Back to Journals » Drug Design, Development and Therapy » Volume 8
Formulation development for the orexin receptor antagonist almorexant: assessment in two clinical studies
Authors Dingemanse J, Gehin M, Cruz HG, Hoever P
Received 7 February 2014
Accepted for publication 10 March 2014
Published 28 April 2014 Volume 2014:8 Pages 397—403
DOI https://doi.org/10.2147/DDDT.S62118
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Jasper Dingemanse, Martine Gehin, Hans Gabriel Cruz, Petra Hoever
Department of Clinical Pharmacology, Actelion Pharmaceuticals Ltd, Allschwil, Switzerland
Abstract: Almorexant, a dual orexin receptor antagonist, was investigated for the treatment of insomnia. The following observations initiated further formulation development: the active pharmaceutical ingredient (API) was sticking to the apparatus used during tablet compression; almorexant has an absolute bioavailability of 11.2%; and almorexant modestly decreased the latency to persistent sleep by 10.4 minutes in patients. Two randomized crossover studies were performed to investigate the pharmacokinetics of several new formulations in healthy subjects. In study I, the old “sticky” tablet was compared to two new formulations developed to prevent sticking: a qualitatively similar tablet but with a larger API crystal size and a tablet with 30% more excipients as well as a larger API crystal size. This latter formulation was available in two strengths. The geometric mean ratios and 90% confidence interval of the area under the curve (AUC) were within the bioequivalence range of 0.80–1.25 for the different comparisons between formulations. In study II, 100 mg of the reference tablet was compared to 25 and 50 mg of a liquid-filled hard gelatin capsule developed to increase the bioavailability of almorexant. The geometric mean ratios of the maximum concentration and AUC comparing the new 25 and 50 mg capsule formulations to the reference tablet did not exceed 0.25 and 0.50, respectively, indicating that the new capsule formulation did not increase the maximum concentration of or the total exposure to almorexant. In conclusion, a new tablet was developed but formulation development aimed at increasing the bioavailability of almorexant failed.
Keywords: almorexant, orexin receptor antagonist, pharmacokinetics, formulation development, healthy subjects
Introduction
The orexin system, which was discovered in the late nineties,1,2 has been shown to play a central role in the regulation of arousal and sleep–wake balance,3–5 and antagonism of this system is a new approach for the treatment of insomnia.6,7 Almorexant was the first orexin receptor antagonist investigated in patients for the treatment of insomnia. In the proof-of-concept study, almorexant significantly improved sleep efficiency and effects on secondary endpoints indicated that almorexant enabled and maintained sleep in these patients. However, the latency to persistent sleep (LPS), which was 46.9 minutes in placebo-treated subjects, was only reduced by 10.4 minutes in patients treated with a dose of 100 mg almorexant,8 ie, the dose used in Phase III. At this dose, the median time to the Cmax (maximum plasma concentration), tmax, was 1.0 hour after single-dose administration to healthy subjects.9 A new formulation could potentially result in more rapid absorption of almorexant, leading to a more pronounced effect on LPS. In order to achieve this goal, further formulation work was undertaken.
Almorexant is a lipophilic compound with a log P-value >5 (Actelion Pharmaceuticals Ltd, data on file) and its absolute oral bioavailability is 11.2%, most likely due to extensive first-pass metabolism.10,11 Together, these features make this compound a good candidate for a lipid-based formulation. Such formulations have the potential to enhance the overall extent of absorption through improved dissolution and solubilization in the intestinal tract and increased mucosal permeability. In addition, the proportion of absorbed drug transported to the systemic circulation via the intestinal lymph may be increased. The latter results in bypassing the liver and thus a decrease of first-pass metabolism, thereby increasing absolute oral bioavailability12,13 and potentially reducing tmax.14 This could lead to a quicker onset of action. Therefore, a new lipid-based formulation of almorexant was developed. The use of a lipid-based formulation aimed at increasing oral bioavailability was successful for a number of drugs such as cyclosporine and some HIV protease inhibitors.15 Prior to testing in humans, the new self-emulsifying capsule formulation of almorexant was first investigated in animals. In dogs, exposure to almorexant was 3-fold higher with the new capsule formulation when compared to the reference tablet formulation, but tmax was similar (Actelion Pharmaceuticals Ltd, data on file).
A capsule formulation was used in early clinical pharmacology studies of almorexant, whereas later studies used a tablet formulation. A relative bioavailability study showed a near identical pharmacokinetic profile for both formulations.16 However, during the compression stage of the manufacturing of the tablet formulation, it was noted that the active pharmaceutical ingredient (API) was sticking to the compression punches on the tableting machine and so the formulation needed to be changed as it was not commercially viable. Therefore, two different tablet formulations were developed.
The present report describes the results of two relative bioavailability studies performed in healthy subjects in which the pharmacokinetics of different newly developed tablet and capsule formulations were compared to a reference tablet formulation.
Methods
Subjects
Twenty and 24 healthy subjects were enrolled in studies I and II, respectively. They were male only in study I whereas both sexes participated in study II. Each subject had to be between 18 and 45 years of age with a body mass index of 18–28 kg/m2 and judged to be in good health based on medical history, physical examination, vital signs, electrocardiogram (ECG), and clinical laboratory tests. Female subjects were required to use a reliable method of contraception from screening until 30 days after the last study drug administration. Participating subjects had to be non-smokers and given that almorexant is a substrate of cytochrome P450 3A4,17 consumption of grapefruit and grapefruit juice was forbidden from screening until the end-of-study examination. The latter took place after the last blood sample for pharmacokinetics was withdrawn, ie, 120 hours after the last study drug administration in treatment period 3 (study II) or 4 (study I).
A priori, no statistical hypothesis was formulated, and therefore, the sample size in both studies was based on empirical considerations. A precision estimate approach was applied in both studies comparing the variables area under the curve (AUC0–∞) and Cmax of the test formulations versus the reference tablet formulation. Using data from a previous study and a mixed-model analysis, intra-subject standard deviation values on a log scale of 0.31 for AUC0–∞ and 0.55 for Cmax of the reference tablet formulation were estimated. With a sample size of 18 evaluable subjects, the 90% confidence interval (CI) for the point estimate of the true ratio on the original scale (dose-corrected for study II) would have been (0.83, 1.20) for AUC0–∞ and (0.73, 1.37) for Cmax if the real ratio was 1. In order to have at least 18 evaluable subjects, 20 subjects were enrolled in study I and 24 in study II.
The protocol and other study-related documents of studies I and II were approved by the ethics committees of the Bayerischen Landesärztekammer, Munich, Germany and Landesärztekammer Baden-Württemberg, Stuttgart, Germany, respectively. Both studies were compliant with the Declaration of Helsinki. The studies were performed in sequence and the results from the first study dictated the choice of the reference formulation in the second study.
Study design
Both studies had a single-center, open-label, randomized, 3- (study II) or 4-way (study I) crossover design. In order to prevent sticking of the API during the manufacturing process, API drug loading needed to be decreased. This was done by increasing the crystal size alone or by increasing both the excipients in the extra-granular phase of the tablet as well as the crystal size when compared to the tablet formulation previously used in clinical studies. In study I, the four treatments consisted of A) a single 200 mg dose of a tablet formulation with increased crystal size administered as 2×100 mg, B) a single 200 mg dose of a tablet formulation with increased crystal size and excipients in the extra-granular phase administered as 2×100 mg, C) a single 200 mg dose of the old formulation used in previous studies administered as 2×100 mg, and D) a single 200 mg dose of a tablet formulation with increased crystal size and excipients in the extra-granular phase administered as one 200 mg tablet.
In study II, which was designed and performed after the results of study I had become available, the three treatments consisted of A) a tablet formulation with increased crystal size and excipients in the extra-granular phase administered as 2×100 mg which was identical to treatment B in study I, B) a single 25 mg dose of a liquid-filled hard gelatin capsule developed to increase the absolute bioavailability of almorexant, administered as one 25 mg capsule, and C) a single 50 mg dose of the liquid-filled hard gelatin capsule administered as two 25 mg capsules. The liquid-filled hard gelatin capsule contained ascorbyl palmitate, PEG40 hydrogenated castor oil (Cremaphor® RH40) as emulsifier, and propylene glycol monolaurate (Lauroglycol® 90) as co-emulsifier. In both studies, almorexant was administered to subjects in the fasted state, treatment periods were separated by a washout of about 10 days (which justified the assumption of no carryover effect), and subjects were confined to the study center from the evening before until 24 hours after each almorexant administration. All other assessments were performed on an ambulatory basis for which the subjects returned to the study center.
Blood samples for the determination of almorexant were withdrawn predose at each administration and at 0.17, 0.33, 0.5, 0.67 (study II only), 0.75 (study I only), 0.83 (study II only), 1, 1.25 (study II only), 1.5, 2, 3, 4, 6, 8, 10, 12, 24, 36, 48, 60, 72, 96, and 120 hours thereafter. Safety was evaluated by monitoring adverse events, clinical laboratory variables, vital signs, ECG, and physical examination. Assessments were performed throughout the study.
Bioanalytical method
Plasma concentrations of almorexant were determined using a validated liquid chromatography method with a limit of quantification of 0.05 ng/mL as previously described.10 The performance of the method was monitored using quality control samples. In study I, inter-assay precision was ≤8.3% and inaccuracy was ≤4.7%, whereas in study II inter-assay precision was ≤8.6% and inaccuracy was ≤4.3%.
Data analysis
Non-compartmental analysis was performed using WinNonlin (v5.2.1; Pharsight Corporation, Mountain View, CA, USA). AUC0–t was calculated according to the linear trapezoidal rule using the measured concentration–time values above the limit of quantification. AUC0–∞ was calculated by combining AUC0–t and AUCextra. AUCextra represents an extrapolated value obtained by Ct/λZ, where Ct is the last plasma concentration measured above the limit of quantification and λZ represents the elimination rate constant determined by log-linear regression analysis of the measured plasma concentrations of the terminal elimination phase. The half-life of almorexant was calculated as follows: t1/2=ln 2/λZ. AUC0–24h was determined as a measure of exposure in study II because no reliable estimate of t1/2 could be obtained due to the fact that the concentration of almorexant in most plasma samples taken 48 hours and later after the administration of 25 mg of the liquid-filled hard gelatin capsule was below the limit of quantification. The variables Cmax and tmax were directly read from the individual plasma concentration–time profiles. Pharmacokinetic parameters were calculated on the basis of the scheduled blood sampling time points. The real time points were only used if they deviated more than 5% from the theoretical ones.
Pharmacokinetic variables were summarized with geometric mean and 95% CI or for tmax with median and minimum and maximum values. Differences between treatments were explored by calculating geometric mean ratios and 90% CI for the variables Cmax and AUC (AUC0–∞ for study I and AUC0–24h for study II). Cmax and AUC were assumed to be log normally distributed. Differences between treatments for tmax were explored using the median difference and its 90% CI. A non-parametric 90% CI for the difference between the reference and test formulations was calculated for tmax using the Hodges–Lehmann estimate.18 This latter analysis ignored any possible period effect.
Results
Subjects
In study I, the 20 enrolled men had a mean age of 31 years (range 23–44 years) and a mean body mass index of 24.1 kg/m2 (range 20.1–28.4 kg/m2). Nineteen completed the study as per protocol and were included in the pharmacokinetic analysis whereas one subject prematurely withdrew from the study because of an adverse event (cold with nasal congestion). The population in study II consisted of 24 subjects, 12 men and 12 women. Their average age and body mass index were 35 years (range 22–45 years) and 23.6 kg/m2 (range 19.4–27.6 kg/m2) and all but two completed the study according to protocol and were included in the pharmacokinetic analysis. One male subject withdrew for personal reasons, whereas a pregnancy was discovered in a female subject. All subjects in both studies were included in the safety analyses.
Pharmacokinetic results
Irrespective of the treatment administered in study I, almorexant plasma concentrations quickly increased and reached a maximum 0.8 hours after drug intake. Thereafter, plasma concentrations decreased rapidly to approximately 10% of Cmax over the course of 8 hours (Figure 1 and Table 1). Exploratory statistical analysis showed no difference between study I formulations for the variables tmax and AUC0–∞; the geometric mean ratio and its 90% CI were entirely within the bioequivalence range of 0.80–1.25 (Table 2). Compared to the old “sticky” formulation (treatment C), the Cmax of tablet formulation with increased crystal size (treatment A) was 15% lower, whereas that of the tablet with increased crystal size and excipients in the extra-granular phase (treatment B) was 22% lower (Table 2). The 90% CI of the geometric mean ratio for Cmax was not entirely within the bioequivalence range for both comparisons (Table 2). Comparing the different strengths of the tablet formulation with increased crystal size and excipients in the extra-granular phase (treatment D vs B) indicated similar exposure as evidenced by a 90% CI of the ratio of the geometric mean for AUC0–∞ within the bioequivalence range, but Cmax was about 19% lower after administration of 200 mg as a single 200 mg tablet compared to 2×100 mg tablets (Tables 1 and 2).
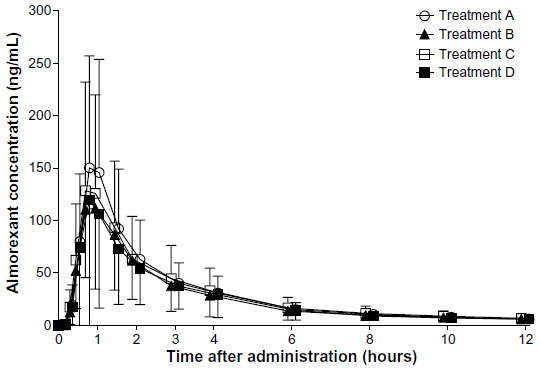 | Figure 1 Arithmetic mean plasma concentration–time profiles of almorexant in healthy male subjects (N=19). |
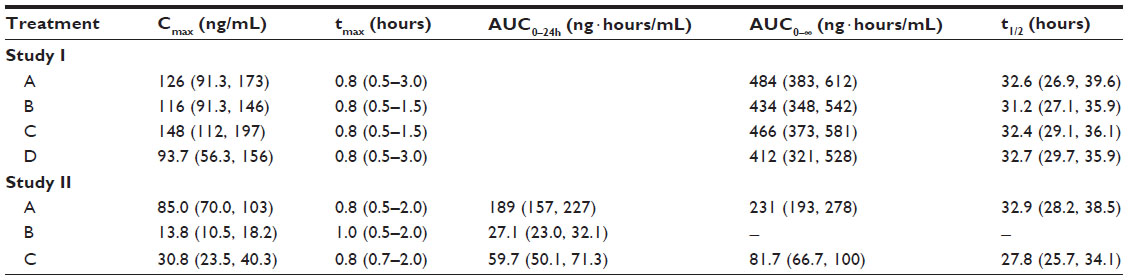 | Table 1 Geometric mean and 95% confidence interval (or median and range for tmax) for pharmacokinetic variables of almorexant after single-dose administration in study I (N=19) and study II (N=22) |
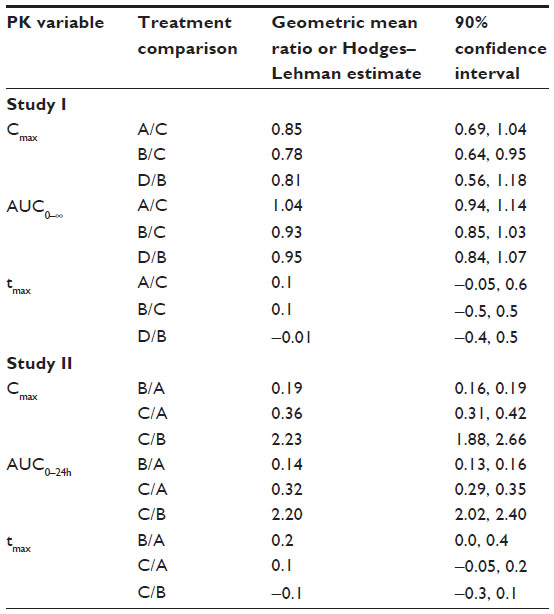 | Table 2 Geometric mean ratios for Cmax and AUC and Hodges–Lehman estimate for tmax and their corresponding 90% confidence intervals for the comparisons between test and reference formulations in studies I (N=19) and II (N=22) |
In study II, the plasma concentrations of almorexant were lower after administration of the new lipid-based capsule formulation when compared to the reference tablet formulation. The shape of the plasma concentration–time profile was similar for both formulations (Figure 2). Doubling the dose of the capsule formulation resulted in an approximate doubling of exposure to almorexant based on both Cmax and AUC0–24h as evidenced by geometric mean ratios of 2.23 and 2.20, respectively (Table 2).
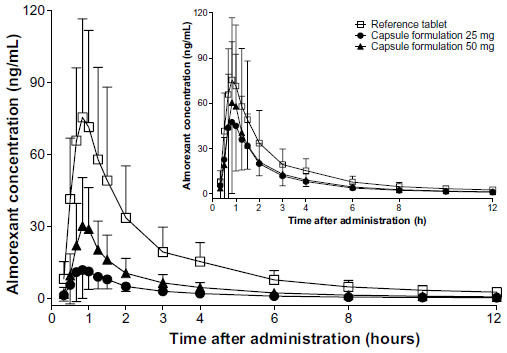 | Figure 2 Arithmetic mean plasma concentration–time profiles of almorexant in healthy male subjects (N=22) comparing a single dose of 100 mg of the tablet formulation with increased crystal size and excipients in the extra-granular phase to doses of 25 and 50 mg of the liquid-filled hard gelatin capsule formulation. The insert shows the profiles normalized to a dose of 100 mg. |
Safety results
Adverse events that were reported by more than one subject in each of the two studies are shown in Table 3. In both studies, fatigue, somnolence, and headache were the most frequently reported adverse events. The incidence of these adverse events was similar for all four treatments in study I, whereas in study II, the incidence of fatigue and somnolence but not that of headache appeared lower after administration of the test formulation when compared to the reference tablet. There were no clinically relevant treatment-related changes of laboratory variables, vital signs, body weight, body mass index, or ECG variables in either study (data not shown).
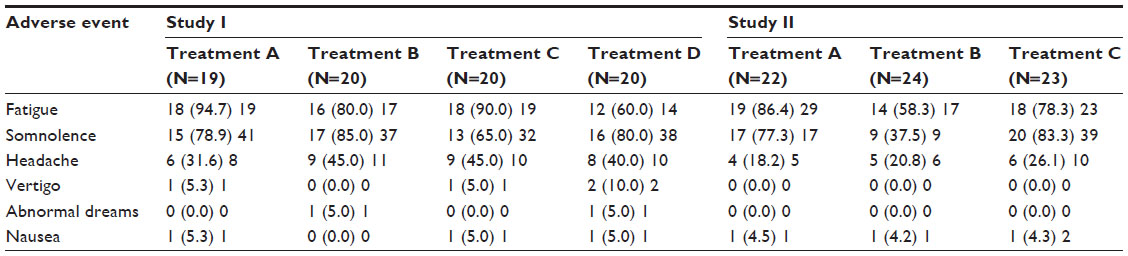 | Table 3 Adverse events occurring in more than one subject in studies I and II by treatment |
Discussion
Here we report the results of two clinical studies in which the pharmacokinetics of different almorexant formulations were compared. The pharmacokinetic results in study I are in good agreement with results obtained previously, although t1/2 was longer in the present study (geometric mean of 32.6 hours vs 21.7 hours).16 This latter finding may be readily explained by the blood sampling scheme which was extended to 120 hours after drug administration in the present study as compared to 72 hours in the previous study resulting in a better characterization of the terminal elimination phase. Another feature which may have contributed is the use of a more sensitive bioanalytical assay.
In study I, the pharmacokinetics of almorexant were similar irrespective of the formulation administered. This result provides a perfect bridge between previous studies performed with the old tablet formulation and future studies with a new formulation. During the up-scaling of the manufacturing process to produce larger batches of the tablet formulation with only increased crystal size, it was noted that API was still sticking, and therefore, this formulation was abandoned. Thus, for later studies, the tablet formulation with both increased excipients in the extra-granular phase and crystal size was chosen, and in fact, was used as the reference formulation in study II. Based on the AUC, the 100 and 200 mg tablets of this formulation, administered as a single 200 mg or as 2×100 mg dose, are similar, indicating dose proportionality, and therefore, both strengths are appropriate for use in clinical studies. Given the large inter-subject variability, the observed difference in Cmax is not considered clinically relevant when comparing the 100 and 200 mg tablets of the tablet formulation with both increased excipients in the extra-granular phase and crystal size.
Most compounds are absorbed by enterocytes and then transported via the portal vein to the liver prior to reaching the systemic circulation. Highly lipophilic drugs may gain access to the intestinal lymphatic system by associating with lymph lipoproteins in the enterocyte and thereby effectively bypassing the liver.12 The use of self-emulsifying drug delivery systems is a fairly recent technology to increase the bioavailability of highly lipophilic drugs.19 Such formulations typically contain a mixture of drug, lipids, emulsifiers, and one or more of hydrophilic co-solvents/co-emulsifiers.19 Based on the promising findings obtained in the dog bioavailability study, testing in humans was initiated in healthy subjects. However, rather than increasing almorexant plasma concentrations, the extent of absorption tended to be smaller with the new formulation. Other than species differences (eg, ratio of surface to lumen area, motility, lymphatic system) no good explanation for these results can be provided. Since clinical usefulness of the new lipid-based formulation could not be demonstrated, this avenue of research was not further pursued. Differences in gastric acidity between dogs and humans could also have contributed.
In both studies, almorexant showed a safety/tolerability profile in accordance with previously reported data8,9 and no new safety findings were detected. The most frequently reported adverse events (fatigue and somnolence) are those expected from a sleep-inducing compound. The lower incidence of these adverse events observed in study II with the new capsule formulation are in line with the observed lower exposure to almorexant when compared to the reference tablet which was administered at a higher dose. However, it should be noted that subject numbers are small, no placebo treatment was included, and neither study was powered to detect a difference in any safety variable.
In conclusion, a new tablet formulation was developed that has similar pharmacokinetic properties as the old formulation but which does not have the same sticking problem of the API during manufacturing as the old formulation. However, the development of a formulation designed to increase bioavailability and/or reduce the tmax of almorexant did not succeed.
Acknowledgments
Study I was conducted at Harrison Clinical Research Deutschland GmbH, Munich, Germany with Stephan de la Motte as principal investigator. Study II was conducted at PHAROS GmbH Clinical Research, Ulm, Germany with Rudolf A Theodor as principal investigator. Almorexant concentrations were measured at Inovalab, Reinach, Switzerland under the supervision of Stefan König and Mirko Glinski. The authors would like to thank Paul van Giersbergen for providing editorial assistance. Both clinical studies were funded by Actelion Pharmaceuticals Ltd, Allschwil, Switzerland.
Disclosure
All authors had support from Actelion Pharmaceuticals Ltd for the submitted work. No competing interests have been raised.
References
de Lecea L, Kilduff TS, Peyron C, et al. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A. 1998;95:322–327. | |
Sakurai T, Mieda M, Tsujino N. The orexin system: roles in sleep/wake regulation. Ann N Y Acad Sci. 2010;1200:149–161. | |
Bonnavion P, de Lecea L. Hypocretins in the control of sleep and wakefulness. Curr Neurol Neurosci Rep. 2010;10:174–179. | |
Inutsuka A, Yamanaka A. The regulation of sleep and wakefulness by the hypothalamic neuropeptide orexin/hypocretin. Nagoya J Med Sci. 2013;75:29–36. | |
Sakurai T, Amemiya A, Ishii M, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. | |
Nishino S. The hypocretin/orexin receptor: therapeutic prospective in sleep disorders. Expert Opin Investig Drugs. 2007;16:1785–1797. | |
Winrow CJ, Renger JJ. Discovery and development of orexin receptor antagonists as therapeutics for insomnia. Br J Pharmacol. 2013; 171(2):283–293. | |
Hoever P, Dorffner G, Beneš H, et al. Orexin receptor antagonism, a new sleep-enabling paradigm: a proof-of-concept clinical trial. Clin Pharmacol Ther. 2012;91:975–985. | |
Hoever P, de Haas S, Winkler J, et al. Orexin receptor antagonism, a new sleep-promoting paradigm: an ascending single-dose study with almorexant. Clin Pharmacol Ther. 2010;87:593–600. | |
Hoch M, Hoever P, Zisowsky J, Priestley A, Fleet D, Dingemanse J. Absolute oral bioavailability of almorexant, a dual orexin receptor antagonist, in healthy human subjects. Pharmacology. 2012;89:53–57. | |
Dingemanse J, Hoever P, Hoch M, et al. Elucidation of the metabolic pathways and the resulting multiple metabolites of almorexant, a dual orexin receptor antagonist, in humans. Drug Metab Dispos. 2013;41:1046–1059. | |
Porter CJ, Charman WN. Transport and absorption of drugs via the lymphatic system. Adv Drug Deliv Rev. 2001;50:61–80. | |
Yáñez JA, Wang SW, Knemeyer IW, Wirth MA, Alton KB. Intestinal lymphatic transport for drug delivery. Adv Drug Deliv Rev. 2011;63:923–942. | |
Song WH, Yeom DW, Lee DH, et al. In situ intestinal permeability and in vivo oral bioavailability of celecoxib in supersaturating self-emulsifying drug delivery system. Arch Pharm Res. 2013. DOI 10.1007/s 12272-013-0202-7. | |
O’Driscoll CM. Lipid-based formulations for intestinal lymphatic delivery. Eur J Pharm Sci. 2002;15:405–415. | |
Hoch M, Hoever P, Haschke M, Krähenbühl S, Dingemanse J. Food effect and biocomparison of two formulations of the dual orexin receptor antagonist almorexant in healthy male subjects. J Clin Pharmacol. 2011;51:1116–1121. | |
Shakeri-Nejad, Hoch M, Hoever P, Dingemanse J. Influence of mild and moderate liver impairment on the pharmacokinetics and metabolism of almorexant, a dual orexin receptor antagonist. Eur J Pharm Sci. 2013;49:836–844. | |
Hodges JL, Lehman EL. Estimation of location based on ranks. Ann Math Stat. 1963;34:598–611. | |
Singh B, Bandopadhyay S, Kapil R, Singh R, Katare O. Self-emulsifying drug delivery systems (SEDDS): formulation development, characterization, and applications. Crit Rev Ther Drug Carrier Syst. 2009;26:427–521. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.