Back to Journals » Journal of Multidisciplinary Healthcare » Volume 15
Food Regime for Phenylketonuria: Presenting Complications and Possible Solutions
Authors Kumar Dalei S, Adlakha N ![]()
Received 23 July 2021
Accepted for publication 11 October 2021
Published 18 January 2022 Volume 2022:15 Pages 125—136
DOI https://doi.org/10.2147/JMDH.S330845
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Sudipt Kumar Dalei, Nidhi Adlakha
Regional Center for Biotechnology, NCR Biotech Science Cluster, Faridabad, Haryana, India
Correspondence: Nidhi Adlakha Email [email protected]
Abstract: In the category of rare inherited genetic disorders, phenylketonuria is a prominent example. Here, the defective phenylalanine hydroxylase enzyme fails to catalyze conversion of phenylalanine to tyrosine. This leads to not only excess deposition of phenylalanine leading to phenylalanine toxicity but also precludes the production of important glutamatergic and cholinergic neurotransmitters, leading to epileptic disorders, microcephaly, low intelligence quotient etc. For long, specialized food products are considered as preferred solution to prevent disease outcome. Different medical diets are developed for managing phenylketonuria includes amino acid mixtures, protein hydrolysates, cofactor-based therapy, large neutral amino acids and glycomacropeptides. However, despite the advent of alternate forms of diet products, the central form of treatment has still been free amino acid mixture. The formulated diet is by and large expensive and in-depth evaluation of several factors which contribute to the expense of medicated diet is requisite to create effective yet affordable avenues for management of disease. For this, we have discussed the role of various factors involved in increasing price of medicated diet and presented possible solutions to it. We have also extensively reviewed prevalence of disease, commercial diet for PKU patients, and their associated limitations. Overall, this is the first attempt to present a holistic view of balance between the overall impact of diet associated therapy and weighing it against the associated finances incurred.
Keywords: phenylalanine hydroxylase, phenylketonuria, cofactor, large neutral amino acids, glycomacropeptides, protein engineering, turnover number, specificity
Introduction
Phenylketonuria (PKU) is an autosomal recessive metabolic disorder where blood phenylalanine is not metabolized to tyrosine resulting in toxic accumulation of phenylalanine in the body causing low intelligence quotient, epileptic seizures, intellectual disability, microcephaly, growth failure, poor skin pigmentation and severe intellectual impairments.1–11 It has a prevalence ratio of 1 in 12,000 babies overall, however the numbers vary from region to region. PKU incidence was reported to be 1.5 per 10,000 and 1.7 per 10,000 for girls and boys respectively12 suggesting that male and female genders are affected almost equally.
Based on phenylalanine blood levels, PKU is categorized into three types viz. classical PKU, mild PKU and mild hyperphenylalaninemia. The phenylalanine blood levels in classical PKU patients is more than 1200 μmol/L, for patients having mild PKU it ranges between 600–1200 μmol/L and for those having mild hyperphenylalaninemia the levels are less than 600 μmol/L,1,10,13 which is 10–20 folds greater than normal blood phenylalanine levels.14
A strict diet plan has to be followed by the PKU patients in order to maintain normal phenylalanine levels in the body where high content proteinaceous foods such as fish, meat, eggs and dietary products should be avoided.1,10,13,15 The condition is critical especially for neonates born with PKU as their sole diet includes proteinaceous mother’s milk and early intervention is critical to avoid any form of irreversible brain damage. Evidence supporting the importance of medicated diet in PKU management includes collaborative study done in USA where it has been shown that discontinuation of low-phenylalanine diet leads to decline in academic performance as well as rise in behavioural and psychosocial problems.1 Hence, special PKU formulations are crucial to counter the problems associated with phenylketonuria and a thorough research in this field is required for enhancing the lifestyle of the patients suffering from phenylketonuria. It has been demonstrated that early diagnosis and carefully planned diet regimen in consultation with paediatricians and healthcare professionals helps drastically reduce the ill-effects of phenylketonuria in children.16,17
Synthetic free amino acid cocktails (without phenylalanine) and protein hydrolysates containing oligopeptides with reduced phenylalanine are currently available diets products for PKU patients providing protein equivalents worth fifty to ninety percent.10 The other sources of low phenylalanine diet are glycomacropeptides and large neutral amino acids (LNAA).10,15,18,19 Despite this, its extensive production and usage is limited owing to (1) its tedious chemical processes and sophisticated purification techniques; (2) palatability of phenylalanine free diet; (3) nutritional deficiencies in commercial mixtures; and (4) expensive formulated end product.20–35 To this end, this review article discusses efforts laid by different research groups in developing palatable and affordable diet for PKU patients. Furthermore, the aim is to provide an overall vision regarding the current situation of commercial PKU diet in terms of cost and availability and more importantly how we as researchers and society as a whole should progress towards tackling the burning issues of creating cost effective and sustainable solutions for improving the lifestyle of PKU patients mentally, physically and financially.36,37
Phenylketonuria Epidemiology
Variations exist in the PKU prevalence around the world. In Europe, it is one in ten thousand and is higher for some particular areas of Europe such as Germany, Estonia, Ireland, Spain and Scotland.38–41 One newborn is detected in every two thousand and six hundred births in Turkey because of high consanguinity within the population (Figure 1A). On the other hand, Finland has the lowest prevalence with one in one hundred and thousand cases. The estimates of prevalence rates in Asia varies from about one per fifteen thousand to one per one hundred and fifteen thousand and in USA the prevalence rate is one in 15,000.19,38,39,41–43 Some countries like Iran and Turkey are not only known to have high prevalence ratio but along with that are known to display extreme proportions of consanguinity who are suffering from PKU.44,45 Studies conducted between 1964–2017 report the highest PKU prevalence in Turkey (38.13), while the lowest was observed in Thailand (0.3) (Figure 1B). Overall, the highest prevalence belonged to eastern Mediterranean region and the lowest observed in southeast Asia.43. The high incidence ratio across the globe clearly indicates the need for development of affordable treatment solutions for appropriate disease management.
 |
Figure 1 Incidence of phenylketonuria worldwide. (A) Heat map indicates incidence ratio varies between 0–0.4 per every 1000 newborns, with maximum in Turkey. (B) Pooled prevalence ratio indicating highest prevalence in Eastern Mediterranean countries. Notes:Data from Shoraka et al.43 |
Financial Burden Associated with Disease Management
Personal financial toll of PKU is daunting problem owing to expensive formulated diet and also the time spent in visiting clinical centers for disease management and consultations. Studies report annual costs for special diet for both children and adults (formula or low-protein diet), tantamount to $6400 and $9000 respectively (Table 1). Additionally, the disease management demands over three hundred hours annually in shopping and preparing special PKU diets46 (Figure 2). A lumpsum amount of $42,830 worth spending by PKU patient was reported to have been calculated by a team of modelling analysts at Wisconsin, USA in the year 1982 which would amount to $92,000 in 2018.21,47 Moreover, the expenditures incurred by addition of LNAAs also adds to the existing cost of medicated food stuffs (Table 2). On top of that, third vendors and pharmacists charge 200 to 300% of the original manufacturing price35 which overall accounts for huge financial burden on families. Annual estimates for cost expenditure on PKU medical formulations over four age groups categorised under: (1) Two year old, (2) Four year old, (3) Fifteen year old and (4) Adults gamuts from €4273 to €21,590 per patient.35 This is a very grave matter and this price issue has to be resolved by employing more cost-effective measures to increase affordability and accessibility of diet formulations.16 An integrated and inter-disciplinary approach that comprises of healthcare professionals such as pediatricians scientists and counsellors are required for testing and screening neonates for PKU and corresponding diet regimen must be employed to avoid severe effects to accrue in later stages that can exponentially increase the expenses in the future.16 The counsellors need to train parents of the PKU affected children about their care and management of their diet in order to avoid future complications and it’s corresponding expenses.36,37
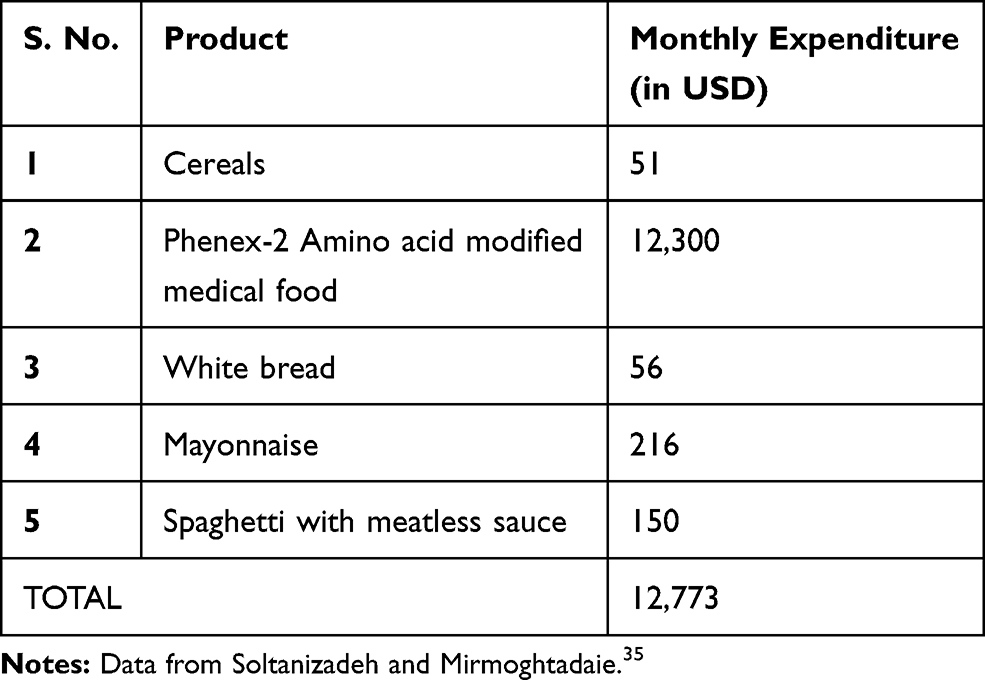 |
Table 1 Typical Cost of Monthly Expenditure of Basic PKU Foodstuff |
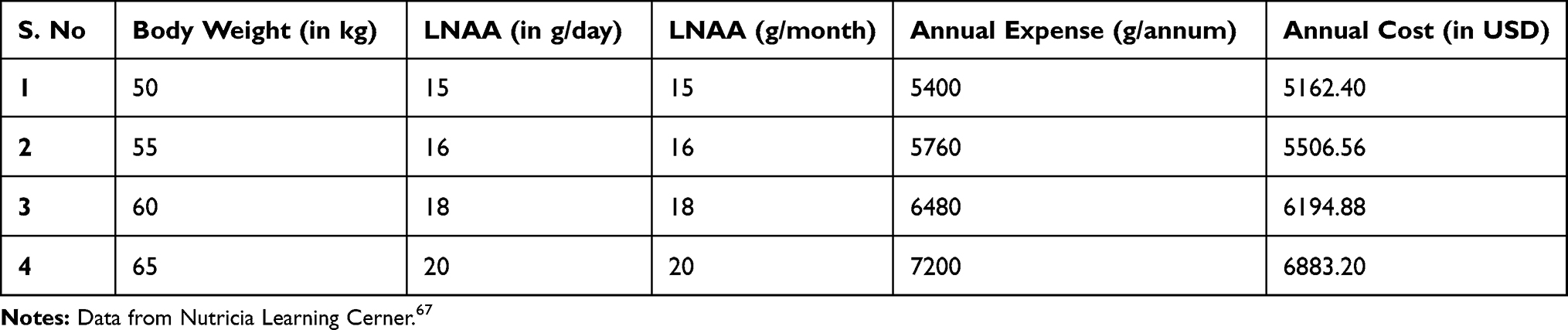 |
Table 2 Cost Economics of 30% LNAA Requirement |
 |
Figure 2 Average annual expenditure of diet associated therapy of different age groups reflecting increased expenditure with age. Notes: Bar graphs displaying metadata analysis of finances associated with PKU expenditures across four age groups. Data from Rose et al.46 |
Therapeutic Diet for PKU
It has been the central form of treatment procedure for the PKU patients for many years. This involves abstemious diet regime to avoid phenylalanine rich diet. An abstemious diet regime is required to be followed where deviation from schedule is considered to be detrimental to the mental and physical well-being of the patient. For this, phenylalanine-free formulated medicated diet are designed to ensure minimal consumption of phenylalanine.1,3,10,43 With diet therapy, the intelligence quotient (IQ) with PKU patient was reported to be 101±11 which is considered to be average IQ according to Woodcock-Johnson test,1 suggesting the metabolic controlling of PKU patients through diet therapy is clearly effective in promoting intellectual development. Overall, this mode of treatment is non-invasive, effective, feasible, accessible and harmless. Different types of diet therapies have evolved over time and its utility depends on intensity of disease. Here, we will discuss different diet therapies, their limitation (Table 3) and associated cost.
 |
Table 3 Pros and Cons of Available Diet Therapies |
Large Neutral Amino Acids (LNAAs)
LNAAs are another important form of protein supplement manufactured, distributed and marketed to the PKU patients across the world.10,48 It is known that amino acid transportation process across blood-brain barrier is common between LNAAs, tyrosine, phenylalanine, tryptophan and branched chain amino acids. Studies on utilization of branched chain amino acids such as valine leucine, methionine and isoleucine on few PKU patients were reported to competitively inhibit the entry of phenylalanine to the brain.49 Therefore, LNAAs are known as ideal competitor for phenylalanine absorption in the blood brain barrier.10,49,50 LNAAs have demonstrated to not only reduce phenylalanine deposition in the brain but also ameliorate neurotransmitter concentration and tyrosine levels too.50
However, clinical data for long term use is unavailable. Moreover, LNAAs compete with blood phenylalanine levels only in the blood-brain barrier, having no impact on the blood phenylalanine levels in other parts of the body.10,19 Generally 20–30% of the protein from LNAAs is co-supplemented along with traditional formulations of diet.
LNAA powder per kilogram costs $956, which is additional cost that will be incurred along with the traditional formulations. According to the guidelines for LNAA usage prescribed in Nutricia metabolics, 20–30% of the LNAA is consumed in addition to the conventional formulated diet. An average PKU patient requiring 30% LNAA supplementation weighing 60–65 kilograms has an annual expense of $6000–7000 (USD), which is a titanic amount adding to the already expensive diet therapy regimen (Table 2). The possible remedy is to employ a protein hydrolysate which is particularly rich in LNAA so that this additional requirement can be met. Canonical composition of amino acids in LNAA powder mix is provided (Table 4).
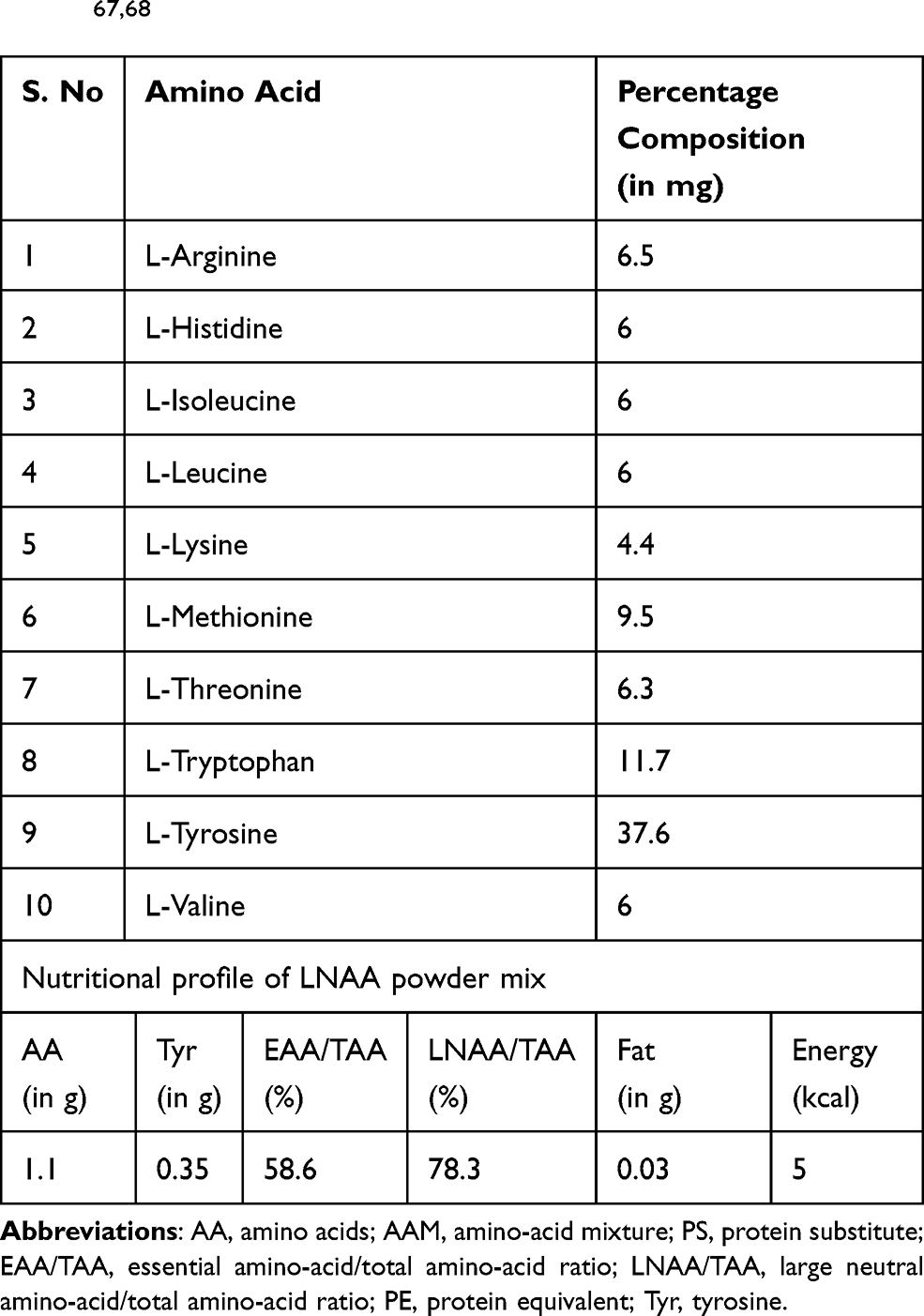 |
Table 4 LNAA Powder Mix Composition and Nutritional Profile67,68 |
Glycomacropeptides (GMP)
Glycomacropeptides, a cheese whey protein, is one such protein which are particularly rich in LNAAs and importantly it has very low phenylalanine content, making it suitable for generation of diet therapy for PKU patients.23 PKU mouse model studies have reported sufficiency in GMP supplementation with limiting amount of integral amino acids is sufficient to ameliorate excess phenylalanine in plasma and brain.35,51 Moreover, studies suggest that PKU patients prefer to have diet that contain GMP based diet as it has been observed to improve palatability of medicated diet. Moreover, it not only provides substitute of L-amino acids but also demonstrated greater absorption in the body.35
However, commercially available GMP is generally adulterated with phenylalanine owing to presence of residual whey proteins, which is particularly detrimental to patients suffering from classical PKU. Many processes for large-scale production of GMP including complex purification and processing conditions adds up to the cost of the product.52
The compositional analysis of GMP indicates 20.5% α-lactalbumin, 56%, β-lactoglobulin, 18% GMP, and 5.5% other whey proteins.52 Composition of different components of a typical GMP food recipe is given (Table 5). Glycomacropeptides are extremely expensive in their refined formulations costing approximately $73 per kg of product.53 The other major obstacles in realizing its commercial potential are (1) insufficient clinical trials and data analysis; (2) arduous process to remove trace contaminants from the glycomacropeptide formulated product making the end product very expensive;23,35 and (3) The appropriate dosage of glycomacropeptides depends upon various different factors such as age, health and several other conditions and we still lack scientific information for determination of appropriate dosages.53
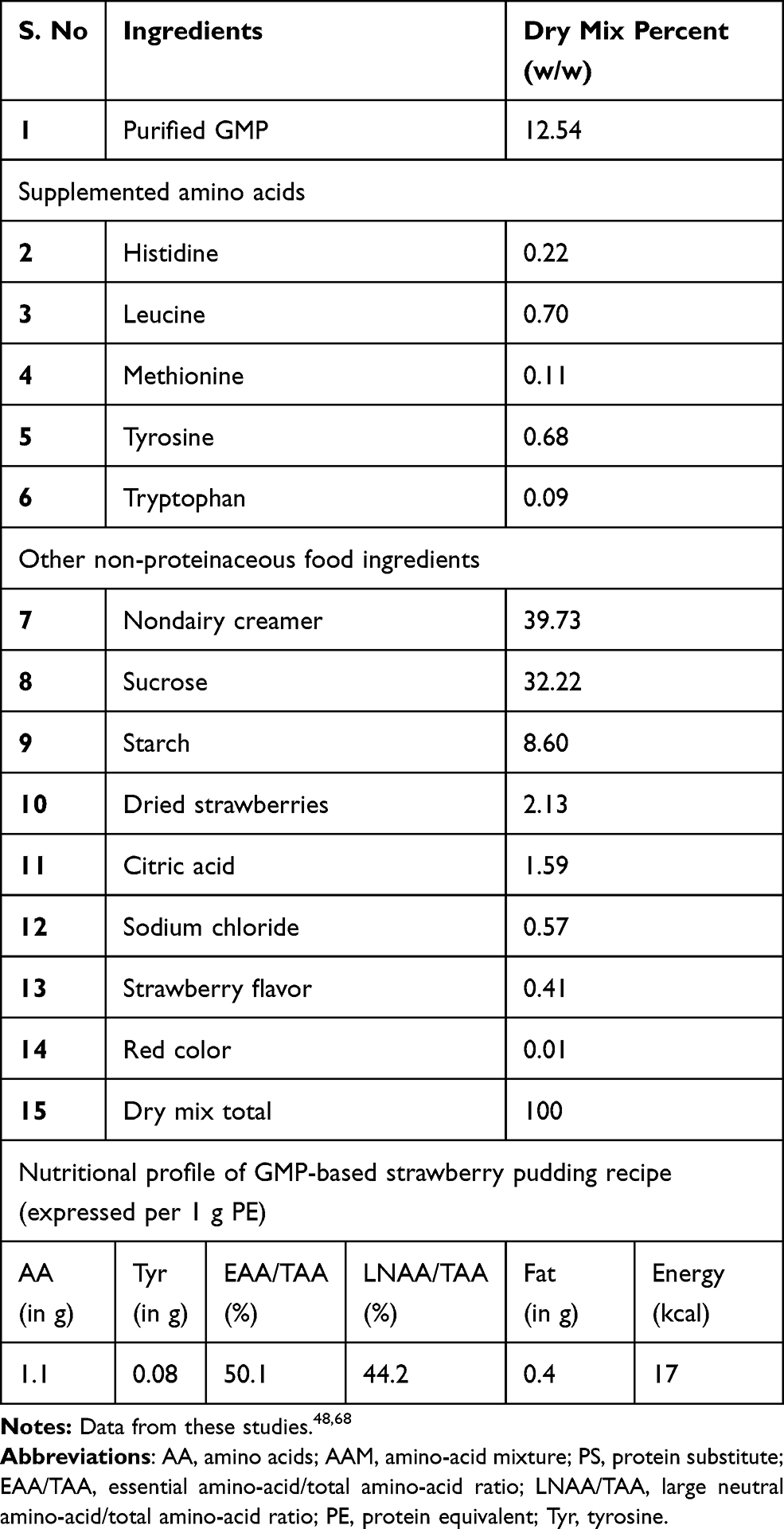 |
Table 5 Typical GMP Strawberry Pudding Recipe and Associated Nutritional Profile |
The possible alternative to glycomacropeptide will be synthesis of artificial protein sequence, which is devoid of phenylalanine and enriched with LNAAs. This alternate protein can be generated by expressing synthetic gene in heterologous system and the encoded protein can then be hydrolysed to generate diet supplements.48
Amino Acid Mixture
In order to facilitate supplementation of essential nutrients with the abstemious diet plan, free amino acid mixtures are marketed across the world such as Europe and the Mediterranean countries like Turkey.54 Since 1960s, synthetic amino acids have been employed for dietary treatment of phenylketonuria, and it fulfilled 80% of protein needs of PKU patients.1 In India, such amino acid formulation for PKU patients is manufactured by Pristine Organics in Bangalore, Karnataka whose price of amino acid mixture product ranges tentatively from $600 for infants up to three years of age to $1390 for PKU children aged 12-17 years per annum (https://pristineorganics.com/metabollic-diets-for-iem-inborn-errors-of-metabollism/). The typical amino acid composition in diet formulation based on synthetic mixture of amino acid is listed in Table 6. While the annual income of an average Indian is $1740 (https://www.statista.com/statistics/802122/india-net-national-income-per-capita/). This small example itself shows the treatment using medicated food is unaffordable from a financial standpoint and that it is high time that critical steps are required to be taken for bringing the cost down to reasonable amount for the general masses.
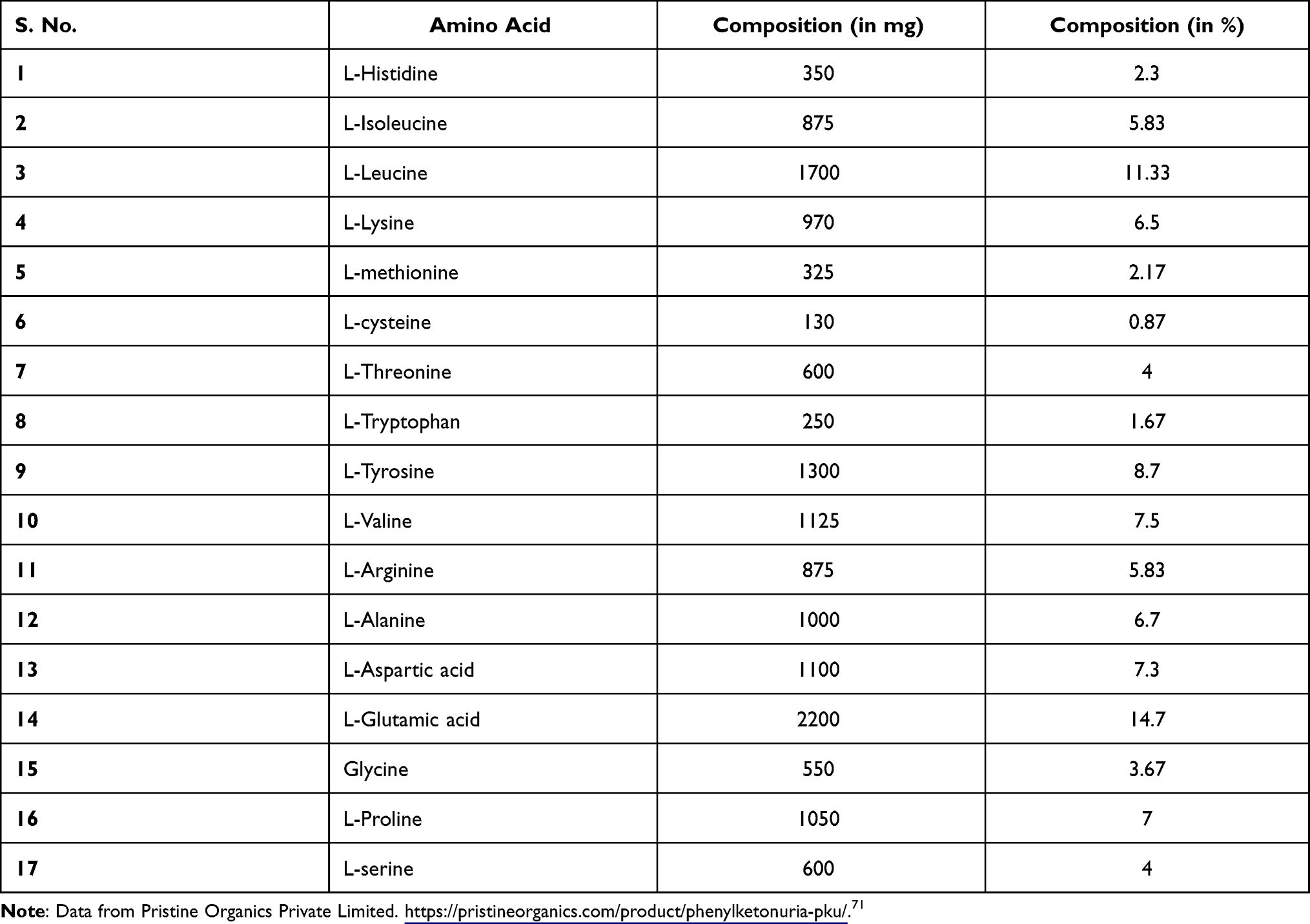 |
Table 6 Typical Amino Acid Composition in Amino Acid Mixture |
Challenges in PKU Diet Production
Currently organoleptic properties, expensive cost, deficiency in few nutritional values, shelf-life, color and texture are the upfront challenges posed to food scientists. Also, nutrition is somewhat a disadvantage as these formulated diets are deficient in certain nutrition like vitamins, omega-3-fatty acids, folic acids, micronutrients like iron, and dietary fibre and protein.10,35 Deficiency in n-3 long chain polyunsaturated fatty acids (LCPUFA) specifically docosahexaenoic acid (DHA) as well as n-6 LCPUFA has been reported to immensely reduce both red blood cells and phospholipids present in the plasma35,55–57 of patients on abstemious routine of PKU diet. PKU patients also show selenium deficiency affecting anti-oxidative properties by decreasing anti-oxidants resulting in high lipid and protein oxidative damage.55–58 About a third of PKU affected individuals have high plasma homocysteine levels, which increases the risk of thrombosis, stroke and atherosclerosis.35,55–58 This is the result of deficiency in seafood and meat consumption leading to brain related disorders.35,54 Anomalies in changes in vitamin B6 and vitamin B12 metabolism along with high homocysteine concentration causes above-mentioned complications. As a result of low absorption of phosphorus and calcium, more than forty percent of young PKU patients have been reported to display low bone mineral density.1
Therefore, special consideration is given to improve nutritional quality of diet by supplementing the missing components, however it increases the expense of available diet therapies.10,35 The other challenge in preparation of diet for PKU patients include palatability; any medically formulated diet have to be artificially processed and mixed with many different flavours and many times excessive sweetening flavors in order to mask its original repugnant taste and odour. The degree of bitterness is proportional to quantity of hydrophobic amino acids which are present in protein hydrolysates or synthetic mixtures. Some peptides produced for formulated mixtures have bitter and unpleasant taste that can easily turn to rebuff by many consumers.35
Leads for Development for Cost Effective and Efficacious Diet for PKU Patients
The advent of affordable diet solution for PKU patients has become need of an hour. Research is ongoing towards finding alternative solutions which ranges from development of oligopeptide based diet to designing phenylalanine-free protein. Here, we have listed so far developed strategies for making efficacious medicated diet for PKU patients.
Heterologous Expression of Phe Free Synthetic Protein
Heterologous expression of phenylalanine free proteins in microbial systems offers an alternative, cost effective solutions for mass production of phenylalanine free protein pertinent to PKU patients.69 For this, prokaryotic microorganisms known for high protein productions were selected such as Bacillus subtilis and Bacillus licheniformis which also come under “Generally regarded as safe” (GRAS) category. Along with that these two microbes have excellent protein secretion capacities making it an ideal host for scaling up of commercial proteins.
Efficacious process for Phe-limited diet production should necessarily involve two major steps: (1) design of Phe free sequence with balanced composition of amino acids; and (2) selection of best heterologous system for expression of target protein.
The strain for heterologous protein production is selected based on its lucid growth conditions, tendency for copious protein production and ease of strain manipulation. The pipeline flowchart for employing phenylalanine free synthetic protein is shown (Figure 3).
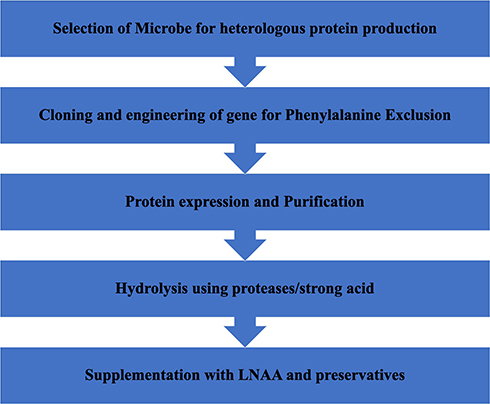 |
Figure 3 Flow chart of steps involved in formulation of Phe free synthetic protein using molecular biology approach. Notes: Data from U.S. Patent 10,174,354.69 |
Employing Oligopeptide Based Therapeutic Diet
Recent studies indicated that absorption of oligopeptides is much better than amino acid cocktail.23,59 Therefore, consumption of phe-free oligopeptides can effectively alleviate the PKU symptoms, better than free amino acid based formulations. Other advantages over free amino acids include thermostability; resistant to degradation and precipitation and quicker absorption of nitrogen of amino acid from oligopeptides.20,23,59 Low phenylalanine peptides (LPP) have been developed as medical diet for patients suffering from phenylketonuria. The procedure is also suitable to be developed in industrial scale. For this, papain, pronase E, α-chymotrypsin and pepsin were used as enzymes for proteolytic degradation. Hydrolysation processing involved single-step hydrolysis with pronase only and double-step with pronase, papain-pronase, chymotrypsin and pepsin-pronase were applied for the effective emancipation of phenylalanine from the peptide chain.20–28,30–34,60 The application of this process resulted in 10-fold reduction in phenylalanine content in whey protein.23 This process established in the oligopeptide design and production has received practical applications, and similar low protein peptide products are being manufactured in an industrial scale. Adding other critical amino acids to the hydrolysed products have paved the way for manufacturing of low phenylalanine peptide formulations for infants to be used in clinical trials at university hospitals with PKU victims.23
Few existing enzymes used for preparation of protein hydrolysates are listed in Table 7 and their potential sources are listed in Table 8. Efficient development of such strategy will not only help in management of PKU but also diseases that rely on artificial protein supplement such as Crohn’s disease, pancreatic, ulcerative colitis, short bowel syndrome, liver diseases and food allergy.23 Other major medical applications of protein hydrolysates include it’s use as anti-coagulants, high blood pressure controller, anti-oxidant, anti-cancer properties, preventing diseases and maintaining good health.61–63 Protein hydrolysates have also found applications in clinical treatment of patients with pertinent ailments pertaining to absorption, digestion and amino acid metabolism.23,
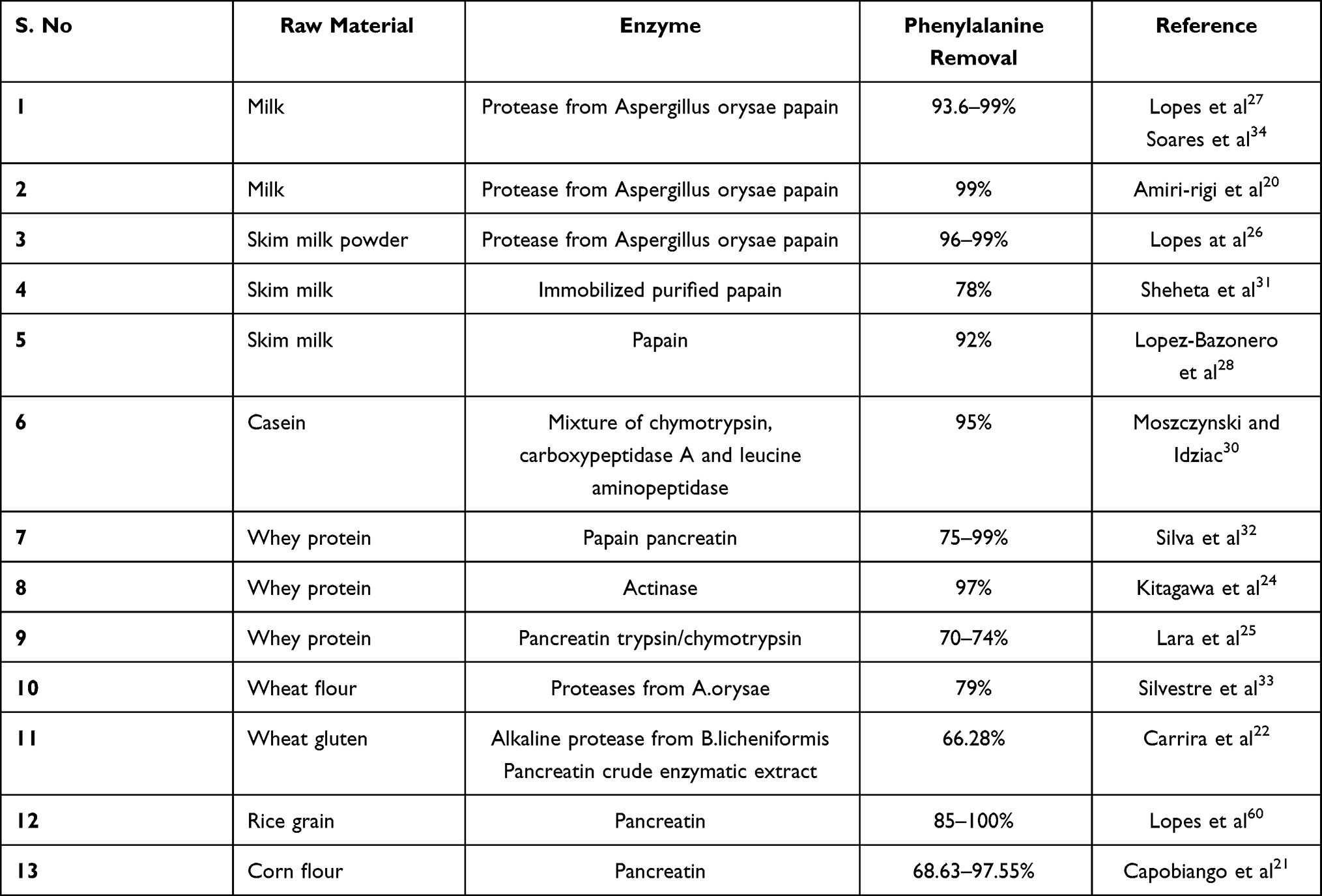 |
Table 7 Brief Summary of the Studies on the Production of Phenylalanine-Free Hydrolysates |
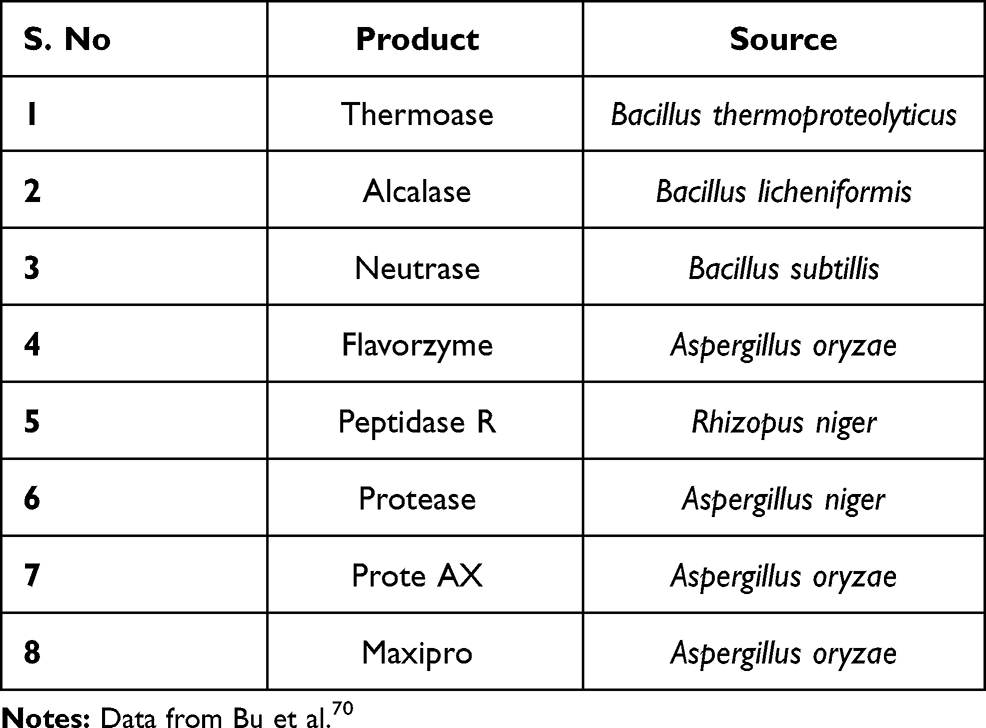 |
Table 8 List of Endopeptidases and Exopeptidases in Cleaving Phenylalanine from Whey Protein |
Advent of Palatable Diet Solutions
Many procedures such as exopeptidases treatment for hydrolyzation, adsorption, extraction, flavozyme treatment for removing bitterness, have been employed.35 Food taste and aroma have been observed to ameliorate with the help of enzymes like glucanase, esterase and lipolytic enzymes that secrete phenolic, ester or fatty acid flavors.23 Moreover, 0.5% sodium bicarbonate and 1% glucono-δ-lactone potentiates in making a significant contribution in ameliorating appearance and food flavor.35 Studies report desirable results on structure quality with low phenylalanine content and gluten free by applying binding agents like Tragant, Xanthan, Guar gum and locust bean.18,59 Further efforts are also laid to transform non palatable diets to flavored medical pouch and drinks. In conclusion, several strategies can be employed to cater to the need of the PKU patients where each method has its own fair share of limitations. Further improvement is possible only via effective communication and collaboration amongst the scientific community working or associated with the field of therapeutic diet development.51
Possibility of Canonical PKU Meal Plan?
There is no typical meal layout for patients suffering from PKU.64 Prescriptions for phenylalanine are based on the level of tolerance that varies from person to person influenced by phenylalanine hydroxylase activity, sex, age, blood phenylalanine level, growth in height and weight.64 The PKU patients need regular monitoring, adjusting and recalculations in their diet plan. According to the recommendations of the pediatric nutrition care manual the assessment of the meal layout calculations: 1) intake of total energy and energy percentage of medical diets and others; 2) consumption of phenylalanine and tyrosine from medical diets; and 3) mineral and vitamin input (including Calcium, Vitamin D, iron and others) from medical diet and others.57 Nutritionists need to draw comparison in the phenylalanine intake and condition it between the levels of 120 to 360 mmol/L or 2 to 6 mg/dL.57 Medical diet opulent with protein levels are usually excluded from the meal plans of patients suffering from PKU. Special expertise and collaborative teamwork between scientists, frontline health workers and families of affected PKU individuals is the need of the hour for efficient dietary management to achieve, sustain and maintain appropriate metabolic control and wholistic growth57 http://www.nutritioncaremanual.org/topic/pediatric-nutrition-care (http://www.nutritioncaremanual.org/topic/pediatric-nutrition-care).
Case Studies for Importance of Early Intervention of Low-Phenylalanine Food
A boy aged twenty months from south Karnataka, India was brought to hospital because he witnessed noticeable procrastinations in achieving certain mental milestones. Clinical exams report average build and nourishment. Green precipitate was observed in urinary ferric chloride test. Dinitrophenylhydrazine test was conducted in his urine to detect aldehydes and ketones which was found to be positive. Ascending paper chromatography confirmed high presence of phenylalanine. Quantification of phenylalanine was reported to be 42 mg/dL (normal value: 3–4 mg/dL) using amino acid analyzer in National Institute of Nutrition, Hyderabad. Improvement in his behaviour was noted by his parents after putting the patient on low phenylalanine diet regimen for two months. His weight gain was also satisfactory. Subsequently, within two months the plasma phenylalanine levels dropped from 24 mg/dL to 18 mg/dL.65 A study was conducted in early and continuously treated PKU subjects based on neuropsychological tests that included Wisconsin card, Visual search, Rey-osterreith test, sorting test, Weigl’s sorting test, Elithhorn’s perceptual Maze test, Motor learning and Tower of London test by Department of child neurology and psychiatry, Rome, Italy. It was reported that the executive cognitive functions scores in the tests conducted demonstrate better results in thirteen tests out of fourteen tests where PKU children were precociously treated under abstemious diet therapy routine.54 This shows early treated PKU patients using diet therapy display better cognitive response than continuously treated PKU patients.
Conclusion
Low phenylalanine diet have served to be the mainstay treatment for majority of the patients suffering from phenylketonuria in not only maintaining blood phenylalanine levels within safe limits but also maintaining average levels of intelligence quotient in children and adults. However, there are issues not only regarding organoleptic properties but also regarding the cost of the end products. There exist four categories of medicated diets viz. amino acid therapy, glycomacropeptides, large neutral amino acids and cofactor therapy. Leaving amino acid formulation, others display some serious pitfalls due to which they are ineffective against severe categories of patients suffering from phenylketonuria. Glycomacropeptides contain impurity issues, because of which is arduous to purify the end product, hence the product is highly expensive. Large neutral amino acids competitively inhibit phenylalanine concentrations in the blood-brain barrier, but do not have any impact whatsoever with blood phenylalanine levels in the rest of the body. Co-factor therapy is applicable to only mild cases where there are minor deficiencies or defects in concentration for tetrahydrobiopterin in the body. Recent studies have also shown importance of slow-release LNAA as supplement for treatment of PKU, which was shown to improve adherence to treatment and improved Quality of Life (QoL).66 Medically formulated free amino acid mixtures are most effective, however the expenses are again quite high for an average person and therefore unaffordable.
Protein hydrolysates have served to be an ideal alternative to free amino acid formulated mixtures, which have improved palatability and organoleptic properties. In addition, peptide based formulations are highly advantageous because it is relatively easily absorbed by our body and also it is resistant to acidic and alkaline degradation. The peptide cocktail is additionally resistant to precipitation. Although, diet based therapy has been a successful treatment, expenditures of hundreds and thousands of dollars along with deficiency and problems related to organoleptic properties owing to the shift in glycomacropeptide based formulations which are more expensive than the combination of low protein diet and amino acid mixtures/protein hydrolysates contributes to the financial, physical and mental distress. It is high time that cheaper and more sustainable forms of therapeutic formulations of low phenylalanine diet products are developed that can address these distressing issues and help relieve the financial burden.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, agreed to the submitted journal, and agree to be accountable for all aspects of the work.
Disclosure
No conflict of interest is reported by the authors in this work.
References
1. Macleod EL, Ney DM. Nutritional management of phenylketonuria. Ann Nestle Eng. 2010;68(2):58–69. doi:10.1159/000312813
2. Burnett JR. Sapropterin dihydrochloride (Kuvan/phenoptin), an orally active synthetic form of BH4 for the treatment of phenylketonuria. IDrugs. 2007;10(11):805–813.
3. Acosta PB, Matalon KM. Nutrition management of patients with inherited disorders of aromatic amino acid metabolism. In: Nutrition Management of Patients with Inherited Metabolic Disorders Boston. Jones and Bartlett Publishers; 2010;119–174.
4. Blau N, Van Spronsen FJ, Levy HL. Phenylketonuria. Lancet. 2010;376(9750):1417–1427.
5. Channon S, German E, Cassina C, Lee P. Executive functioning, memory, and learning in phenylketonuria. Neuropsychology. 2004;18(4):613.
6. Ho G, Christodoulou J. Phenylketonuria: translating research into novel therapies. Trans Pediatr. 2014;3(2):49.
7. Marcdante K, Kliegman RM. Nelson Essentials of Pediatrics e-Book. Elsevier Health Sciences; 2014.
8. Rocha JC, MacDonald A. Dietary intervention in the management of phenylketonuria: current perspectives. Pediatric Health Med Ther. 2016;7:155–163.
9. Scriber C. Hyperphenylalaninemia: phenylalanine hydroxylase deficiency. The metabolic and molecular bases of inherited disease; 2001:1667–1724.
10. Al Hafid N, Christodoulou J. Phenylketonuria: a review of current and future treatments. Transl Pediatr. 2015;4(4):304–317. doi:10.3978/j.issn.2224-4336.2015.10.07
11. Giovannini M, Verduci E, Salvatici E, Paci S, Riva E. Phenylketonuria: nutritional advances and challenges. Nutr Metab. 2012;9(1):7. doi:10.1186/1743-7075-9-7
12. Habib A, Fallahzadeh MH, Kazeroni HR, Ganjkarimi AH. Incidence of phenylketonuria in Southern Iran. Iran J Med Sci. 2010;35(2):137–139.
13. Azen CG, Koch R, Friedman EG, et al. Intellectual development in 12-year-old children treated for phenylketonuria. Am J Dis Child. 1991;145(1):35–39. doi:10.1001/archpedi.1991.02160010037012
14. Williams RA, Mamotte CD, Burnett JR. Phenylketonuria: an inborn error of phenylalanine metabolism. Clin Biochem Rev. 2008;29(1):31–41.
15. Ramsay J, Morton J, Norris M, Kanungo S. Organic acid disorders. Ann Transl Med. 2018;6(24):472. doi:10.21037/atm.2018.12.39
16. Abadie V, Berthelot J, Feillet F, et al. Neonatal screening and long-term follow-up of phenylketonuria: the French database. Early Hum Dev. 2001;65(2):149–158. doi:10.1016/S0378-3782(01)00223-7
17. Dhondt J, Farriaux J, Briard M, Boschetti R, Frezal J. Neonatal screening in France. Screening. 1993;2(2–3):77–85. doi:10.1016/0925-6164(93)90021-A
18. Acs E, Kovacs Z, Matuz J. Bread from corn starch for dietetic purposes I. Structure Formation. Cereal Res Commun. 1996;24:441–449.
19. Castañeda MT, Adachi O, Hours RA. Reduction of l-phenylalanine in protein hydrolysates using l-phenylalanine ammonia-lyase from Rhodosporidium toruloides. J Ind Microbiol Biotechnol. 2015;42(10):1299–1307. doi:10.1007/s10295-015-1664-z
20. Amiri-Rigi A, Mohammadi M, Emam-Djomeh Z, Mohammadifar M. The effect of type of enzyme and activated carbon concentration on phenylalanine removal from milk. Iran J Nutr Sci Food Technol. 2012;7(1):1–9.
21. Capobiango M, Lopes DCF, Carreira RL, de Oliveira Afonso W, Segall SD, Silvestre MPC. Optimization of enzyme assisted processes for extracting and hydrolysing corn proteins aiming phenylalanine removal. Int J Food Eng. 2007;3(6). doi:10.2202/1556-3758.1268
22. Carreira RL, Silva MR, Starling ALP, Aguiar MJ, Januario JN, Silvestre MP. Association of two enzymes for obtaining low phenylalanine protein hydrolysates from wheat flour. Int J Food Eng. 2008;4(7). doi:10.2202/1556-3758.1544
23. Clemente A. Enzymatic protein hydrolysates in human nutrition. Trends Food Sci Technol. 2000;11(7):254–262. doi:10.1016/S0924-2244(01)00007-3
24. Kitagawa T, Owada M, Aoki K, et al. Treatment of phenylketonuria with a formula consisting of low-phenylalanine peptide. Enzyme. 1987;38(1–4):321–327. doi:10.1159/000469222
25. Lara MG, Izumi C, Greene LJ, Vilela L, Freitas O. Preparation and scaling up of a low phenylalanine enzymatic hydrolysate of bovine whey proteins. Rev Bras Ciênc Farm. 2005;41(4):459–466. doi:10.1590/S1516-93322005000400008
26. Lopes DCF, Delvivo FM, Silvestre MPC. Use of activated carbon for removing phenylalanine from reconstituted skim milk powder hydrolysates. LWT. 2005;38(5):447–453. doi:10.1016/j.lwt.2004.07.021
27. Lopes DCF, Delvivo FM, Silvestre MPC. Dietary supplements for phenylketonuria: removing Phe by activated carbon. Nutr Food Sci. 2006. doi:10.1108/00346650610652303
28. Lopez‐Bajonero L, Lara‐Calderon P, Galvez‐Mariscal A, Velazques‐Arellano A, Lopez‐Munguia A. Enzymatic production of a low‐phenylalanine product from skim milk powder and casemate. J Food Sci. 1991;56(4):938–942. doi:10.1111/j.1365-2621.1991.tb14610.x
29. Matalon R, Michals-Matalon K, Bhatia G, et al. Large neutral amino acids in the treatment of phenylketonuria (PKU). J Inherit Metab Dis. 2006;29(6):732–738. doi:10.1007/s10545-006-0395-8
30. Moszczynski P, Idziac J. Preparation of enzymatic hydrolysates of casein depleted in phenylalanine. Appl Biochem Microbiol. 1993;29(3):302–306.
31. Shehata AE, El-Magdoub MN, Kamal TM, Mohamed HA. enzymatic preparation of low-phenylalanine formula derived from skim milk hydrolysate for phenyl ketonuric patients. Egypt J Med Hum Genet. 2008;9(1):51–71.
32. Silva VD, De Marco LM, Afonso WO, Lopes DC, Silvestre MP. Comparative study of the immobilization of pancreatin and papain on activated carbon and alumina, using whey as protein substrate. World Appl Sci J. 2007;2:175–183.
33. Silvestre MP, Da silva MC, de Souza MW, Silva VD, de Aguiar MJ, Silva MR. Hydrolysis degree, peptide profile and phenylalanine removal from whey protein concentrate hydrolysates obtained by various proteases. Int J Food Sci Technol. 2013;48(3):588–595.
34. Soares RD, Biasutti EA, Capobiango M, et al. Preparation of enzymatic skim milk hydrolysates with low phenylalanine content. Acta Farmaceutica Bonaerense. 2007;25(3):325.
35. Soltanizadeh N, Mirmoghtadaie L. Strategies used in production of phenylalanine‐free foods for PKU management. Compr Rev Food Sci Food Saf. 2014;13(3):287–299.
36. National Institutes of Health Consensus Development Panel. National institutes of health consensus development conference statement: phenylketonuria: screening and management, October 16–18, 2000. Pediatrics. 2001;108(4):972–982.
37. Singh RH, Rohr F, Frazier D, et al. Recommendations for the nutrition management of phenylalanine hydroxylase deficiency. Gene Med. 2014;16(2):121–131.
38. Mathias D, Bickel H. Follow-up study of 16 years neonatal screening for inborn errors of metabolism in West Germany. Eur J Pediatr. 1986;145(4):310–312.
39. Missiou‐Tsagaraki S, Soulpi K, Loumakou M. Phenylketonuria in Greece: 12 year’s experience. J Intell Disabil Res. 1988;32(4):271–287.
40. Õunap K, Lilleväli H, Metspalu A, Lipping-Sitska M. Development of the phenylketonuria screening programme in Estonia. J Med Screen. 1998;5(1):22–23.
41. Smith I, Cook B, Beasley M. Review of neonatal screening programme for phenylketonuria. Br Med J. 1991;303(6798):333–335.
42. Motamedi N, Goodarzi E, Pordanjani SR, et al. Incidence of phenylketonuria in Lorestan province, West of Iran (2006–2016). In. J Pediatr. 2017;5:4713–4721.
43. Shoraka HR, Haghdoost AA, Baneshi MR, Bagherinezhad Z, Zolala F. Global prevalence of classic phenylketonuria based on Neonatal Screening Program Data: systematic review and meta-analysis. Clin Exper Pediatr. 2020;63(2):34.
44. Özalp I, Coşkun T, Tokatli A, et al. Neonatal PKU screening in Turkey: 7 years experience in a developing country. Screening. 1995;4(3):139–147.
45. Koochmeshgi J, Bagheri A, Hosseini-Mazinani S. Incidence of phenylketonuria in Iran estimated from consanguineous marriages. J Inherit Metab Dis. 2002;25(1):80.
46. Rose AM, Grosse SD, Garcia SP, et al. The financial and time burden associated with phenylketonuria treatment in the United States. Mol Gene Metab Rep. 2019;21:100523.
47. Barden HS, Kessel R, Schuett VE. The costs and benefits of screening for PKU in Wisconsin. Soc Biol. 1984;31(1–2):1–17.
48. LaClair CE, Ney DM, MacLeod EL, Etzel MR. Purification and use of glycomacropeptide for nutritional management of phenylketonuria. J Food Sci. 2009;74(4):E199–E206.
49. Berry HK, Bofinger MK, Hunt MM, Phillips PJ, Guilfoile MB. Reduction of cerebrospinal fluid phenylalanine after oral administration of valine, isoleucine, and leucine. Pediatr Res. 1982;16(9):751–755.
50. Burlina AP, Cazzorla C, Massa P, et al. Large neutral amino acid therapy increases tyrosine levels in adult patients with phenylketonuria: a long-term study. Nutrients. 2019;11(10):2541.
51. Ney D, Gleason S, Van Calcar S, et al. Nutritional management of PKU with glycomacropeptide from cheese whey. J Inherit Metab Dis. 2009;32(1):32–39.
52. Bonnaillie LM, Qi P, Wickham E, Tomasula PM. Enrichment and purification of casein glycomacropeptide from whey protein isolate using supercritical carbon dioxide processing and membrane ultrafiltration. Foods. 2014;3(1):94–109.
53. Leuzzi V, Pansini M, Sechi E, et al. Executive function impairment in early‐treated PKU subjects with normal mental development. J Inherit Metab Dis. 2004;27(2):115–125.
54. Robinson M, White FJ, Cleary MA, Wraith E, Lam WK, Walter JH. Increased risk of vitamin B12 deficiency in patients with phenylketonuria on an unrestricted or relaxed diet. J Pediatr. 2000;136(4):545–547.
55. Hanley W, Feigenbaum A, Clarke J, Schoonheyt W, Austin V. Vitamin B 12 deficiency in adolescents and young adults with phenylketonuria. Eur J Pediatr. 1996;155(1):S145–S147.
56. Schulpis K, Karikas G, Papakonstantinou E. Homocysteine and other vascular risk factors in patients with phenylketonuria on a diet. Acta Paediatr. 2002;91(8):905–909.
57. Hvas A, Nexo E, Nielsen J. Vitamin B 12 and vitamin B 6 supplementation is needed among adults with phenylketonuria (PKU). J Inherit Metab Dis. 2006;29(1):47–53.
58. Procházková D, Jarkovský J, Vinohradská H, Konečná P, Machačová L, Doležel Z. Controlled diet in phenylketonuria and hyperphenylalaninemia may cause serum selenium deficiency in adult patients: the Czech experience. Biol Trace Elem Res. 2013;154(2):178–184.
59. van Spronsen FJ, Enns GM. Future treatment strategies in phenylketonuria. Mol Genet Metab. 2010;99:S90–S95.
60. Lopes DCF, Bizzotto CS, Carreira RL, Afonso W, Lopes JC, Silvestre MPC. Removal of phenylalanine from protein hydrolysates prepared with rice. J Food Technol. 2008;6(2):57–65.
61. Etemadian Y, Ghaemi V, Shaviklo AR, Pourashouri P, Mahoonak ARS, Rafipour F. Development of animal/plant-based protein hydrolysate and its application in food, feed and nutraceutical industries: state of the art. J Clean Prod. 2021;278:123219.
62. Allaoui A, Gascón S, Benomar S, et al. Protein hydrolysates from fenugreek (Trigonella foenum graecum) as nutraceutical molecules in colon cancer treatment. Nutrients. 2019;11(4):724.
63. Al-Shamsi KA, Mudgil P, Hassan HM, Maqsood S. Camel milk protein hydrolysates with improved technofunctional properties and enhanced antioxidant potential in in vitro and in food model systems. J Dairy Sci. 2018;101(1):47–60.
64. Marcason W. Is there a standard meal plan for phenylketonuria (PKU)? J Acad Nutr Diet. 2013;113(8):1124.
65. Bajaj R, Patel Z, Ambani L, Rao BN, Merchant S. Dietary therapy in a case of phenylketonuria in India. Indian J Pediatr. 1981;48(2):175–178.
66. Burlina AP, Cazzorla C, Massa P, Loro C, Gueraldi D, Burlina AB. The impact of a slow-release large neutral amino acids supplement on treatment adherence in adult patients with phenylketonuria. Nutrients. 2020;12(7):2078.
67. Nutricia Learning Cerner. Guidelines for the usage of PhenylAde® PheBLOC™. Available from: https://www.nutricialearningcenter.com/globalassets/pdfs/metabolics/phebloc-guidelines_101617.pdf Accessed December 22, 2021.
68. Pena MJ, De Almeida MF, Van Dam E, et al. Protein substitutes for phenylketonuria in Europe: access and nutritional composition. European Journal of Clinical Nutrition. 2016;70(7):785–789.
69. Inventors: Li Q, Kämpe O. Assignee: NEXTTOBE AB. Recombinant Phe-free proteins for use in the treatment of phenylketonuria. U.S. Patent 10,174,354. 2019 Jan 8.
70. Bu T, Zhou M, Zheng J, et al. Preparation and characterization of a low-phenylalanine whey hydrolysate using two-step enzymatic hydrolysis and macroporous resin adsorption. LWT. 2020;132:109753.
71. Pristine Organics Private Limited. Phenylketonuria (PKU)-1, 400 g. Available from: https://pristineorganics.com/product/phenylketonuria-pku/. Accessed December 21, 2021.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.