag time (FLt) and floating duration for all formulations. The test was performed by placing each of the tablets or 10 mL raft systems (placed into watch glass) in a 250 mL beaker containing 200 mL of 0.1 N HCl (pH 1.2), and the system was maintained at 37°C ±0.5°C in a water bath. Their physical state was observed for 12 h. The time between introduction of the dosage form and its buoyancy on the 0.1 N HCl (FLt) and the time during which the dosage form remains floating (duration of floating) were noted. Three replicate measurements for each formula were performed.22
Measurement of in vitro MbH release
The release of MbH from FT and FRS was determined using USP dissolution test apparatus II (USP 24). The temperature was maintained at 37°C with a paddle stirrer at 50 rpm. The dissolution medium used was 900 mL of 0.1 N HCl (pH 1.2). One FT of MbH or 10 mL of FRS (placed into watch glass) was kept in the dissolution vessel without much disturbance. At each predetermined time interval, a precisely measured sample of the dissolution medium was removed and replenished with the same volume of a prewarmed (37°C) fresh medium. Absorbance of Mebeverin in withdrawn samples was measured at 362 nm using a UV spectrophotometer (UV-1601; Shimadzu, Kyoto, Japan). All dissolution runs were performed in triplicate.2
Mechanism and kinetics of drug release
Drug release data were analyzed according to zero-order, first-order, Higuchi, Hixon–Crowell, Peppas, and Weibull kinetic equations.28 DDSolver, which is an add-in program for Microsoft Excel for modeling and comparison of drug release profiles was used. The model with the highest coefficient of determination (R2) was considered to be the best fitting one.29
Statistical analysis
Results were analyzed by using the software SPSS 17.0 (SPSS Inc., Chicago, IL, USA) applying one-way analysis of variance (ANOVA) and paired Student’s t-test. Differences between formulations were considered to be significant at P<0.05.
In vivo pharmacokinetic study in beagle dogs
An in vivo pharmacokinetic study was conducted in accordance with the ethical guidelines for investigations in laboratory animals and was approved by the Institutional Animal Ethics Committee, Faculty of Pharmacy, Cairo University, (number [PI/1535]). All procedures and care of the beagle dogs were in accordance with institutional guidelines for animal use in research. Six male beagle dogs weighing 11–14.5 kg were used and divided into three groups randomly (Figure 1), and the study was carried out in a crossover experimental design in three phases with a washout period of 1 week to eliminate the effect of the prior dose before the next drug administration. All dogs were fasted overnight prior to the experiment; no food was allowed until a standard meal was served 2 h after dosing. Water was available ad libitum throughout the study period. During each period, dogs received orally the following formulations: FT-10 (one floating tablet), FRS-11 (10 mL equivalent to 200 mg of MbH), and one sustained-release MbH marketed product “Duspatalin® retard 200 mg” capsule. Five milliliter of blood samples were taken into a heparinized blood collection tube via a detaining needle at predose and at 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h postdose. The plasma fraction was obtained by centrifuging the samples at 3,500 rpm for 10 min, and this was stored at −20°C until analysis. The plasma concentration was assayed using HPLC.30
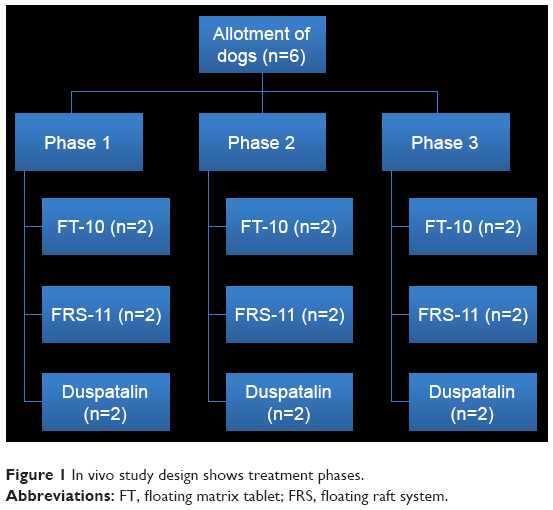 | Figure 1 In vivo study design shows treatment phases. |
Chromatographic system
Drug concentrations in plasma were determined by a previously reported HPLC method.31 The method allowed the determination of Mebeverine metabolite (veratic acid) using Sulpiride (SUL) as internal standard. The analysis was carried out on a Waters Acquity HPLC™ (Waters Corp., Milford, MA, USA). The separation of these compounds was achieved on a Waters® C18 column (250 mm, 4.6 mm i.d., 5 m particle size) using isocratic mobile phase containing a mixture of acetonitrile and 0.01 M dihydrogen phosphate buffer 45:55 (v/v). The analysis was performed at pH 4, flow rate of 1 mL·min−1, and with fluorescence detection at excitation 300 nm and emission 365 nm. Prior to any analysis, the mobile phase was filtered using 0.45 μm filters. The system was equilibrated with the mobile phase before injection. All determinations were made at ambient temperature.
All data were collected and analyzed using Lynx TMV 4.1 software (Waters Corp.). The method was validated for selectivity, linearity, precision, accuracy, carryover, extraction recovery, and stability, briefly before the beginning of this study, according to a previously published assay study.31
Drug analysis
Mebeverine metabolite (veratic acid) concentrations in plasma were measured using a validated specific and sensitive HPLC method. Briefly, sample aliquots of 200 μL were added to a 1.8 mL Eppendorf tube and were spiked with 20 μL of SUL, the internal standard. The mixture was vortexed (Paramix II; Julabo, Seelbach, Germany) for 30 sec, and then 800 μL of acetonitrile was added and the mixture was vortexed for 1 min and centrifuged at 20,000 rpm for 15 min at 10°C. The supernatant was transferred into a clean glass tube and evaporated to dryness under a gentle stream of nitrogen. The residue was reconstituted in 200 μL HPLC eluent, vortexed for 1 min, and 20 μL of this was injected into the HPLC system.31,32
Calculation of MbH parameters and statistical analysis
Pharmacokinetic parameters were estimated from the individual plasma concentrations versus time profiles. Plasma concentrations of veratic acid are presented as the mean ± standard deviation. The peak plasma concentration (Cmax), the time to reach the maximum peak (tmax), and the time the veratic acid first appeared in the plasma (tlag) were obtained as directly measured values. The terminal elimination rate constant (Kel) was estimated by linear regression analysis of the terminal portion of the log-linear plasma concentration–time profile of veratic acid. The extent of absorption (AUC0–t) was calculated using linear trapezoidal rules. Extrapolated AUCs (AUC0–∞) were determined by the following equation:
AUC0–∞ = AUC0–t + Ct/Kel | (1) |
The Wagner–Nelson model was used to calculate the percentage of Mebeverine dose-absorbed profiles.33
Fat = (Ct + Kel × AUC0–t) 1/Kel × AUC0–∞, | (2) |
where Fat is the fraction of drug absorbed at time t, Ct is the concentration of drug in the plasma at time t, and Kel is the elimination rate constant. The elimination rate constant, Kel, was calculated from the mean plasma concentration–time profile of MbH.33
The relative bioavailability values (F) were calculated using the following formula with the market product as a reference.
F = AUCtest/AUCref × 100 | (3) |
Statistical evaluation of the results
The in vivo experiment was preplanned to compare the difference between the mean pharmacokinetic parameters obtained after administration of each of the three treatments into each group of dogs in a crossover model. All statistical differences in data were evaluated by IBM SPSS Statistics 20 (Armonk, NY, USA) using one-way ANOVA with extended LSD post hoc test for the determined pharmacokinetic parameters, and P-value <0.05 was considered significant. Nonparametric Kruskal–Wallis test was done to compare the data of tmax obtained from different treatments.
Results and discussion
Assessment of FT and FRS for delivery of MbH
Powder mixture characterization and physicochemical characters of FT of MbH
The FTs were prepared by a single compression method. To extend the drug residence time in the stomach, the tablets with a density lower than the gastric fluids were developed by incorporating a CO2-generating agent (sodium bicarbonate). One of the following lipid polymers (Cr/Pr/GMS) with HPMC K15M or HPMC K100M were used in different ratios as sustained release agents and entrapped the produced gas to maintain buoyant capacities. The details of the formulations are depicted in Table 1.
All formulation components powders were free flowing. The angle of repose of the powder mixture for all formulations (FT-1–FT-12) was ≤29°C, indicating excellent flow properties. Hausner’s ratios and compressibility indices ranged from 1.2 to 1.27 and 8.52% to 11.15%, respectively. The results of flow properties are acceptable for all powder mixtures; data not shown.34
Floating tablets were whitish-buff or white in color, all were round, concave, with smooth surface on both sides, and no visible cracks were observed. The mean diameter of floating tablets was 10.0±0.0 mm, while mean thickness ranged from 3.2 to 3.4 mm. Mean hardness was in the range of 4–6 kg/cm2, indicating that the floating tablets are of sufficient strength to withstand physical abrasion.35 The percentage friability for all formulations was less than 1%, which is an indication of satisfactory mechanical resistance. All the formulated products lay within the pharmacopoeial requirement of ±5% for weight variation. The percentage of mean drug content ranged from 98.6% to 100.5%, which met the pharmacopeial standard.36
Physicochemical characters of FRS of MbH
The compositions of the eleven formulations investigated are presented in Table 2. A visual estimation of the formulations indicated that they were fluid upon preparation and would not cause any issue for swallowing. Low-molecular-weight alginate LFR5/60 used in this study produced gels with larger volume and thickness than other alginates.37
To meet the expected floating requirements, in situ formed raft system should preserve its integrity without dissolving or eroding, while floating for a prolonged period to facilitate sustained release of drugs. In the presence of either divalent or monovalent cations in the medium, sol to gel transformation of alginate occurs; the mechanism involved in sol to gel transition by these polymers has been reported by many authors.38 In an ion-free aqueous medium, sodium alginate forms double helices, and the helices are only weakly associated with each other by van der Waals attraction and the solution has low viscosity. When gel-promoting cations are present, some of the helices associate into cation-mediated aggregates, which cross-link the polymer. Tang et al’s study,15 reported that divalent ions such as calcium are superior to monovalent cations in promoting the gelation of the polysaccharide. In the present study, CaCO3 was used as a source of calcium, and also as a gas-generating agent. Upon contact of the formulation with acidic pH of the stomach, the dispersed CaCO3 dissolves and releases carbon dioxide and enhances the gel buoyancy.
Gelling properties of FRS of MbH
The gelling properties of raft systems are of importance for their proposed oral administration. The concentration of the gelling polymer should be sufficiently high for the formation of gels of satisfactory gel strength for use as a delivery vehicle and sufficiently low to maintain an acceptable viscosity for ease of swallowing.
Calcium carbonate being present in the formulation as insoluble dispersion releases calcium ions in acidic media, which combine with the polymer sol, causing gelation of the FRS.39 Studies reported that sodium bicarbonate is preferred as the gas-generating agent and that CaCO3 showed internal ionotropic gelation during storage.2 Thus, in our study, sodium citrate was added to prevent premature gelation which may occur during storage, as sodium citrate complexes the free Ca2+ ions and only releases them in the acidic environment of the stomach. The formulation thus remains in liquid form until it reaches the stomach, where gelation is instantaneous. The optimum amount of sodium citrate that maintained fluidity of the formulation before administration and then gelation after administering the formulation has been previously reported.23,39 HPMC K100 M was incorporated to improve the gelation capacity of alginate, and as sustained-release materials amphiphilic lipids (Cr, Pr) were incorporated into alginate-based formulations to increase retardation of drug release.
The gelation study was conducted in 0.1 N HCl, pH 1.2 (simulated gastric fluid [SGF]). All the formulations except FRS-1 showed rapid gelation when in contact with the SGF. Increasing the alginate concentration from 1% to 3% increased the gelation capacity as the gelation time decreased and gel strength increased. Formulations with low content of sodium alginate (FRS-1 and FRS-2) formed weak gels, leaving turbid solutions below that dissolved rapidly within 2 and 4 h, respectively. Such systems are not suitable as oral liquid formulations as they will be removed earlier from the stomach by the peristaltic movements.24 FRS-3 with a higher concentration of alginate forms a rigid gel in short gelation time. Results also showed that incorporation of HPMC in the system resulted in adequate gel strength when pressed with a pair of fine forceps, indicating that formed gel will withstand the shear forces likely to be encountered in the stomach. As shown in Table 2, incorporation of lipids in raft system did not show significant change in gelation capacity.
In vitro floating properties
In vitro floating properties of FTs
The floating ability of the prepared formulations was evaluated in SGF (Figure 2). Faster onset and continuous flotation can prevent the dosage forms from premature evacuation from the stomach. Thus, an investigation into the floating properties of the dosage form in vitro would help to develop an optimized drug product with the desired characteristics. The time the formulation took to emerge on the medium surface (FLt) and the time the formulation constantly floated on the dissolution medium surface (duration of floating) were evaluated and are shown in Table 1. Sodium bicarbonate (NaHCO3) and citric acid were used in the ratio of 2:1 as a gas forming mixture.40 Citric acid was added to negate the effect of difference in acidity in vivo. Sodium bicarbonate generates CO2 in the presence of dissolution medium (0.1 N HCl). The gas generated is trapped and protected within the gel formed by hydration of the polymer, thus decreasing the density of the tablet, and so the tablet becomes buoyant.
 | Figure 2 Gelling and floating ability of FRS-11 (A), FRS-6 (B), and FRS-2 (C). |
As can be seen from Table 1, all tablets could float for more than 12 h, but their FLt was different. The result indicated that CO2 bubbles generated could be entrapped efficiently in the polymer gel layers. HPMC used a sustained-release material, and its density became lower when the polymer was swelling in the fluid.41 Results showed that FLt ranged between 32 and 41 sec, and an increase was observed in FLt with the increase in HPMC viscosity, suggesting that the hydration and the gas forming process of the tablet prolonged, but it was not significant at P<0.5. Incorporation of lipid polymer showed no significant difference (P<0.5) in FLt or floating duration between different formulations.
In vitro floating properties of FRS
In our study calcium carbonate was used as gas forming agent, in the acidic medium of stomach carbon dioxide will be released and entrapped in formed gel network producing buoyant formulation.41 An increase in alginate concentration resulted in decreasing FLt and an increase in floating duration of the prepared systems.42 Using HPMC in concentration 1 and 2% (FRS-4, FRS-5) showed no significant effect (P<0.05) on FLt or duration. Increasing lipid polymer content enhanced the floating ability, where a decrease in FLt was achieved in FRS-10 and FRS-11 (Table 2). This enhancement in floating ability was attributed to the low density of the used lipids in addition to the three-dimensional network of the cubic phase of the used lipids, which further reduces the permeability of the formed gel, leading to a reduction in diffusion of the entrapped carbon dioxide, thus resulting in excellent buoyancy.43
In vitro release study
In vitro release study of FTs
In vitro drug release experiments were done to investigate the probability of employing HPMC K15M and HPMC K100M as matrix for intragastric floating drug delivery and also to evaluate the effect of using Cr, Pr, and GMS in two different ratios on the retardation of drug release. The in vitro dissolution profiles of MbH from FTs in 0.1N HCl for 8 h are shown in Figures 3–7. Release profiles are plotting the percent cumulative amount of drug released in 0.1 N HCl against time. Floating tablets formulations (FT-1–FT-12) kept their integrity throughout the release studies, with a slow diminution of the matrix thickness due to polymer dissolution. It was observed that polymers in the matrix undergo simultaneous swelling, dissolution, and diffusion into the bulk medium, resulting in erosion and reduction of the matrix strength. It is also considered that the gas bubbles dissipating from the inside to the outside of the matrix debilitate the matrix structure.44 The reduction in HPMC K15M-based FT size was greater than that in HPMC K100M FTs.
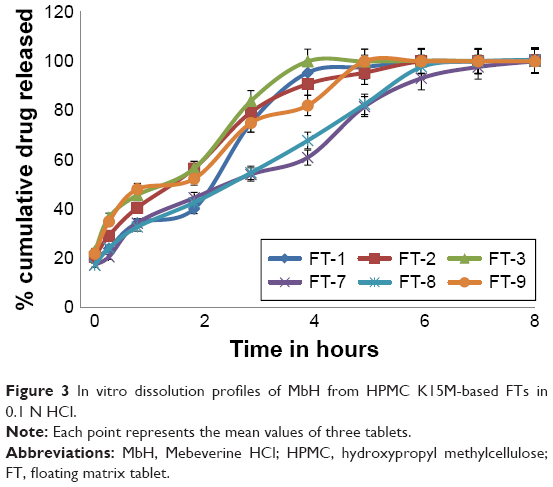 | Figure 3 In vitro dissolution profiles of MbH from HPMC K15M-based FTs in 0.1 N HCl. |
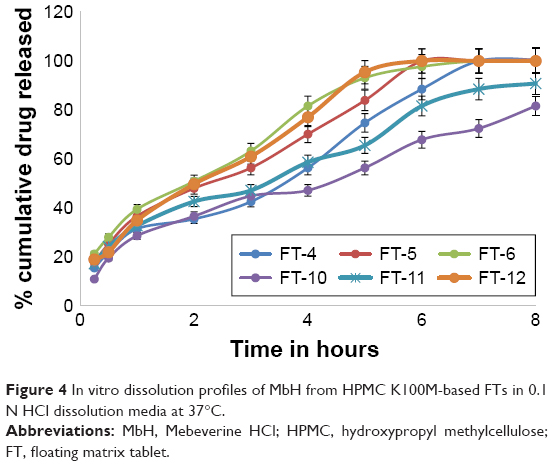 | Figure 4 In vitro dissolution profiles of MbH from HPMC K100M-based FTs in 0.1 N HCl dissolution media at 37°C. |
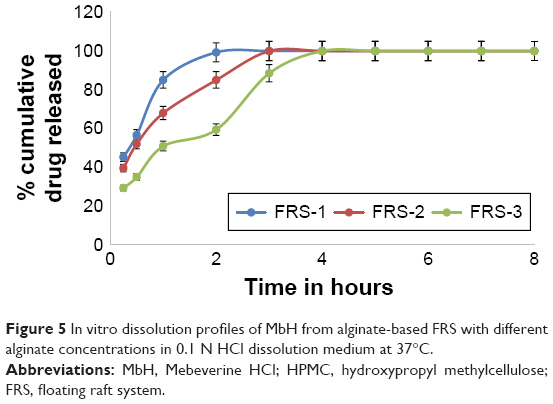 | Figure 5 In vitro dissolution profiles of MbH from alginate-based FRS with different alginate concentrations in 0.1 N HCl dissolution medium at 37°C. |
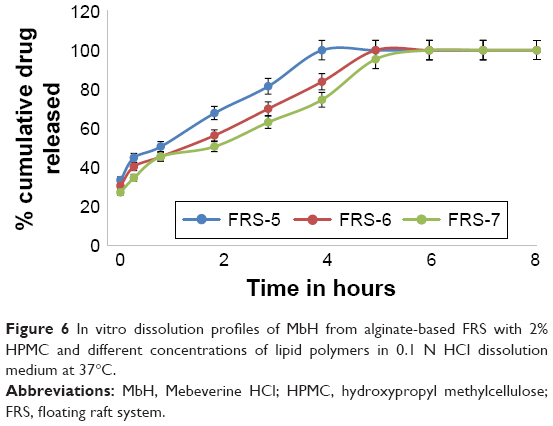 | Figure 6 In vitro dissolution profiles of MbH from alginate-based FRS with 2% HPMC and different concentrations of lipid polymers in 0.1 N HCl dissolution medium at 37°C. |
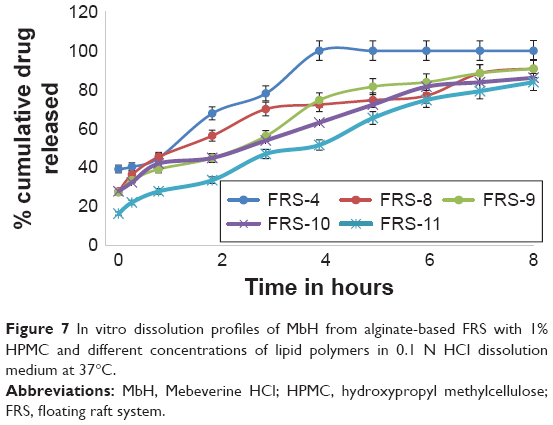 | Figure 7 In vitro dissolution profiles of MbH from alginate-based FRS with 1% HPMC and different concentrations of lipid polymers in 0.1 N HCl dissolution medium at 37°C. |
Figure 3 shows the release profiles of MbH from the HPMC K15M-based floating matrix. FT-1, FT-2, and FT-3 exhibited burst release since about 23%, 28%, and 32% of MbH released in 30 min, respectively. The initial burst effect could be due to rapid dissolution of the drug from the surface while the HPMC K15M undergoes hydration to form a protective gel layer.45 T50 (time required to release 50% of the drug) was 2.1 h, 1.1 h, and 1.00 h for FT-1, FT-2, and FT-3, respectively, and almost 100% of the drug was released within 4 h. So, F1, F-2, and F-3 did not show the expected sustained release characteristics, whereas, formulas F7, F8, and F9 containing higher lipid polymer ratio released 20%, 24%, and 30% of MbH, respectively, in 30 min. The decrease in HPMC content with the accompanying increase in lipid polymer content in the matrix tablets (FT-7, FT-8) lead to a significant decrease in the drug release (P<0.5) in the acidic medium owing to the formation of a hydrophobic insoluble mass that acts as a barrier to drug diffusion,46 and consequently the initial burst effect and release rate were decreased. The results showed that the release of MbH from the prepared formulations was in the following order FT-3> FT-1> FT-2> FT-9> FT-8> FT-7. The slower MbH release rate with the decreased HPMC content could be due to slower penetration of water into the matrix and/or more matrix erosion in addition to the increase in hydrophobic mass. This may be explained by an axial expansion of the tablets.47 Close examination of tablets showed that the extent of their deformation was greater for those of higher HPMC content. That drug dissolution makes more pores in matrix structure facilitated the penetration of water into the matrix interior, promoting diffusion of more drug from the tablets. This might be the main reason why decreasing HPMC content in tablets prolonged drug dissolution significantly and thus met our design objectives. The difference in release between Cr, Pr, and GMS tablets could be interpreted as a result of the Hydrophile-Lipophile Balance (HLB) value of the lipid polymers. GMS, the one with higher HLB value of 3.8, had less capability for sustaining drug release, compared to Pr and Cr, which had lower HLB values of 2. Lipids with lower HLB value readily transform from the lamellar phase to the cubic phase, which is one main cause for sustaining drug release.48
Replacing HPMC K15M with HPMC K100M with the same ratio resulted in FT-4, 5, and 6 and FT-10, 11, and 12 that produced significantly (P<0.05) lower release rate compared to HPMC K15M based tablets (Figure 4). It can be suggested that the relative higher viscosity of the polymer can lead to formation of more viscous matrix, which provides greater diffusional resistance compared to that in the case of the less viscous polymer. A similar explanation was also reported by others.49
Concerning FT4, FT-5, and FT-6 release profiles, it was observed that the rate of drug release was different; 50% of the drug was released after 4.0 h, 2.7 h, and 2 h from FT-4, FT-5, and FT-6, respectively, that could be explained on the basis of HLB values of lipid polymers (as previously discussed).
The in vitro release profiles of FT-10, FT-11, and FT-12 are shown in Figure 4. As can be seen from the Figure, GMS-based FT (FT-12) showed the highest release rate (t50, 2.9 h and t85,4 h), while the Cr-based one showed the slowest release (t50, 5.2 h and t85, 8 h). The release of MbH was in the following order FT-12> FT-11> FT-10. The faster release rate from FT-11 as compared with FT-10 could be explained on the basis of melting temperature of both polymers, and also because Cr has higher melting range than Pr and because greater loss of structure and weakening of bonds between particles at 37°C occur during compression of Pr-based matrices. Our study showed that combined use of lipid-based polymer and HPMC K100M sustained drug release significantly. Hydrophilic polymers such as HPMC K100M rely on water absorption to produce gel swelling and matrix relaxation, which subsequently facilitate drug dissolution and diffusion from the matrix. When a lipid-based excipient is concurrently present in the same matrix, its lipophilicity is able to reduce water uptake rate by the matrix. Consequently, drug dissolution and diffusion from the tablet matrix is reduced to produce a sustained release pattern for a prolonged period of time. Tiwari and Rajabi-Siahboomi45 found that combined use of hydrophilic and hydrophobic excipients was not desirable because immediate tablet disintegration and drug dissolution took place. We did not observe such phenomena in our dissolution studies. Another study by Xiaochen et al50 reported that combined use of lipid-based Cr and hydrophilic Methocel sustained the highly soluble pseudoephedrine release significantly compared to any formulation that was composed of only single matrix excipients. Considering the floating behavior together with the release pattern, FT-10 was considered as the optimum formulation. The matrix of this formulation was used to further investigate in vivo pharmacokinetic parameters in beagle dogs.
In vitro release study from raft system
In this study, FRSs with different combinations of sodium alginate with HPMC K100 M and lipid polymers (Cr and Pr) were prepared. Sodium alginate was used in three different concentrations: 1, 2, and 3% w/v. HPMC K100 was used in 1% and 2% concentrations, while Cr and Pr were used at two levels, 1 and 2%, to prepare eleven floating raft formulations (FRS-1–FRS-11). The release of the drug from the alginate FRS is shown in Figure 5. A significant decrease (P<0.05) in the rate and extent of drug release was observed with the increase in alginate concentration from 1 to 3%, which could be attributed on the basis of polymer matrix density and the increase in the diffusional path length the drug molecules have to traverse with increasing polymer concentration. Preparations formed of alginate and HPMC K100M (FRS-4 and FRS-5) did not show significant retardation in drug release when compared with FRS-3. T50, T85 were (1.0 h, 3 h), (1.2 h, 3.2 h), and (1.1 h, 3.5 h) for FRS-3, FRS-4, and FRS-5, respectively, which could be interpreted on the basis of the hydrophilic nature of both alginate and HPMC and the fact that the matrix formed was not able to produce the intended retardation.
Figures 6 and 7 show the effect of Cr and Pr incorporation at two levels in alginate FRS on MbH release. FRS-6 and FRS-7 release profiles showed significant retardation (P<0.5) in MbH release as compared with FRS-5, which proved that the use of lipid polymers significantly decreased the rate and extent of drug release. Studies reported that when polar amphiphilic lipids placed in gastric fluids reorganize into lipid bilayers forming a reversed micellar phase, the cubic phase as the dominate phase. The stiffness and high viscosity of the cubic phase can provide a slow sustained release of the incorporated drug by slowing its diffusion.43 Pr exhibited more pronounced effect on both rate and extent of MbH release, which could be explained based on viscosity of the polymer. Although both Cr and Pr are polar amphiphilic lipid polymers and both have the ability to control drug release through formation of cubic structure, the higher drug release rate obtained from Cr- compared to Pr-based formulation is attributed to its lower viscosity and incomplete transformation to cubic phase gel.51
Use of 1% of Cr and Pr while decreasing HPMC to 1% in FRS-8 and FRS-9 showed better control of drug release, which could be interpreted on the basis of rapid hydration of HPMC and more rapid penetration of water into the matrix with faster release rate of MbH. Moreover, by increasing Cr to 2% (FRS-10), the rate of drug release decreased, but this decrease was not significant at P<0.05 when compared to FRS-8, while the retardation of drug release was significant when Pr content increased from 1% (FRS-9) to 2% (FRS-11). Results also revealed that the release from FRS-11 is significantly slower than release from FRS-10 (P<0.5), which could be explained on the basis of lipid viscosity as mentioned earlier. Our investigation showed that dissolution of MbH was well prolonged to a large extent with the use of hydrophilic matrix HPMC K100M with lipophilic polymer (Pr). In addition, the increase in Pr concentration with the decrease of HPMC content produced a sustained-release pattern for a prolonged period of time. Based on these results, FRS-11 was used for further investigation in beagle dogs.
Kinetics of release
In order to investigate the release kinetics of MbH from FT and FRS, the data of in vitro release experiments of optimum formulations were treated according to the model-dependent methods, zero order, first order, Higuchi model, Korsmeyer–Peppas model, and Hixson–Crowell model (Table 3). The criteria for selecting the most appropriate model was based on best fit indicated by the value of coefficient of determination (R2) nearer to 1.52 Concerning FT-10, FT-11, and FT-12, the highest values of R2 were obtained after fitting the data into Peppas equation. The value of “n” was calculated to characterize the release as either Fickian diffusion n≤0 or anomalous diffusion (non-Fickian), 0.5< n <1.52 The n values for FT-11and F-12 were 0.59 and 0.61, respectively, which indicates a non-Fickian diffusion mechanism and that drug release was governed by both diffusion and matrix erosion,53 whereas, the value of n in the case of FT-10 (n=0.43) revealed a Fickian diffusion release mechanism of MbH from this FT.
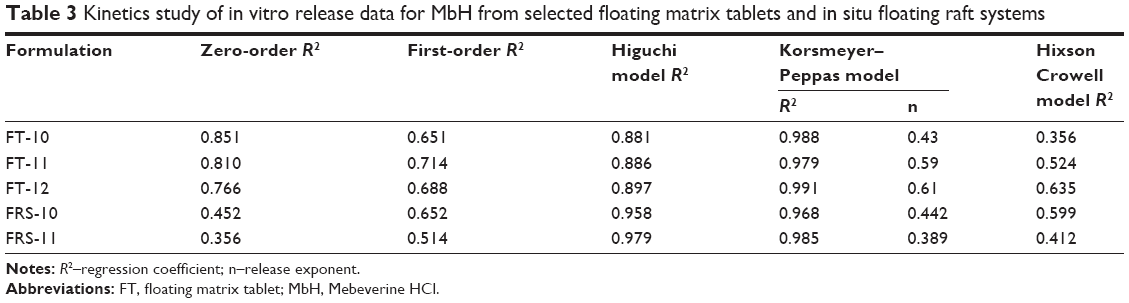 | Table 3 Kinetics study of in vitro release data for MbH from selected floating matrix tablets and in situ floating raft systems |
The regression coefficient (R2) of FRSs FRS-10 and FRS-11 indicated that the Peppas power law equation had the best fit to the experimental data for both formulations evaluated. The values for the release exponent (n) were 0.44 and 0.39 for FRS-10 and FRS-11, respectively, indicating that the mechanism of MbH release from the gels is controlled by Fickian diffusion and that the release rate of MbH from these systems is controlled by diffusion through channels within the structure of the hydrogels. These results are in agreement with the kinetic model reported by Rao and Shelar.54 Abou Youssef et al2 reported the same results regarding the release of metronidazole from sodium alginate and gelan gum in in situ gelling systems.
Pharmacokinetic study
A pharmacokinetic study of the optimized FT (FT-10) and in situ raft system (FRS-11) of MbH compared with marketed MbH product (Duspitaline® retard 200 mg) was done following oral administration of 200 mg of the drug in beagle dogs in three phases. The mean concentration–time profiles for the FT (FT-10), FRS (FRS-11), and commercial product are shown in Figure 8. The pharmacokinetic parameters are shown in Table 4. Studies showed that after oral administration of MbH, only traces of Mebeverine were found in plasma with simultaneous appearance of veratic acid; hence, veratic acid concentrations in plasma were determined for monitoring the therapeutic efficacy of MbH. HPLC chromatograms of veratic acid and SUL (internal standard) after 12 h of oral administration of FRS FRS-11 is shown in Figure 9.
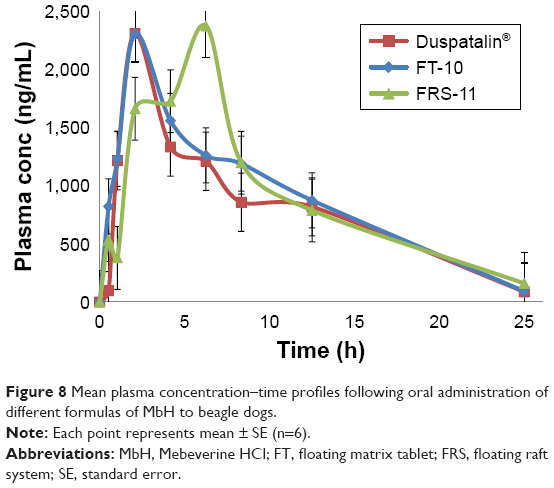 | Figure 8 Mean plasma concentration–time profiles following oral administration of different formulas of MbH to beagle dogs. |
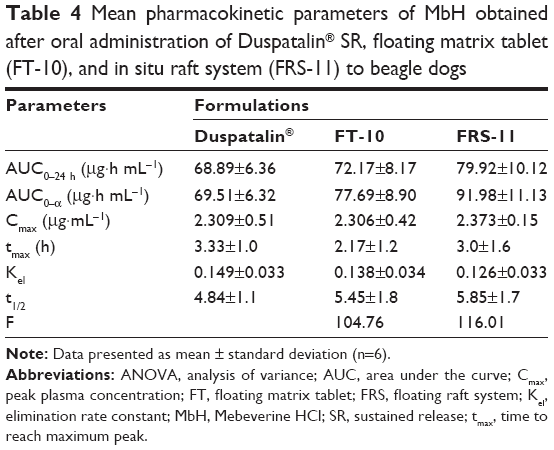 | Table 4 Mean pharmacokinetic parameters of MbH obtained after oral administration of Duspatalin® SR, floating matrix tablet (FT-10), and in situ raft system (FRS-11) to beagle dogs |
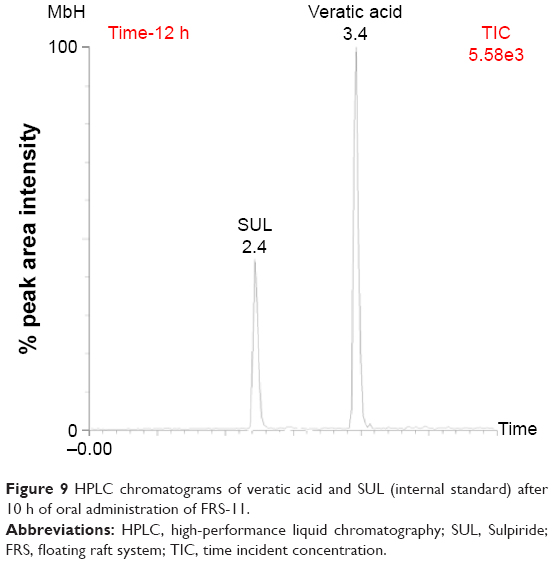 | Figure 9 HPLC chromatograms of veratic acid and SUL (internal standard) after 10 h of oral administration of FRS-11. |
Results revealed that after oral administration of FT-10, FRS-11, and market product to beagle dogs, the drug appeared in plasma after 0.53±0.12 h, 0.51±0.1 h, and 0.70±0.24 h, respectively. Mean peak drug concentration of FRS-11 Cmax (2.37±0.15 μg/mL) was higher than that of the market product “Duspatalin® SR” (2.309±0.51 μg/mL) and FT-10 (2.306±0.42 μg/mL). The mean time to reach the peak concentration (tmax) was comparable, and no statistically significant difference (P>0.05) was observed among the tmax values of the three samples. Moreover, there was no significant difference (P>0.05) in the terminal elimination rate constant among the three products. The AUC0–24 value was 68.89±6.36, 72.17±8.17, and 79.92±10.12 (μg·h mL−1) for the market product, FT-10, and FRS-11, respectively, suggesting that FRS (FRS-11) showed the highest rate and extent of drug absorption, whereas the market product showed the lowest rate and extent of drug absorption. The relative bioavailability of FT-10 and FRS-11 were 104.76% and 116.01%, respectively.
The higher bioavailability of the raft system may be interpreted on the basis of, 1) higher viscosity of Precirol polymer and the formation of dense internal structure matrix, which increases the ability of the raft system to entrap drug and decrease the loss of drug in the curing medium, and 2) the higher polymer concentration in the raft system, which increases the viscosity and swelling of the polymer matrix as well as the formation of a swollen gel with longer diffusion path length that could substantially reduce the penetration of the dissolution medium, so the drug release was retarded. ANOVA applied to log AUC0–∞ and log Cmax data as recommended by USP 24 are shown in Table 5. There are no statistical significant differences between the values of AUC0–∞ and Cmax calculated for both formulations.
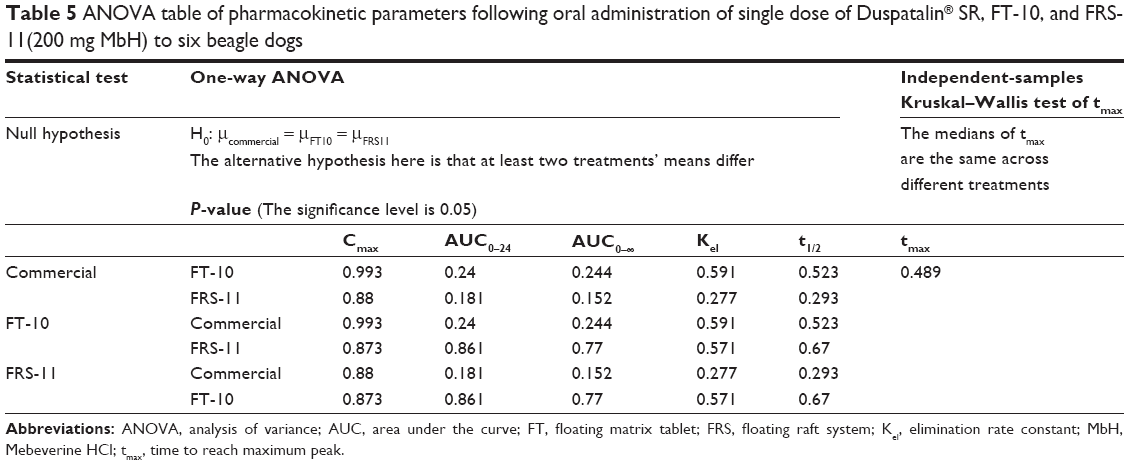 | Table 5 ANOVA table of pharmacokinetic parameters following oral administration of single dose of Duspatalin® SR, FT-10, and FRS-11(200 mg MbH) to six beagle dogs |
Conclusion
This study has demonstrated the suitability of using hydrophilic polymers with lipid polymer to sustain drug release from FTs and raft systems. According to the above results, optimum formulations from both FTs and FRSs were able to control drug release along with acceptable FLt and total floating time >12 h. The in vitro drug release profile was modified by using different lipid polymers in different ratios. Analysis of the release profiles indicated that drug release from optimum formulations occurred through diffusion according to Fickian diffusion release mechanism. Both floating matrix and raft system showed higher relative bioavailability of MbH than the reference “Duspatalin®200 SR.” These results demonstrated that both controlled-released FT and raft system using hydrophilic and lipid polymer would be promising gastroretentive delivery systems for prolonging drug action. However, the raft floating system showed higher concentration and extent of drug absorption in vivo.
Disclosure
The authors report no conflicts of interest in this work.
References
Mudie DM, Amidon GL, Amidon GE. Physiological parameters for oral delivery and in vitro testing. J Mol Pharm. 2010;7:1388–1405. | ||
Abou Youssef NA, Kassem AA, El-Massik MA, Boraie NA. Development of gastro-retentive metronidazole floating raft system for targeting Helicobacter pylori. Int J Pharm. 2015;486:297–305. | ||
Malik R, Garg T, Goyal AK, Rath G. Polymeric nanofibers: targeted gastro-retentive drug delivery systems. J Drug Target. 2015;23:109–124. | ||
Gopalakrishnan S, Chenthilnathan A. Floating drug delivery systems: a review. J Pharm Sci Technol. 2011;3:548–554. | ||
Fukuda M, Peppas NA, McGinity JW. Floating hot-melt extruded tablets for gastro-retentive controlled drug release system. J Control Release. 2006;115:121–129. | ||
Ponchel G, Irache J. Specific and non-specific bio-adhesive particulate system for oral delivery to the GI tract. Adv Drug Del Rev. 1998;34:191–219. | ||
Davies NM, Farr SJ, Kellaway IW, Taylor G, Thomas M. A comparison of the gastric retention of alginate containing tablet formulations with and without the inclusion of excipient calcium ions. Int J Pharma. 1994;105:97–101. | ||
Tadros M. Controllrd-release effervescent floating matrix tablets of ciprofloxacin drochloride: development, optimization and in vitro-in vivo evaluation in health human volunteers. Eur J Pharm Biopharm. 2010;74:332–339. | ||
Sharma S, Prashar M, Sahu R. Floating drug delivery system: incredible revolution. J Pharmacol online. 2011;3:1039–1054. | ||
Arora S, Ali J, Ahuja A, Khar RK, Baboota S. Floating drug delivery systems: a review. J AAPS Pharm Sci Tech. 2005;6:372–390. | ||
Bardonnet PL, Faivre V, Pugh WJ, Piffaretti JC, Falson F. Gastro retentive dosage forms: overview and special case of Helicobacter pylori. J Control Release. 2006;111:1–18. | ||
Kotreka U, Adeyeye M. Gastro retentive floating drug-delivery systems: a critical review. J Crit Rev Ther Drug Carrier Syst. 2011;28:47–99. | ||
Akbari J, Adrangr M, Farid D, Siahi-shadbad M, Saeedi M, Nokhodchi A. The effect of various factors on the release rate of poorly soluble drug (carbamazepine) from hydroxypropylmethylcellulose matrices. J STP Pharma Sci. 2000;10:473–478. | ||
Padmavathy J, Saravanan D, Rajesh D. Formulation and evaluation of ofloxacin floating tablets using HPMC. Int J Pharm Pharm Sci. 2010;3(1):170–173. | ||
Tang M, Alvani K, Tester R. Production and utilization of gastric rafts from polysaccharide combinations to induce satiety: a preliminary study. Nutr Food Sci. 2010;40:155–165. | ||
Dettmar PW, Little SL, Baxter T. The effect of omeprazole pretreatment on rafts formed by reflux suppressant tablets containing alginate. J Int Med Res. 2005;33:301–308. | ||
Maderuelo C, Zarzuelo A, Lanao M. Critical factors in the release of drugs from sustained release hydrophilic matrices. J Control Release. 2011;154:2–19. | ||
Rosenzweig O, Lavy E, Gati I. Development and in vitro characterization of floating sustained release drug delivery systems of polyphenols. J Drug Deliv. 2013;20:180–189. | ||
Longmei W, Ruihua F, Jinhua G, Yanwei X, Guihua H. Generic sustained release tablets of trimetazidine hydrochloride: preparation and in vitro–in vivo correlation studies. Asian J Pharm Sci. 2016;11:417–426. | ||
Hamdani J, Moës AJ, Amighi K. Development and evaluation of prolonged release pellets obtained by the melt pelletization process. Int J Pharm. 2002;45:167–177. | ||
Mayur M, Avani F. Optimization and characterization of mebeverine hydrochloride loaded guar gum microspheres for irritable bowel syndrome. J Carbohydrate Polymers. 2011;86:536–545. | ||
Patel A, Modasiya M, Shah D, Patel V. Development and in vivo floating behavior of verapamil hcl intra gastric floating tablets. J AAPS Pharm SciTech. 2009;10(1):310–315. | ||
Rajinikanth PS, Mishra B. Floating in situ gelling system for stomach site-specific delivery of clarithromycin to eradicate H. pylori. J Control Release. 2008;125(1):33–41. | ||
Ibrahim H. A novel liquid effervescent floating delivery system for sustained drug delivery. Drug Discov Ther. 2009;3(4):168–175. | ||
Hadi MA, Babu VL, Pal N. Formulation and evaluation of sustained release matrix tablets of glimepiride based on combination of hydrophilic and hydrophobic polymers. J Appl Pharm Sci. 2012;2:101–107. | ||
Rao NGR, Hadi MA, Panchal H, Reddy B. Formulation and evaluation of biphasic drug delivery system of Montelukast sodium for chronotherapy. World J Pharm Res. 2012;1:757–775. | ||
Al Remawia M, Al-akayleh F, Salem M, Al Shami M, Badwan A. Application of excipient made from Chitosan-Xanthan as a single component for the controlled release of ambroxol tablet. J Excipients Food Chem. 2013;4:48–57. | ||
Dash S, Murthy PN, Nath L, Chowdhury P. Kinetic modeling on drug release from controlled drug delivery systems. J Acta Pol Pharm. 2010;67:217–223. | ||
Zhang Y, Huo M, Zhou J, et al. DDSolver: an add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010;12:263–271. | ||
Lee S, Zhang W, Liu X, Sun J. Preparation and evaluation of sustained-release azithromycin tablets in vitro and in vivo. Asian J Pharm Sci. 2014;9:155–161. | ||
Walash MI, Sharaf El-din K, El-enan N, Eid M, Shalan S. Simultaneous determination of sulpiride and mebeverine by HPLC method using fluorescence detection: application to real human plasma. Chemist Cent J. 2012;6:1–13. | ||
Radwan M, Abou el Ela A, Hassan M, El-Maraghy D. Pharmacokinetics and analgesic effect of ketorolac floating delivery system. J Drug Deliv. 2015;22(3):320–327. | ||
Wagner J, Nelson E. Kinetic analysis of blood levels and urinary excretion in the absorptive phase after single dose of drug. J Pharm Sci. 1964;53:1392–1404. | ||
Trivedi P, Verma A, Garud N. Preparation and characterization of aceclofenac microspheres. Asian J Pharm. 2008;2:110–115. | ||
Manivannan R, Chakole V. Formulation and development of extended release floating tablet of atenolol. Int J Recent Adv Pharm Res. 2011;3:25–30. | ||
Garg A, Gupta M. Taste masking and formulation development & evaluation of mouth dissolving tablets of levocetrizine dihydrochloride. J Drug Deliv Ther. 2013;3:123–130. | ||
Johnson F, Craig D, Mercer A, Chauhan S. The effects of alginate molecular structure and formulation variables on the physical characteristics of alginate raft systems. Int J Pharma. 1999;159:35–42. | ||
Siew CK, Williams PA, Young NW. New insights into the mechanism of gelation of alginate and pectin: charge annihilation and reversal mechanism. Biomacromolecules. 2005;6(2):963–969. | ||
Kubo W, Miyazaki S, Attwood D. Oral sustained delivery of paracetamol from in situ-gelling gellan and sodium alginate formulations. Int J Pharm. 2003;258(1):55–64. | ||
Pare A, Yadav S, Patil U. Formulation and evaluation of effervescent floating tablet of amlodipine besylate. Res J Pharm Tech. 2008;1:526–530. | ||
Basak S, Jayakumar Reddy B. Formulation and release behaviour of sustained release ambroxol hydrochloride HPMC matrix tablet. Indian J Pharm Sci. 2006;68:594–598. | ||
Singh B, Kim K. Effect of monovalent and divalent cations on the rheological properties of gellan gels. J Control Release. 2000;63:235–259. | ||
Shah J, Sadhale Y, Chilukuri D. Cubic phase gels as drug delivery systems. J Adv Drug Deliv Rev. 2001;47(2):229–250. | ||
Lara-Herna B, Herna A, Villafuerte-Robles L. Effect of stearic acid on the properties of metronidazole/methocel K4M floating matrices. Braz J Pharm Sci. 2009;45:497–505. | ||
Tiwari S, Rajabi-Siahboomi A. Applications of complementary polymers in HPMC hydrophilic extended release matrices. Drug Deliv Technol. 2009;9:20–27. | ||
Tatavarti A, Mehta K, Augsburger L, Hoag S. Influence of methacrylic and acrylic acid polymers on the release performance of weakly basic drugs from sustained release hydrophilic matrices. J Pharm Sci. 2004;93:2319–2331. | ||
Ghosal K, Rajabalaya R, Chakraborty S, Nanda A. Formulation and characterization of both hydrophilic and hydrophobic HPMC based hydrogels containing diclofenac potassium. Latin Am J Pharm. 2010;29:1137–1143. | ||
Lynch M, Spicer P. Functionalized cubic liquid crystalline phase materials and methods for their preparation and use. Google patents. 2003. | ||
Kumar R, Patil S, Patil M. Formulation and evaluation of effervescent floating tablet of famotidine. Int J Pharm Tech Res. 2009;1(3):754–763. | ||
Xiaochen G, Daryl J, Estelle R, Keith J. Evaluation and comparison of five matrix excipients for the controlled release of acrivastine and pseudoephedrine. Drug Dev Ind Pharm. 2004;30:1009–1017. | ||
Duclos R, Bourret E, Brossard C. Rheology of polyol behenates and drug release from matrix monolithic capsules. Int J Pharm. 1999;182(2):145–154. | ||
Costa P, Sousa Lobo JM. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13:123–133. | ||
Barakat N, Elbagory I, Almurshedi A. Formulation, release characteristics and bioavailability study of oral monolithic matrix tablets containing carbamazepine. J AAPS Pharm SciTech. 2009;9(3):931–938. | ||
Rao M, Shelar S. Controlled release ion sensitive floating oral in situ gel of a prokinetic drug using gellan gum. Ind J Pharm. 2015;49(2):158–167. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.