Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Folate-modified lipid–polymer hybrid nanoparticles for targeted paclitaxel delivery
Authors Zhang L, Zhu D ![]() , Dong X, Sun H, Song C, Wang C, Kong D
, Dong X, Sun H, Song C, Wang C, Kong D
Received 17 November 2014
Accepted for publication 22 January 2015
Published 16 March 2015 Volume 2015:10(1) Pages 2101—2114
DOI https://doi.org/10.2147/IJN.S77667
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Lei Yang
Linhua Zhang,1 Dunwan Zhu,1 Xia Dong,1 Hongfan Sun,1 Cunxian Song,1 Chun Wang,2 Deling Kong1
1Tianjin Key Laboratory of Biomaterials, Institute of Biomedical Engineering, Peking Union Medical College and Chinese Academy of Medical Sciences, Tianjin, People’s Republic of China; 2Department of Biomedical Engineering, University of Minnesota, Minneapolis, MN, USA
Abstract: The purpose of this study was to develop a novel lipid–polymer hybrid drug carrier comprised of folate (FA) modified lipid-shell and polymer-core nanoparticles (FLPNPs) for sustained, controlled, and targeted delivery of paclitaxel (PTX). The core-shell NPs consist of 1) a poly(ε-caprolactone) hydrophobic core based on self-assembly of poly(ε-caprolactone)–poly(ethylene glycol)–poly(ε-caprolactone) (PCL-PEG-PCL) amphiphilic copolymers, 2) a lipid monolayer formed with 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] (DSPE-PEG2000), 3) a targeting ligand (FA) on the surface, and were prepared using a thin-film hydration and ultrasonic dispersion method. Transmission electron microscopy and dynamic light scattering analysis confirmed the coating of the lipid monolayer on the hydrophobic polymer core. Physicochemical characterizations of PTX-loaded FLPNPs, such as particle size and size distribution, zeta potential, morphology, drug loading content, encapsulation efficiency, and in vitro drug release, were also evaluated. Fluorescent microscopy proved the internalization efficiency and targeting ability of the folate conjugated on the lipid monolayer for the EMT6 cancer cells which overexpress folate receptor. In vitro cytotoxicity assay demonstrated that the cytotoxic effect of PTX-loaded FLPNPs was lower than that of Taxol®, but higher than that of PTX-loaded LPNPs (without folate conjugation). In EMT6 breast tumor model, intratumoral administration of PTX-loaded FLPNPs showed similar antitumor efficacy but low toxicity compared to Taxol®. More importantly, PTX-loaded FLPNPs showed greater tumor growth inhibition (65.78%) than the nontargeted PTX-loaded LPNPs (48.38%) (P<0.05). These findings indicated that the PTX loaded-FLPNPs with mixed lipid monolayer shell and biodegradable polymer core would be a promising nanosized drug formulation for tumor-targeted therapy.
Keywords: lipid–polymer hybrid nanoparticles, paclitaxel, drug delivery, PCL-PEG-PCL, folate
Introduction
Nanoparticles (NPs) have been extensively exploited to improve conventional cancer chemotherapy because of their ability to deliver drugs to the therapeutic targets at relevant times and doses, reduce the side effects of chemotherapeutics, and protect drugs from degradation.1,2 Of all the common nanoparticulate systems, liposomes and polymeric NPs are two dominant kinds of drug nanocarriers accounting for the majority of the clinically approved products.3 Liposomes, spherical lipid vesicles with single or multiple lipid bilayers, have been widely applied due to their superior biocompatibility, favorable pharmacokinetic profile, ease of surface modification, and long circulation time after being surface modified with polyethylene glycol (PEG).4,5 Nevertheless, liposomes suffer from drawbacks of insufficient drug loading, fast drug release, and instability during storage.6 Generally, polymeric nanoparticles are advantageous in terms of improved stability in biological fluids, versatile drug loading, controlled release capability, high cellular internalization efficiency, desired pharmacokinetics, and long circulation half-life.7,8 However, the biocompatibility of nanoparticles formed by most synthetic polymers is not as high as liposomes. Moreover, polymeric nanoparticles have shown moderate circulation half-lives compared with liposomes.9 It is thus ideal if any technology could be developed to combine the advantages and, meanwhile, overcome the disadvantages of these two types of nanocarriers.
More recently, a new class of therapeutic nanocarriers known as lipid–polymer hybrid nanoparticles (LPNPs) has emerged as a potentially more potent alternative to polymeric nanoparticles and liposomes.10–12 LPNPs are polymeric nanoparticles enveloped by lipid layers that combine the highly biocompatible nature of lipids with the structural integrity afforded by polymeric nanoparticles.3 They typically comprise three distinct functional components: 1) an inner hydrophobic core where poorly water-soluble drugs are incorporated with high loading yields; 2) an interfacial lipid layer enveloping the core that acts as a highly biocompatible shell; and 3) a hydrophilic polymer stealth layer outside the lipid shell to enhance nanoparticle stability and circulation time.13–15 Depending on how these hybrid nanoparticles are prepared, the lipid shell can be a lipid monolayer, bilayer, or multilayer.16 These LPNPs have exhibited some unique advantages of both liposomes and polymeric nanoparticles, including high structural integrity, stability during storage, controlled release capability, high biocompatibility, and favorable pharmacokinetic profile, while excluding some of their intrinsic limitations.13,14,17,18
LPNPs are usually prepared through a single-step nanoprecipitation method12,19–21 or a two-step method involving co-incubation of separately preformed nanoparticles and lipid vesicles (or lipid films).22–24 The hydrophobic polymeric core of the LPNPs are commonly made of polylactic-co-glycolic acid (PLGA), poly(ε-caprolactone) (PCL), dextran, or albumin because of their biocompatibility, biodegradability, nontoxicity, and previous use in approved products.3,25 Ruysschaert et al26 reported the preparation of hybrid nanocapsules that consist of amphiphilic ABA triblock copolymers and lipids using a thin film method. Their observations suggested that when the concentration ratio of lipid-to-polymer was below 20 mol%, formation of homogeneous mixed polymer-lipid composites occurred, with the lipid homogeneously dispersed among the ABA molecules of the nanoparticle membrane. In our previous study, flower-like paclitaxel-loaded polymeric micelles were successfully developed from poly(ε-caprolactone)–poly(ethylene glycol)–poly(ε-caprolactone) (PCL-PEG-PCL, PCEC) copolymers by thin film hydration method.27 This method allowed successful loading of paclitaxel (PTX) into the micelles’ hydrophobic PCL cores with high drug loading content and encapsulation efficiency. These micelles could be ideal candidates for preparing hybrid LPNPs.
Herein, we use the thin film hydration method to synthesize folate modified lipid-shell and polymer-core nanoparticles (FLPNPs), thus allowing us to combine the advantages of liposomes and polymeric NPs. PTX, which is one of the most effective chemotherapeutic drugs for the treatment of a wide spectrum of cancers, is used as a model hydrophobic anticancer drug. The solid polymeric core based on PCL-PEG-PCL polymer self-assembly can act as a nanoscale skeleton that provides high drug loading capacity, mechanical stability, controlled morphology, biodegradability, and narrow size distribution. The lipid–PEG conjugate, which can form lipid monolayer shell on the surface of the LPNPs, will also participate in the self-assembly process with its lipid moiety inserting into the hydrophobic polymer core and its PEG moiety extending into the external aqueous environment to form a stabilizing and stealth corona of the LPNPs. Active targeting can be further realized by conjugating molecular probes onto the LPNPs’ surface, which provides a promising approach for the LPNPs to target tumor cells with overexpression of the corresponding receptors on their surface, and then release the encapsulated therapeutics in a controlled and sustained manner. Folate is selected as the model molecular probe for targeted delivery to cancer cells overexpressing folate receptor such as certain breast and ovarian cancer cells.28
In the present work, we focus on the preparation and systematic characterizations of PTX loaded-LPNPs conjugated with folate. The nanoparticles were characterized by dynamic light scattering (DLS) for size and size distribution, by transmission electron microscopy (TEM) for surface morphology and structure, and by high-performance liquid chromatography (HPLC) for drug loading content, encapsulation efficiency, and in vitro drug-release kinetics. Cellular uptake efficiency of FLPNPs in EMT6 cancer cells was evaluated using a microplate reader and by confocal laser scanning microscopy (CLSM) analyses. The antitumor activity of PTX incorporated FLPNPs was also examined both in vitro and in vivo in comparison with that of nontargeted LPNPs and Taxol®.
Materials and methods
Materials
ε-Caprolactone (ε-CL), Poly(ethylene glycol) (PEG, Mn =8000), and Nile Red were purchased from Sigma-Aldrich (St Louis, MO, USA). 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (rhodamine-PE), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] (DSPE-PEG2000), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[folate(polyethyleneglycol)-2000] (DSPE-PEG(2000) Folate) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Cell counting kit-8 (CCK-8) was obtained from Dojindo Laboratories (Kumamoto, Japan). Actin-Tracker Green and 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) solution were purchased from Beyotime Institute of Biotechnology (Jiangsu, People’s Republic of China). Paclitaxel (PTX, >99.5% purity) was purchased from Sichuan Jiufeng Natural Pharmaceutical Co., Ltd. (Sichuan, People’s Republic of China). Paclitaxel injection (Taxol®, 30 mg/5 mL) was purchased from Beijing Union Pharmaceutical Factory (Beijing, People’s Republic of China). Acetonitrile and methanol used as mobile phase in high performance liquid chromatography (HPLC) were purchased from EM Science (HPLC grade; Mallinckrodt Baker, Phillipsburg, NJ, USA). All cell culture medium and supplements were obtained from Gibco/Invitrogen (Grand Island, NY, USA). All other chemicals were reagent grade or higher and were used without further purification.
Preparation of the nanoparticles
PTX or Nile Red-loaded NPs of mixed lipid monolayer shell and biodegradable polymer core were denoted in this research by LPNPs for those of no ligand conjugation and FLPNPs for those of folate conjugation, which were prepared using the thin-film hydration and ultrasonic dispersion method as described previously.29 For PTX-loaded FLPNPs, PCL-PEG-PCL copolymer, paclitaxel, DSPE-PEG2000, and DSPE-PEG(2000) Folate (weight ratio =70:33:7:0.7) were dissolved in dichloromethane in an eggplant-shaped flask. The flask was then connected to rotary evaporator (Eyela N-1001; Tokyo Rikakikai Co., Ltd., Tokyo, Japan) and water bath with temperature maintained at 35°C under the aspirate vacuum. The thin-film layer formed was flushed with nitrogen gas for 5 minutes and maintained overnight under vacuum to remove traces of dichloromethane. Then, the solid thin-film layer was preheated in a warm water bath (65°C) to obtain a transparent gel-like sample. Double-distilled water was taken as an aqueous phase and heated to the same temperature of the gel-like sample. This hot aqueous phase was added to the gel-like sample and hydrated for 60 minutes at 65°C. Then the mixture was sonicated over an ice bath using a microtip probe sonicator (VCX-130-PB; Sonics & Material Inc., Newtown, CT, USA) at 40 kW power output for 10 minutes to obtain a clear solution. The solution was centrifuged at 20,000× g for 30 minutes and then washed three times to remove the unencapsulated drug. PTX-loaded LPNPs were produced by the same process without adding DSPE-PEG(2000) Folate to the lipid phase. The fluorescent LPNPs and FLPNPs were fabricated in the same way with the drug being replaced by Nile Red.
Characterization of the nanoparticles
Particle size, polydispersity, and zeta potential
The particle size, polydispersity, and zeta potential of the drug loaded NPs were determined by dynamic light scattering (DLS) using a NanoZS Zetasizer (Malvern Instruments Ltd., Malvern, UK). The dispersion of NPs was diluted by ultrapure water according to the mass concentration. Each sample was analyzed in triplicate.
Particle morphology
The morphology of the NPs was investigated by TEM (Tecnai-F20; FEI Co., Eindhoven, the Netherlands). A drop of the particle dispersion was deposited onto the carbon-coated copper grid, followed by drawing off the excess with a filter paper. The grid was dried at room temperature before being examined under TEM.
Determination of lipid layer on the surface of PCEP NPs
The distribution of lipid layer outside the PCEP NPs was examined under a confocal laser scanning microscope (Zeiss LSM 710; Carl Zeiss Meditec AG, Jena, Germany). 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (rhodamine-PE) was used to track the lipid layer. In brief, the labeled PTX-loaded FLPNPs containing rhodamine-PE (weight ratio of rhodamine-PE and DSPE-PEG2000 was 1:10) were prepared using the abovementioned method with a slight modification in sonication time. After hydration, the mixture was sonicated only for 1 minute to obtain large nanoparticles for visualization under confocal laser scanning microscope (CLSM). A small drop of rhodamine-PE labeled FLPNPs in water was mounted onto a slide individually and visualized in the rhodamine (543 nm) channel. The representative images were taken and selected from a slice through the midsection of the FLPNPs.
Encapsulation efficiency and loading capacity
Ten milligrams of PTX-loaded NPs were accurately weighed and dissolved in 5 mL of acetonitrile, and then diluted with distilled water to 10 mL. The amount of PTX in the solution was determined by HPLC (Agilent LC 1100; Agilent Technologies, Santa Clara, CA, USA). The HPLC was equipped with a reverse-phase column (Symetry, 150 mm ×4.6 mm, 5 μm) at 40°C and a UV detector. The mobile phase was a mixture of acetonitrile and water (50:50 v/v). The samples were delivered at a flow rate of 1.0 mL/min and PTX was detected at a wavelength of 227 nm. The drug loading (DL) and encapsulation efficiency (EE) were calculated as follows:
|

|
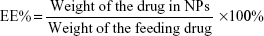
In vitro PTX release
In order to create pseudo-sink conditions, the in vitro release of PTX from NPs was monitored in an aqueous medium containing 1 M sodium salicylate at 37°C using double-chamber diffusion cells on a shaker stand. A Millipore® hydrophilic, low-protein-binding polyvinylidene fluoride membrane (Millipore Co., Bedford, MA, USA) with 0.1 mm pore size was placed between the two chambers. The donor chamber contained 4.5 mL NPs–sodium salicylate suspension, with a NPs concentration of 0.4 mg/mL. The receiver chamber contained 4.5 mL sodium salicylate solution (1 M, pH 7.4). The receiver chamber fluid was replaced with fresh sodium salicylate buffer at predetermined intervals. All the samples collected from the receiver chamber were extracted with dichloromethane to determine the amount of PTX released. The concentration of PTX in samples was determined by HPLC as described in the “Encapsulation efficiency and loading capacity” section.
In vitro cell culture
Cell lines and cell culture
EMT6 mammary carcinoma cells which overexpress folate receptor (FR)30 and normal L929 fibroblast cells, which lack FR expression,28 were employed. The two type cells were cultured in folate-deficient RPMI 1640 culture medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin under a humidified atmosphere of 95% air and 5% CO2 at 37°C. Before the experiment, cells were precultured till they reached 75% confluence.
In vitro cellular uptake
For quantitative cellular uptake analysis, EMT6 cells were seeded (5,000 cells/well) into 96-well black plates (Corning Costar Corp, Corning, NY, USA) and cultured for 24 hours, the medium was changed to the suspension of Nile Red-loaded NPs at an NP concentration of 0.250 mg/mL for 0.5, 2, and 24 hours, respectively. After incubation, the NP suspension in the testing wells was removed and the wells were washed with PBS three times to remove the NPs outside the cells. After that, RIPA Lysis Buffer was added to lyse the cells. Spectral scanning multimode reader (Varioskan Flash 3001; Thermo Scientific Inc., Rockford, IL, USA) was used to measure the fluorescence intensity from Nile Red-loaded NPs in the desired wells with excitation wavelength at 552 nm and emission wavelength at 636 nm. The cellular uptake efficiency was expressed as the percentage of fluorescence of the testing wells over that of the positive control wells.
For the fluorescence microscopy study, EMT6 and L929 cells were seeded in the 8-well cover-glass chamber and incubated for 24 hours at 37°C to encourage cell adhesion and spread. The fluorescent Nile Red-loaded NPs dispersed in the cell culture medium at concentration of 0.250 mg/mL were added into the wells. Cells were washed three times after incubation for 2 hours and then fixed by immunostaining fix solution for 10 minutes. The cells were further washed three times with immunostaining wash buffer, and stained with Actin-Tracker Green (Beyotime Biotechnology, Shanghai, People’s Republic of China) for 1 hour. After washing with immunostaining wash buffer, DAPI solution was added to stain cell nucleus for 5 minutes. The fixed cell monolayer was finally washed thrice with PBS and observed under CLSM (Zeiss LSM 710; Carl Zeiss Meditec AG).
In vitro cytotoxicity assay
For cytotoxicity measurement, EMT6 cells were incubated in 96-well plates (Corning Costar Corp) at a density of 5×103 cells per well and allowed to grow for 24 hours. Cells were then incubated in the media containing Taxol® or PTX-loaded LPNPs, FLPNPs (PTX concentrations of 0.25, 2.5, 12.5, and 25 μg/mL), and drug-free FLPNPs with the same amount of NPs for 24, 48, and 72 hours, respectively. At designated time intervals, the medium was removed, and the wells were washed twice with PBS. CCK-8 solution (10 μL) was added to each well, followed by incubation for 2 hours at 37°C. The absorbance at 450 nm was determined by spectral scanning multimode reader. Cell viability was expressed by the following equation:
Cell viability (%) = Abssample/Abscontrol ×100%, |
where Abssample is the fluorescence absorbance of the cells incubated with Taxol® or PTX-loaded NPs, while Abscontrol is the fluorescence absorbance of the cells incubated with the culture medium only (positive control).
In vivo antitumor efficacy study
BALB/c female mice (18–22 g) were purchased from Tianjin Medical University Laboratory Animals Center (Tianjin, People’s Republic of China). Animals were humanely cared for and supplied with FA-free food (Vital Laboratory Animal Center, Beijing, People’s Republic of China) and water. All the animal experiments were performed in compliance with the Guiding Principles for the Care and Use of Laboratory Animals, Peking Union Medical College, People’s Republic of China.
The tumor growth-inhibitory activities were assessed in EMT6 tumor-bearing BALB/c mice. EMT6 is a PTX-sensitive mouse mammary carcinoma cell line and can be grown as a solid tumor in BALB/c mice.30 In brief, EMT6 cells (0.1 mL) in the culture medium were implanted into the subcutaneous space of the right limb armpit of mice at a dosage of 2×107 cells/mouse. After inoculation of EMT6 cells, the tumor growth in each mouse was closely observed. When the tumor volume achieved about 100 mm3, the mice were randomly divided into four groups (each group, n=10), which were subject to intratumoral injection of the PTX-loaded FLPNPs, PTX-loaded LPNPs, Taxol®, and saline, respectively. Each mouse received the same dose of 20 mg/kg of PTX at days 0, 2, 4, 6, and 8 for five consecutive cycles. All mice were tagged, and tumors were measured every other day with an electronic caliper during the period of study. The tumor volume (V) was calculated using the formula V = (ab2)/2, where a and b are the largest and the smallest diameter of the tumor. The individual relative tumor volume (RTV) was defined as Vx/V1, where Vx is the volume in mm3 at a given time and V1 at the start of treatment. Drug efficacy was expressed as the percentage tumor growth inhibition (% TGI), calculated using the equation 100− (T/C ×100), where T is the mean RTV of the treated tumor and C is the mean RTV in the control group at the time of sacrifice of the mice. Furthermore, in order to estimate the side effects of the PTX-loaded LPNPs and FLPNPs, body weight of the mice was recorded during the treatment.
Statistical analysis
All data were expressed as the mean ± standard deviation. Statistical analysis (independent t-test) was performed with SPSS 10.0 software (SPSS Inc., Chicago, IL, USA). The statistical difference was considered to be significant when the P-value was less than 0.05.
Results and discussion
Preparation of the nanoparticles
Folic acid conjugated LPNPs comprise three distinct functional components: PCL-PEG-PCL, DSPE-PEG2000, and DSPE-PEG(2000) Folate. In our previous work, flower-like polymeric micelles based on PCL-PEG-PCL triblock copolymers were successfully produced by thin-film hydration and ultrasonic dispersion method. As shown in Figure 1, PCL-PEG-PCL triblock copolymers can form core-shell polymeric nanoparticles (PCEP NPs), which are characterized by a hydrophobic PCL core surrounded by a flower-like PEG hydrophilic corona. Both ends of the PEG blocks are anchored at the core/shell interfaces through the terminals, which results in the formation of a closed surface conformation and consequently limits the possible opsonization. Hydrophobic PTX can be readily incorporated into the hydrophobic core of the nanoparticles through hydrophobic interactions. Drug-loaded LPNPs in which a lipid layer coats the PCEP NPs can be formed using the same method, where the hydrophobic tails of the lipids self-assemble and attach onto the core of the PCEP NPs, whereas the hydrophilic heads extend outward toward the aqueous phase. The schematic structure of the novel self-assembled LPNPs is represented in Figure 2A. DSPE-PEG2000, the PEGlyated DSPE, was selected to form the lipid monolayer shell on the surface of the LPNPs. DSPE-PEG(2000) Folate, a PEGylated DSPE chain functionalized by folic acid conjugation, was employed for providing the LPNPs for targeted delivery capability.
 | Figure 1 (A) The formation of PTX-loaded polymeric nanoparticles from PCL-PEG-PCL. (B) TEM image of PTX-loaded PCEP NPs. |
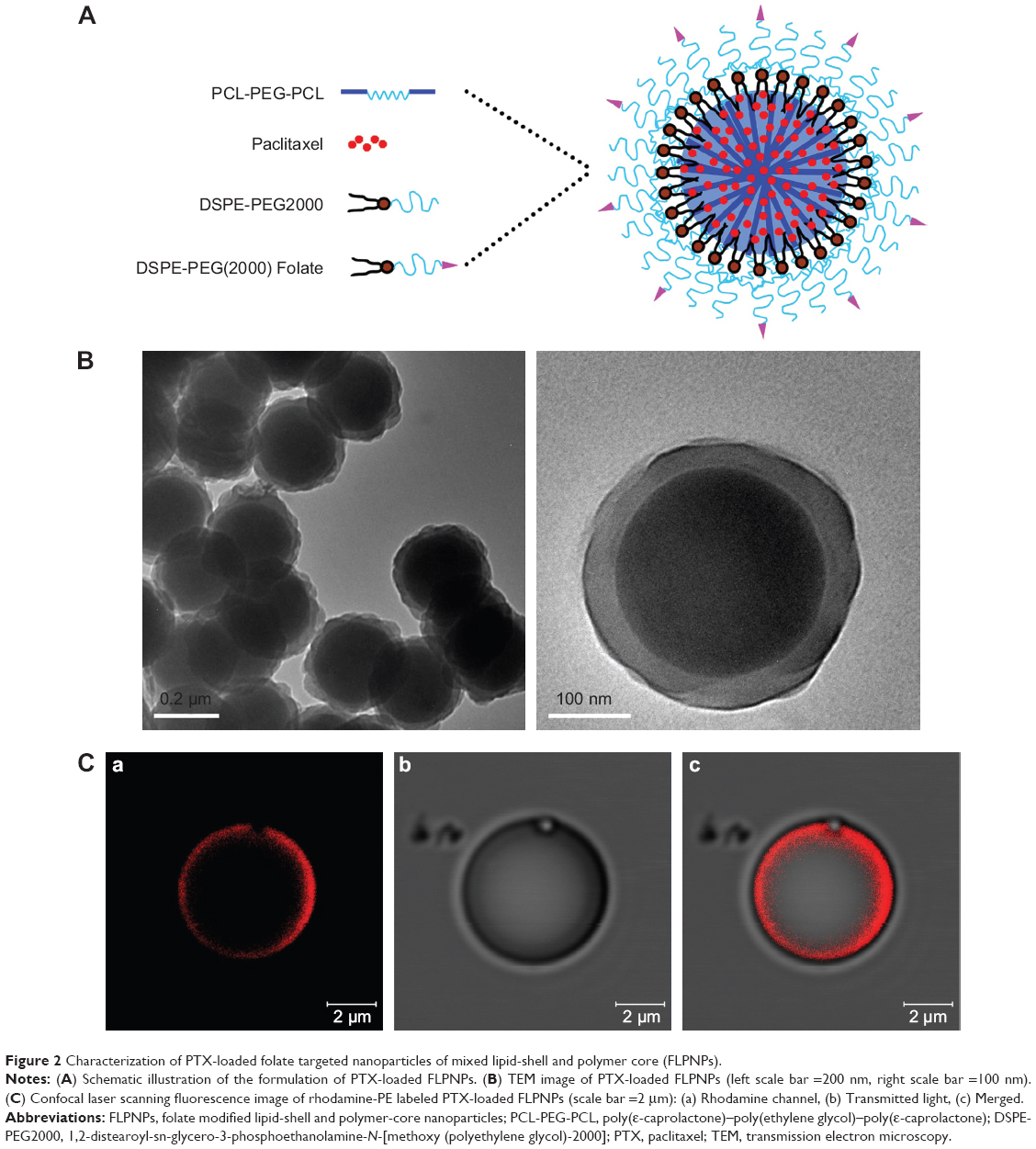 | Figure 2 Characterization of PTX-loaded folate targeted nanoparticles of mixed lipid-shell and polymer core (FLPNPs). |
Characterization of the nanoparticles
The morphology of the prepared FLPNPs was observed by TEM. As shown in Figure 2B, the FLPNPs exhibited homogeneous spherical shapes with an apparent lipid monolayer on the surface. It is noticeable that the morphology of FLPNPs is very complex, as the hydrophobic PCL core is surrounded by two layers resulting in the “core–shell–shell” structure. We speculated that the possible mechanism might be that the core is formed with PCL and PTX, while the inner and outer layers are formed with DSPE and extended PEG molecules, respectively. Figure 2C shows the confocal laser scanning fluorescence image of PTX-loaded FLPNPs, where the lipid shell was labeled with rhodamine-PE. A red shell was clearly observed in the inner lipid layer, confirming the “core–shell–shell” structure created by this procedure.
Table 1 illustrates the characteristics of the drug-loaded PCEP NPs, LPNPs (without folate conjugation) and FLPNPs (with folate conjugation). The particle size and polydispersity of various nanoparticles determined by DLS are shown in Table 1. The resulting FLPNPs were sized at 279.9±8.7 nm, which was larger than that of PCEP NPs (235.2±12.6 nm), presumably owing to the presence of a lipid monolayer shell on the PCEP NPs’ surface. Additionally, there was an approximate 8 nm increase in the particle size of FLPNPs as compared to the diameter of LPNPs. The polydispersity of PCEP NPs, LPNPs, and FLPNPs was 0.233, 0.146, and 0.173, respectively, demonstrating narrow size distribution.
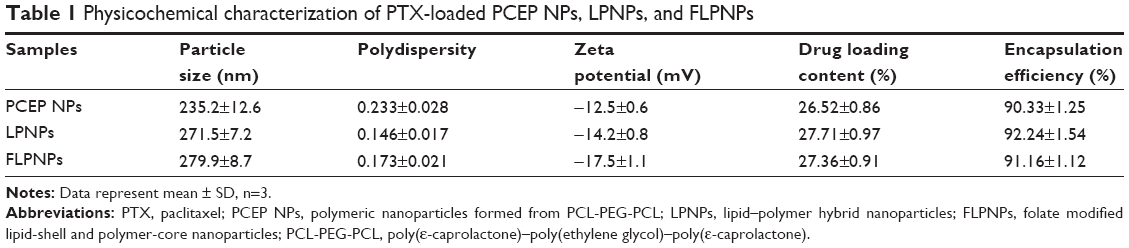 | Table 1 Physicochemical characterization of PTX-loaded PCEP NPs, LPNPs, and FLPNPs |
The drug loading (DL) and encapsulation efficiency (EE) of the NPs are important indexes for drug delivery systems. The DL and EE of PCEP NPs, LPNPs, and FLPNPs determined from three batches were 26.52%±0.86%, 27.71%±0.97%, 27.36%±0.91% and 90.33%±1.25%, 92.24%±1.54%, 91.16%±1.12%, respectively, with satisfactory reproducibility (Table 1). Using the thin-film hydration and ultrasonic dispersion method, high DL of more than 26% (w/w) in all of the three NPs could be achieved with high EE of more than 90% when the PTX feed ratio was 30% (w/w). The high DL and EE values prove the effectiveness of the NPs of lipid monolayer shell and polymeric core to load anticancer drugs. In addition, all PTX formulations showed similar DL and EE, indicating that drug encapsulation mainly depends on the core properties and on the affinity between the polymer and the drug. The hydrophobic core of FLPNPs was based on self-assembly of PCL-PEG-PCL amphiphilic copolymers. Therefore, hydrophobic PTX can be readily incorporated into the hydrophobic core of the nanoparticles through hydrophobic interactions in aqueous media.
Zeta potential is of significance for the characterization of the prepared NPs since it reflects the surface charge of colloidal dispersions and has been often used to predict and control dispersion stability.31,32 In addition, from the zeta potential measurement, we can estimate the dominant component on the particles’ surface.33 The zeta potential of the drug loaded PCEP NPs, LPNPs, and FLPNPs (Table 1) indicates the negative charges on the nanoparticle surface. LPNPs and FLPNPs showed higher negative zeta potential value than that of PCEP NPs. The increased zeta potential values may be explained by the location of the lipid on the particle surface since the absolute value of the negative charge from the phosphate group is larger than the positive charge from the amino group in the lipid molecule.34 The zeta potential of PTX-loaded FLPNPs was −17.5 mV (Table 1). The higher negative zeta potential value of the FLPNPs rather than that of the LPNPs results from the deprotonation of the carboxylic groups of folate.
In vitro drug release
Maintaining sink condition for poorly water-soluble drugs has been one of the difficulties in designing in vitro release experiments. In this study, the sink condition was achieved by using 1 M sodium salicylate solution35 and frequent replacement of fresh buffer during the in vitro release experiment. The in vitro drug release profiles of the PTX-loaded LPNPs and FLPNPs in 28 days are shown in Figure 3. PTX was released from both of the formulations in a controlled and sustained fashion with no apparent initial drug burst, indicating that there was almost no drug located at the surface of the NPs and the drugs were entrapped well in the hydrophobic core of the NPs. In the first 5 days, the cumulative release percent reached 70.28%±1.4% and 68.05%±1.6% for the FLPNPs and LPNPs, respectively. The sustained release of PTX provides the possibility to continually suppress cancer cells. The cumulative release achieved 90% after 14 days, showing almost complete drug release from the NP formulation. The drug release from the FLPNPs showed similar behavior to that of LPNPs, probably due to their similar polymer composition, percentage of drug loading, and particle size.
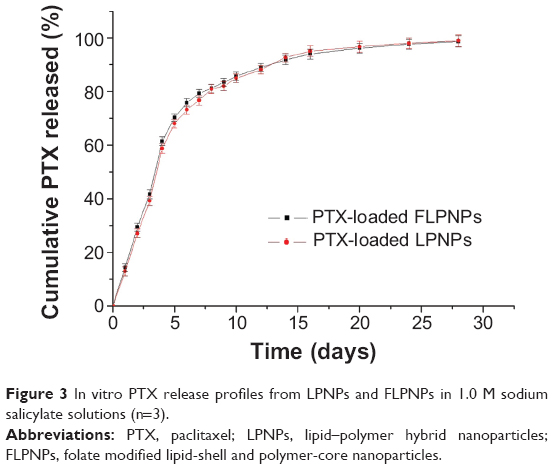 | Figure 3 In vitro PTX release profiles from LPNPs and FLPNPs in 1.0 M sodium salicylate solutions (n=3). |
In vitro cellular uptake
In order to evaluate the penetration of the NPs into the cells and the targeting effects of the NPs conjugated with folate, cellular uptakes of the LPNPs and FLPNPs were performed using folate receptor (FR) overexpressed EMT6 breast cancer cells and FR-deficient L929 fibroblast cells. The lipophilic stain, Nile Red, was chosen in this study because the water solubility of Nile Red is poor (less than 1 μg/mL) and it can be easily encapsulated into the hydrophobic core of the NPs.36 The internalization of Nile Red-loaded NPs incubated for 2 hours was visualized by CLSM. To precisely observe the cellular distributions of NPs, we performed triple fluorescence-labeling experiments and visualized red fluorescence from Nile Red, green fluorescence from Actin-Tracker Green labeling the actin, and blue fluorescence from DAPI labeling the nucleus. As described in Figure 4, the fluorescence from the NPs internalized in EMT6 cells and L929 cells was shown in Row 1, 2 and 3, 4, respectively. Row 1 and 3 show the images of the cells incubated with the LPNPs of no targeting effect, and Row 2 and 4 show the FLPNPs of folate conjugation. Thus, it can be seen from the figures that the blue fluorescence representing the nucleus stained by DAPI was circumvented by red fluorescence representing the Nile Red-loaded NPs internalized in the cytoplasm. Meanwhile, the cytoskeleton of EMT6 and L929 cells can be seen clearly from the green fluorescence representing the actin stained by Actin-Tracker green. Hence, the qualitative cellular uptake can be visually verified by the CLSM images. In addition, the folate receptor targeted behavior of the FLPNPs can also be examined. Folate is an attractive target ligand, which is easily recognized by FR that is overexpressed in a large number of cancer cells.29 In this regard, the PEG end group of the lipid-PEG layer of the LPNPs is particularly suitable for conjugation of the targeting moieties. Thus, we introduced DSPE-PEG(2000) Folate into the PTX-loaded LPNPs to promote the cellular uptake by receptor-mediated endocytosis (RME) and increase their therapeutic efficiency. As revealed in Figure 4, the fluorescence of FLPNPs in the cytoplasm (row 2) is much brighter than that of the LPNPs (row 1) after incubating for 2 hours. It can be explained that RME facilitates and promotes the entry of NPs into cells when folate targeted NPs meet the overexpressed FR on EMT6 cells. Nevertheless, in FR-deficient L929 cells, the fluorescence intensity in the cytoplasm does not show significant difference between the LPNPs (row 3) and the FLPNPs (row 4). These results directly demonstrated that the cellular uptake of the NPs can be enhanced by FR-mediated endocytosis process, which are consistent with previous reports regarding FR-dependent cellular uptake of folate-conjugated nanoparticles of mixed lipid-shell and polymer-core for anticancer therapy.21,22
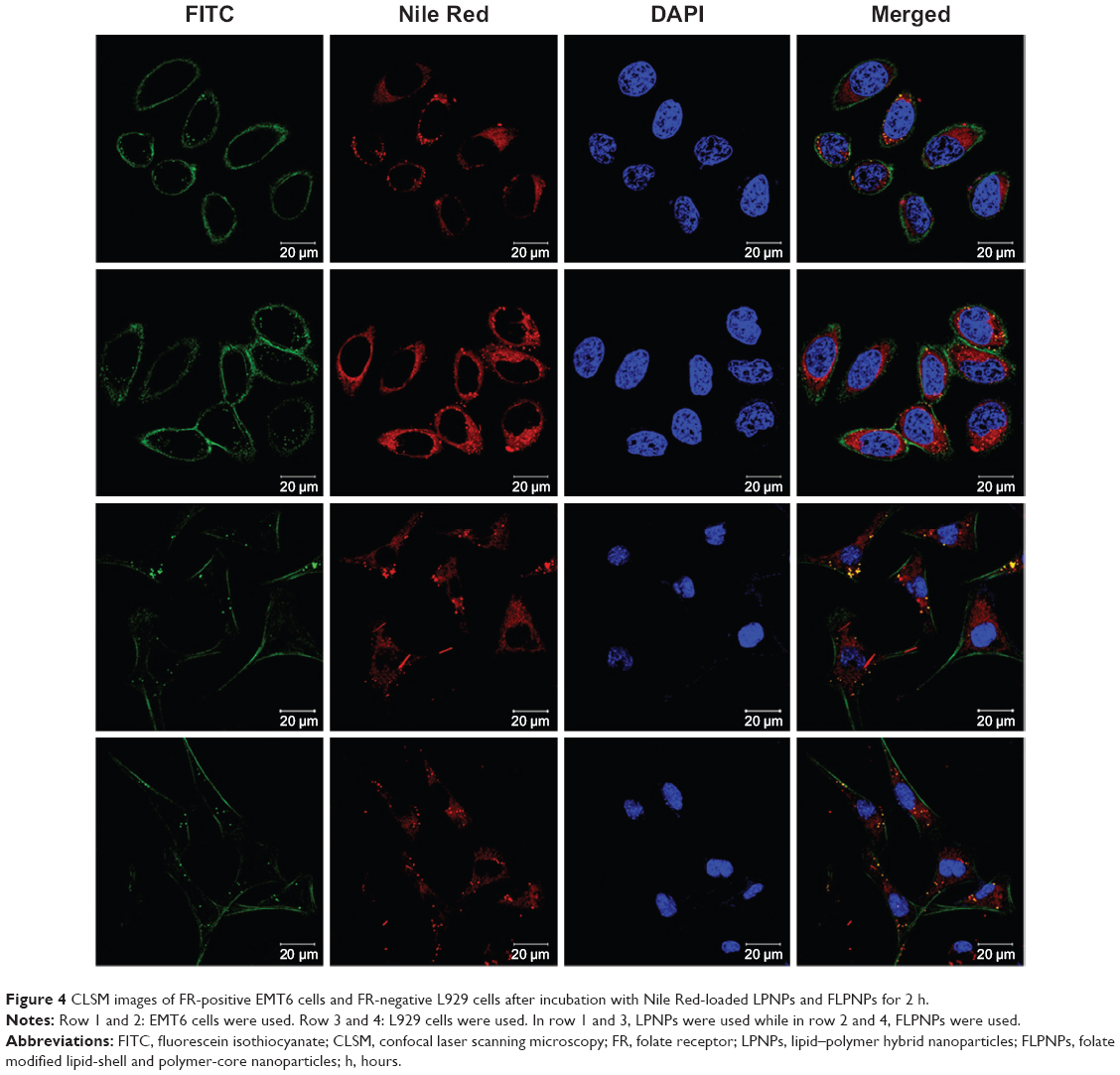 | Figure 4 CLSM images of FR-positive EMT6 cells and FR-negative L929 cells after incubation with Nile Red-loaded LPNPs and FLPNPs for 2 h. |
The quantitative cellular uptake efficiency of the Nile Red-loaded NPs was conducted by measuring percentage of the NPs used in incubation which have been entrapped in the EMT6 cells. The same concentration of dispersed NPs (0.250 mg/mL) as used in the previous CLSM was applied in this investigation. The cellular uptake efficiency of the LPNPs was measured to be 26.46%±1.92% and 31.42%±2.35% for 0.5 hours and 2 hours incubation time, respectively (Figure 5). Instead, the cellular uptake efficiency of the FLPNPs was measured to be 53.89%±2.64% and 60.34%±3.69% for 0.5 hours and 2 hours incubation time, respectively (Figure 5). The targeting effect of folate conjugation is thus significant of 103.7% increment for 0.5 hours incubation and 92.0% increases for 2 hours incubation, respectively (P<0.05). After 24 hours of incubation, the FLPNPs and LPNPs showed a similar degree of cellular uptake efficiency (74.72±5.29 vs 69.50±5.63, P>0.05), presumably due to the nonspecific uptake resulting from saturation of FR-mediated internalization of FLPNPs.
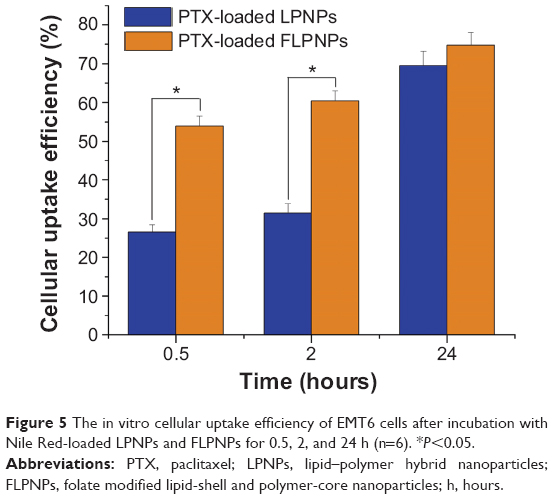 | Figure 5 The in vitro cellular uptake efficiency of EMT6 cells after incubation with Nile Red-loaded LPNPs and FLPNPs for 0.5, 2, and 24 h (n=6). *P<0.05. |
These qualitative and quantitative results of cellular uptake of the NPs provide strong evidence for the targeting effect of folate decoration for EMT6 cells. The potential mechanism of the in vitro cellular uptake of the LPNPs can be thought to be carrier-mediated endocytosis and cell fusion. LPNPs could be transported into cells across the lipid bilayer-structured cellular membranes by a nonspecific endocytosis pathway depending on the particle size and the presence of lipids on the NPs’ surface. The existence of lipid layer on the surface of the LPNPs could facilitate the entry of NPs into cells by the mutual interaction of the LPNPs and the cell membrane. As for the FLPNPs, the receptor-mediated endocytosis can further promote the cellular uptake, resulting in higher cellular uptake efficiency.
In vitro cytotoxicity assay
The in vitro cytotoxicity of PTX-loaded FLPNPs and PTX-loaded LPNPs against EMT6 cells was investigated in comparison with that of Taxol® using CCK-8 method. Figure 6 shows that the cytotoxicity of different PTX formulations was basically in the following order: Taxol® > FLPNPs > LPNPs. Drug-free FLPNPs did not exhibit obvious cytotoxicity to EMT6 cells during the test period, confirming the proposal that the NPs act merely as vehicles for the drug. Any cytotoxicity of the NPs observed was thus mainly attributed to the effects of the released drug alone. The cytotoxicity of Taxol®, PTX-loaded LPNPs, and PTX-loaded FLPNPs all showed time- and dose-dependent cell proliferation inhibition, confirming that higher drug concentration and longer incubation time are essential for the drug to effectively kill tumor cells. During the first 24 hours of incubation, PTX-loaded FLPNPs displayed a slightly higher cytotoxicity compared with PTX-loaded LPNPs in all of the concentration ranges (P>0.05), while Taxol® demonstrated significantly greater cytotoxicity than both of the two NPs at high drug concentrations (12.5 and 25 μg/mL, P<0.05). After 48 and 72 hours of incubation, there was no statistical difference among the three PTX formulations at low drug concentrations (0.25 and 2.5 μg/mL, P>0.05). However, at high drug concentrations (12.5 and 2.5 μg/mL), the cell cytotoxicity of Taxol® was significantly greater than both of the PTX-loaded LPNPs and FLPNPs (P<0.05); meanwhile, PTX-loaded FLPNPs contributed to higher reduction in cell viability than PTX-loaded LPNPs (P<0.05). The reason might be attributed to different cell uptake pathways of free drugs and drug-loaded NPs, and the controlled release manner of drug-loaded NPs. In cell culture medium, free PTX could directly enter cells via passive diffusion and be effective, whereas the two PTX-loaded NPs were mainly taken up by cells via the endocytic pathway and then the drug was released to kill cancer cells. Furthermore, it should be emphasized that in the case of Taxol® significant cytotoxic effect could be partially attributed to the excipient Cremophor® EL.15,37 However, in the case of PTX-loaded NPs the cytotoxicity observed was only induced by PTX itself. The in vitro cytotoxicity data of PTX-loaded FLPNPs and PTX-loaded LPNPs were in accordance with the results of the cellular uptake efficiency. As a result of the much higher cellular uptake for the first several hours, for the same PTX concentration and exposure time, FLPNPs would exhibit higher intracellular PTX release than LPNPs, thus resulting in higher in vitro cytotoxicity. Furthermore, for longer incubation periods, a larger number of EMT6 cells enter the G2 and M phases of the cell cycle during which PTX is more active. Thus, PTX-loaded FLPNPs showed higher cytotoxicity than the LPNPs for longer incubation time (48 and 72 hours, P<0.05). Therefore, it is clear that the FLPNPs formulation demonstrated higher cytotoxicity than the LPNPs formulation at high drug concentration and long exposure time, which demonstrated that folate receptor-mediated endocytosis can further facilitate the cellular uptake and thus enhance the cytotoxic effect of the drug-loaded NPs.
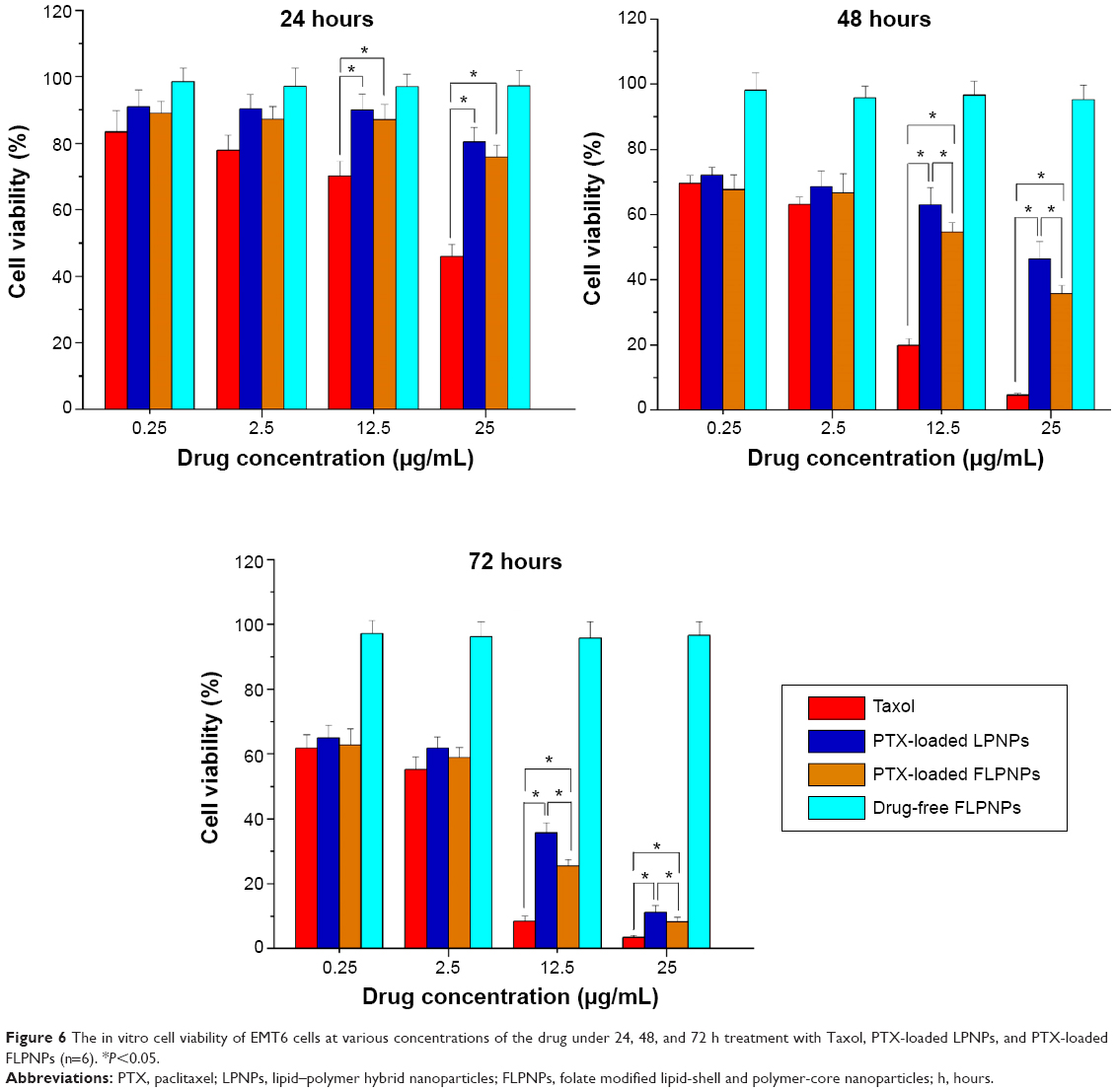 | Figure 6 The in vitro cell viability of EMT6 cells at various concentrations of the drug under 24, 48, and 72 h treatment with Taxol, PTX-loaded LPNPs, and PTX-loaded FLPNPs (n=6). *P<0.05. |
In vivo antitumor efficacy study
The antitumor efficacy of Taxol®, PTX-loaded LPNPs, and PTX-loaded FLPNPs was investigated in mice bearing EMT6 tumors. Figure 7A shows the tumor growth observed for 14 days in the mice received intratumoral injection of PTX-loaded LPNPs, PTX-loaded FLPNPs, Taxol®, and physiological saline. By 14 days after intratumoral injection, the average tumor volume in mice treated with Taxol®, PTX-loaded LPNPs, and PTX-loaded FLPNPs had increased relatively slowly and reached about 379, 424, and 639 mm3 while the average tumor volume in PBS was 1,238 mm3. PTX-loaded FLPNPs showed more efficient antitumor efficacy than did PTX-loaded LPNPs, with tumor growth inhibition of 65.78% and 48.38%, respectively (P<0.05). However, there is no significant difference between PTX-loaded FLPNPs and Taxol® (65.78% vs 69.44%, P>0.05).
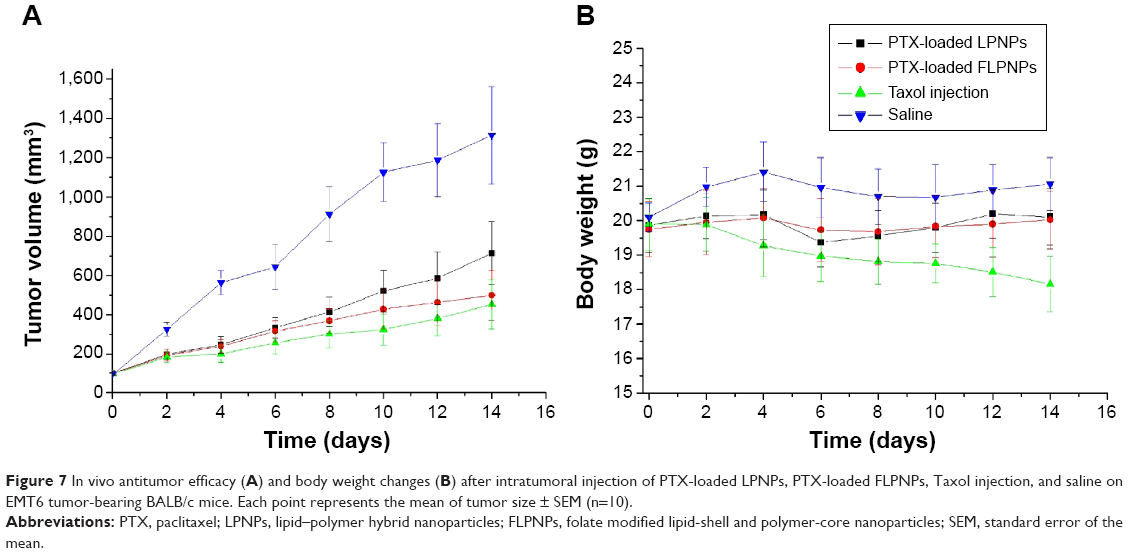 | Figure 7 In vivo antitumor efficacy (A) and body weight changes (B) after intratumoral injection of PTX-loaded LPNPs, PTX-loaded FLPNPs, Taxol injection, and saline on EMT6 tumor-bearing BALB/c mice. Each point represents the mean of tumor size ± SEM (n=10). |
Minimization of the side effects of drug-loaded NPs is one of the main concerns when developing novel drug-delivery vehicles. The change in body weight of the mice is an important index to evaluate systemic adverse effects.38 As shown in Figure 7B, PTX-loaded FLPNPs and PTX-loaded LPNPs demonstrated favorable results without any obvious body weight loss, whereas Taxol® induced obvious weight loss. The mice treated with PTX-loaded FLPNPs and PTX-loaded LPNPs kept a vigorous and healthy appearance throughout the full experiment. Nevertheless, the mice treated with Taxol® showed weakened vitality.
The results of the antitumor-efficacy experiment indicated that PTX could be released from NPs effectively and that the released PTX maintained its bioactivity. PTX-loaded FLPNPs showed significant antitumor efficacy as well as low toxicity in vivo, probably due to the more effective accumulation of FLPNPs in tumor cells via ligand-receptor interactions and the PTX released from NPs maintaining effective therapeutic concentration for a long period of time.
Conclusion
In this study, a novel lipid–polymer hybrid nanocarrier was synthesized that composed of folate (as targeting ligand), DSPE-PEG2000 (as lipid-shell), and PCL-PEG-PCL (as self-assembled core). Sustained, controlled, and targeted delivery of the anticancer drug PTX from the hybrid nanoparticles was successfully demonstrated both in vitro and in vivo. The PTX-loaded LPNPs and FLPNPs with high drug-loading efficiency and capacity were prepared using thin-film hydration and ultrasonic dispersion method. TEM image confirmed that the FLPNPs exhibited homogeneous spherical shapes with apparent hydrophobic polymer core and lipid monolayer shell on the surface. CLSM image further confirmed the “core–shell–shell” structure of the FLPNPs, where the inner lipid-shell was labeled with rhodamine-PE. PTX was released in a controlled and sustained fashion with no apparent initial drug burst, providing the possibility to continually fight against cancer cells. Nile Red-loaded FLPNPs showed higher efficient endocytosis abilities in FR-overexpressing EMT6 cells when compared to nontargeted LPNPs. In addition, in vitro cytotoxicity assays further confirmed that PTX-loaded FLPNPs had a significantly higher cytotoxicity than PTX-loaded LPNPs at high drug concentration and long exposure time. In BALB/c mice bearing EMT6 tumors, PTX-loaded FLPNPs showed similar antitumor efficacy but low toxicity compared to Taxol® via intratumoral chemotherapy. More importantly, PTX-loaded FLPNPs exhibited superior therapeutic efficiency than the PTX-loaded LPNPs, confirming the effective cellular accumulation and anticancer activity of folate targeted NPs via receptor-mediated endocytosis. Based on these findings, it can be concluded that PTX-loaded FLPNPs might have the potential to be developed as an attractive drug delivery system for effective chemotherapy against FA receptor overexpressing cancer.
Acknowledgment
The authors are grateful to the National Natural Science Foundation of China (Grant No 51103180, 81100100, and 51373199) and Tianjin Natural Science Foundation (Grant No 15JCZDJC38300) for financially supporting this work.
Disclosure
The authors report no conflicts of interest in this work.
References
Jia F, Liu X, Li L, Mallapragada S, Narasimhan B, Wang Q. Multifunctional nanoparticles for targeted delivery of immune activating and cancer therapeutic agents. J Control Release. 2013;172(3):1020–1034. | ||
Cooper DL, Conder CM, Harirforoosh S. Nanoparticles in drug delivery: mechanism of action, formulation and clinical application towards reduction in drug-associated nephrotoxicity. Expert Opin Drug Deliv. 2014;11(10):1661–1680. | ||
Mandal B, Bhattacharjee H, Mittal N, et al. Core-shell-type lipid-polymer hybrid nanoparticles as a drug delivery platform. Nanomedicine. 2013;9(4):474–491. | ||
Madni A, Sarfraz M, Rehman M, et al. Liposomal drug delivery: a versatile platform for challenging clinical applications. J Pharm Pharm Sci. 2014;17(3):401–426. | ||
He Y, Zhang L, Song C. Luteinizing hormone-releasing hormone receptor-mediated delivery of mitoxantrone using LHRH analogs modified with PEGylated liposomes. Int J Nanomedicine. 2010;5:697–705. | ||
Karanth H, Murthy RS. pH-sensitive liposomes–principle and application in cancer therapy. J Pharm Pharmacol. 2007;59(4):469–483. | ||
Pridgen EM, Alexis F, Farokhzad OC. Polymeric nanoparticle technologies for oral drug delivery. Clin Gastroenterol Hepatol. 2014;12(10):1605–1610. | ||
Mukherjee B, Satapathy BS, Mondal L, Dey NS, Maji R. Potentials and challenges of active targeting at the tumor cells by engineered polymeric nanoparticles. Curr Pharm Biotechnol. 2013;14(15):1250–1263. | ||
Li Y, Pei Y, Zhang X, et al. PEGylated PLGA nanoparticles as protein carriers: synthesis, preparation and biodistribution in rats. J Control Release. 2001;71(2):203–211. | ||
Wang Y, Kho K, Cheow WS, Hadinoto K. A comparison between spray drying and spray freeze drying for dry powder inhaler formulation of drug-loaded lipid-polymer hybrid nanoparticles. Int J Pharm. 2012;424(1–2):98–106. | ||
Gao J, Xia Y, Chen H, et al. Polymer-lipid hybrid nanoparticles conjugated with anti-EGF receptor antibody for targeted drug delivery to hepatocellular carcinoma. Nanomedicine (Lond). 2014;9(2):279–293. | ||
Zhang L, Chan JM, Gu FX, et al. Self-assembled lipid–polymer hybrid nanoparticles: a robust drug delivery platform. ACS Nano. 2008;2(8):1696–1702. | ||
Hadinoto K, Sundaresan A, Cheow WS. Lipid-polymer hybrid nanoparticles as a new generation therapeutic delivery platform: a review. Eur J Pharm Biopharm. 2013;85(3 Pt A):427–443. | ||
Li Y, Wu H, Yang X, et al. Mitomycin C-soybean phosphatidylcholine complex-loaded self-assembled PEG-lipid-PLA hybrid nanoparticles for targeted drug delivery and dual-controlled drug release. Mol Pharm. 2014;11(8):2915–2927. | ||
Danhier F, Lecouturier N, Vroman B, et al. Paclitaxel-loaded PEGylated PLGA-based nanoparticles: in vitro and in vivo evaluation. J Control Release. 2009;133(1):11–17. | ||
Fang RH, Aryal S, Hu CM, Zhang L. Quick synthesis of lipid-polymer hybrid nanoparticles with low polydispersity using a single-step sonication method. Langmuir. 2010;26(22):16958–16962. | ||
Thevenot J, Troutier AL, David L, Delair T, Ladavière C. Steric stabilization of lipid/polymer particle assemblies by poly(ethylene glycol)-lipids. Biomacromolecules. 2007;8(11):3651–3660. | ||
Salvador-Morales C, Zhang L, Langer R, Farokhzad OC. Immunocompatibility properties of lipid-polymer hybrid nanoparticles with heterogeneous surface functional groups. Biomaterials. 2009;30(12):2231–2240. | ||
Valencia PM, Basto PA, Zhang L, et al. Single-step assembly of homogenous lipid-polymeric and lipid-quantum dot nanoparticles enabled by microfluidic rapid mixing. ACS Nano. 2010;4(3):1671–1679. | ||
Wang AZ, Yuet K, Zhang L, et al. ChemoRad nanoparticles: a novel multifunctional nanoparticle platform for targeted delivery of concurrent chemoradiation. Nanomedicine (Lond). 2010;5(3):361–368. | ||
Liu Y, Li K, Pan J, Liu B, Feng SS. Folic acid conjugated nanoparticles of mixed lipid monolayer shell and biodegradable polymer core for targeted delivery of Docetaxel. Biomaterials. 2010;31(2):330–338. | ||
Zhao P, Wang H, Yu M, et al. Paclitaxel loaded folic acid targeted nanoparticles of mixed lipid-shell and polymer-core: in vitro and in vivo evaluation. Eur J Pharm Biopharm. 2012;81(2):248–256. | ||
Wong HL, Bendayan R, Rauth AM, Xue HY, Babakhanian K, Wu XY. A mechanistic study of enhanced doxorubicin uptake and retention in multidrug resistant breast cancer cells using a polymer-lipid hybrid nanoparticle system. J Pharmacol Exp Ther. 2006;317(3):1372–1381. | ||
Li B, Xu H, Li Z, et al. Bypassing multidrug resistance in human breast cancer cells with lipid/polymer particle assemblies. Int J Nanomedicine. 2012;7:187–197. | ||
Seyednejad H, Ghassemi AH, van Nostrum CF, Vermonden T, Hennink WE. Functional aliphatic polyesters for biomedical and pharmaceutical applications. J Control Release. 2011;152(1):168–176. | ||
Ruysschaert T, Sonnen AF, Haefele T, Meier W, Winterhalter M, Fournier D. Hybrid nanocapsules: interactions of ABA block copolymers with liposomes. J Am Chem Soc. 2005;127(17):6242–6247. | ||
Zhang L, He Y, Ma G, Song C, Sun H. Paclitaxel-loaded polymeric micelles based on poly(varepsilon-caprolactone)-poly(ethylene glycol)-poly(varepsilon-caprolactone) triblock copolymers: in vitro and in vivo evaluation. Nanomedicine. 2012;8(6):925–934. | ||
Bhattacharya D, Das M, Mishra D, et al. Folate receptor targeted, carboxymethyl chitosan functionalized iron oxide nanoparticles: a novel ultradispersed nanoconjugates for bimodal imaging. Nanoscale. 2011;3(4):1653–1662. | ||
Wang F, Chen Y, Zhang D, et al. Folate-mediated targeted and intracellular delivery of paclitaxel using a novel deoxycholic acid-O-carboxymethylated chitosan-folic acid micelles. Int J Nanomedicine. 2012;7:325–337. | ||
Janat-Amsbury MM, Yockman JW, Lee M, et al. Combination of local, nonviral IL12 gene therapy and systemic paclitaxel treatment in a metastatic breast cancer model. Mol Ther. 2004;9(6):829–836. | ||
Liu Y, Pan J, Feng SS. Nanoparticles of lipid monolayer shell and biodegradable polymer core for controlled release of paclitaxel: effects of surfactants on particles size, characteristics and in vitro performance. Int J Pharm. 2010;395(1–2):243–250. | ||
Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–515. | ||
Mei L, Zhang Y, Zheng Y, et al. A novel docetaxel-loaded poly (ε-caprolactone)/pluronic F68 nanoparticle overcoming multidrug resistance for breast cancer treatment. Nanoscale Res Lett. 2009;4(12):1530–1539. | ||
Duncanson WJ, Figa MA, Hallock K, Zalipsky S, Hamilton JA, Wong JY. Targeted binding of PLA microparticles with lipid-PEG-tethered ligands. Biomaterials. 2007;28(33):4991–4999. | ||
Cho YW, Lee J, Lee SC, Huh KM, Park K. Hydrotropic agents for study of in vitro paclitaxel release from polymeric micelles. J Control Release. 2004;97(2):249–257. | ||
Yu Y, Chen CK, Law WC, et al. Well-defined degradable brush polymer-drug conjugates for sustained delivery of Paclitaxel. Mol Pharm. 2013;10(3):867–874. | ||
Bouquet W, Boterberg T, Ceelen W, et al. In vitro cytotoxicity of paclitaxel/beta-cyclodextrin complexes for HIPEC. Int J Pharm. 2009;367(1–2):148–154. | ||
Zhao T, Chen H, Dong Y, et al. Paclitaxel-loaded poly(glycolide-co-epsilon-caprolactone)-b-D-alpha-tocopheryl polyethylene glycol 2000 succinate nanoparticles for lung cancer therapy. Int J Nanomedicine. 2013;8:1947–1957. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.