Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
Fluticasone propionate/formoterol for COPD management: a randomized controlled trial
Authors Papi A ![]() , Dokic D
, Dokic D ![]() , Tzimas W, Mészáros I, Olech-Cudzik A
, Tzimas W, Mészáros I, Olech-Cudzik A ![]() , Koroknai Z
, Koroknai Z ![]() , McAulay K, Mersmann S, Dalvi PS, Overend T
, McAulay K, Mersmann S, Dalvi PS, Overend T
Received 9 March 2017
Accepted for publication 23 May 2017
Published 5 July 2017 Volume 2017:12 Pages 1961—1971
DOI https://doi.org/10.2147/COPD.S136527
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
A Papi,1 D Dokic,2 W Tzimas,3 I Mészáros,4 A Olech-Cudzik,5 Z Koroknai,6 K McAulay,7 S Mersmann,8 PS Dalvi,9 T Overend9
1Department of Internal and CardioRespiratory Medicine, Reseach Center on Asthma and COPD, University of Ferrara, Ferrara, Italy; 2Clinic of Pulmology and Allergy, Clinical Centre, Medical Faculty, Ss. Cyril and Methodius University, Skopje, Macedonia; 3Pneumologische Praxis, München, Germany; 4Coral Szakorvosi Centrum, Budapest, Hungary; 5Ostrowieckie Centrum Medyczne Spólka, Ostrowiec Swietokrzyski, Poland; 6PAREXEL International, Global Medical Services, Budapest, Hungary; 7Medical Operations, Mundipharma Research Limited, Cambridge, UK; 8Biostatistics and Clinical Data Science, Mundipharma Research GmbH & Co. KG, Limburg, Germany; 9Medical Science - Respiratory, Mundipharma Research Limited, Cambridge, UK
Purpose: To evaluate fluticasone propionate/formoterol (FP/FORM) in COPD.
Patients and methods: COPD patients with forced expiratory volume in 1 s (FEV1) ≤50% predicted and ≥1 moderate/severe COPD exacerbation in the last 12 months were randomized to FP/FORM 500/20 or 250/10 µg bid, or formoterol (FORM) 12 µg bid for 52 weeks. The primary outcome was the annualized rate of moderate/severe COPD exacerbations.
Results: In total, 1,765 patients were randomized. There were fewer discontinuations with FP/FORM 500/20 µg (20.6%) and 250/10 µg (24.0%) compared with FORM (26.1%). None of the two FP/FORM doses reduced the moderate/severe exacerbation rate versus FORM (rate ratios [RR]: 0.93; P≤0.402). There was a trend toward a lower moderate/severe exacerbation rate with FP/FORM 500/20 µg versus FORM in patients with ≥2 exacerbations in the preceding year (RR: 0.79; P=0.084). Pre- and post-dose FEV1 and forced vital capacity were greater with FP/FORM 500/20 µg versus FORM (P≤0.039). There was a trend toward a lower EXAcerbations of Chronic pulmonary disease Tool (EXACT) exacerbation rate with FP/FORM 500/20 µg versus FORM (RR: 0.87; P=0.077). There were more St George’s Respiratory Questionnaire for COPD (SGRQ-C) responders with FP/FORM 500/20 µg than FORM (odds ratios [OR] at weeks 6, 23 and 52 ≥1.28; P≤0.054). EXACT-respiratory symptoms total and breathlessness scores were lower with both FP/FORM 500/20 µg and 250/10 µg versus FORM (P≤0.066). Acute β2-agonist-induced effects and 24-hour Holter findings were similar for all treatments. Mean 24-hour urinary cortisol was similarly reduced with both FP/FORM doses. Radiologically confirmed pneumonia was seen in 2.4%, 3.2% and 1.5% of FP/FORM 500/20 µg, FP/FORM 250/10 µg and FORM-treated patients, respectively. Adverse events were otherwise similar across treatment groups.
Conclusion: FP/FORM did not reduce exacerbation rates versus FORM. Numerical benefits were observed with FP/FORM 500/20 µg versus FORM for secondary variables, including lung function, EXACT exacerbations, SGRQ-C and EXACT-respiratory symptoms total and breathlessness scores. Few efficacy differences were evident between FP/FORM 250/10 µg and FORM. Pneumonia was more frequent in FP/FORM-treated patients, although the absolute difference was low. Adverse events were otherwise similar between treatments.
Keywords: flutiform, chronic bronchitis, emphysema, exacerbations, eosinophils
Introduction
The primary goals of COPD management are the improvement of symptoms, exercise tolerance and health status, the prevention of disease progression and exacerbations, and mortality reduction.1 Currently, inhaled corticosteroid/long-acting β2-agonist combinations (ICS/LABAs) are recommended for the treatment of Global Initiative for Chronic Obstructive Lung Disease (GOLD) group C and D patients, that is, those at risk of exacerbations, given evidence of risk reduction from a number of trials.2–8 There is additionally much interest at present in eosinophilic and non-eosinophilic COPD phenotypes and a large ongoing trial may further define the role of ICS/LABAs and other treatment classes in the future.9
Flutiform® is a fixed combination of fluticasone propionate and formoterol fumarate (FP/FORM) in a pressurized metered-dose inhaler (pMDI), which is licensed for use in asthma following a comprehensive series of clinical trials.10–19 The EFFECT trial (Efficacy of Fluticasone propionate/FormotErol in COPD Treatment) was a Phase III study undertaken to evaluate the efficacy and safety of FP/FORM in COPD.
Methods
The methodology of the EFFECT trial has previously been reported.20 The trial is registered with the EU Clinical Trials Register (EudraCT Number: 2012–004162–17). The protocol and other relevant study documentation were formally approved in each country by central and/or local ethics committees (Supplementary materials, Table S1) before subjects were screened, and all subjects provided written informed consent prior to any study-specific procedures being performed.
Patients
Male and female COPD patients aged ≥40 years, with post-bronchodilator forced expiratory volume in 1 s (FEV1) ≤50% predicted and an FEV1/forced vital capacity (FVC) ratio <0.7, a history of at least 1 moderate or severe COPD exacerbation in the last 12 months (requiring systemic corticosteroids and/or antibiotics and/or hospitalization), and a minimum 10 pack-year smoking history were enrolled. Moderate or severe exacerbations at screening (or during the run-in period) rendered a patient ineligible. During the treatment period, prohibited medications included long-acting muscarinic antagonists, phosphodiesterase-4 inhibitors, xanthine derivatives, short-acting β-agonist/muscarinic antagonist combinations, oral β-agonists, non-selective β-blockers, maintenance acetylcysteine or carbocysteine, and systemic steroids (except those required for the short-term treatment of an exacerbation).
Study design
This was a randomized, parallel-group, double-blind study. Patients discontinued their existing COPD medications and received tiotropium dry powder inhaler (Spiriva®) 18 μg once daily during a 2-week run-in period. During the run-in, a baseline EXAcerbations of Chronic pulmonary disease Tool (EXACT) score was determined. An electronic interface (Model 2120 In2itive™ eDiary [Vitalograph, Buckingham, UK]) was used to self-administer the EXACT daily. At the end of the run-in, patients were randomized to 52 weeks of treatment with FP/FORM pMDI 500/20 μg bid or 250/10 μg bid or formoterol (FORM) pMDI 12 μg bid (Atimos® Modulite®). Patients attended post-randomization visits at weeks 2, 6, 13, 23, 33, 43 and 52. The EXACT “baseline” score was continually reset throughout the 12-month study per EXACT user guidelines.21 When changes in EXACT symptom scores met validated exacerbation thresholds,21 alerts were sent to both the investigator (via email) and the patient (via the electronic diary) to prompt patient–physician contact and ascertain whether clinical review was necessary.
Outcomes
The primary outcome was the annualized rate of moderate-to-severe COPD exacerbations during the 52-week treatment period. Moderate events were those requiring treatment with systemic corticosteroids and/or antibiotics. Severe exacerbations were events requiring hospitalization or resulting in death. Events separated by at least 7 days were defined as 2 distinct exacerbations. A standardized regimen of 30–40 mg of prednisolone for 7–14 days (per GOLD 2014 guidelines) was recommended if oral corticosteroid treatment was considered necessary for exacerbation management.
Secondary outcomes included: the average pre- and 1-hour post-dose FEV1 and FVC over 52 weeks; the annualized rate of EXACT exacerbations; the time to first moderate or severe COPD exacerbation; the change in St George’s Respiratory Questionnaire for COPD (SGRQ-C) from baseline to weeks 6, 23, and 52; the proportion of SGRQ-C responders; daily rescue medication use (occasions/day); the percentage change in awakening-free nights from baseline over 52 weeks; and the average EXACT-respiratory symptoms (E-RS) breathlessness and total scores over 52 weeks. Changes from baseline to week 6 in surfactant protein-D (SP-D) and C-C motif chemokine ligand 18 (CCL-18) were also measured and their relationships with clinical outcomes were examined.
A number of post hoc outcomes were also defined to further evaluate the study data including: time to first clinically important deterioration (CID; deterioration defined as either a moderate or severe exacerbation, an increase in SGRQ ≥4 units or a decrease in FEV1 ≥100 mL);22 time to discontinuation; the distribution of baseline blood eosinophil counts; and the annualized rate of moderate or severe exacerbations dichotomized by baseline blood eosinophil counts. Two other exploratory definitions of CID (incorporating differing combinations of FEV1, SGRQ-C, moderate/severe exacerbation events and EXACT exacerbation events) were also evaluated.
Safety outcomes included: adverse event summaries; the incidence of radiologically- and clinically-defined pneumonia per British Thoracic Society (BTS) criteria; 24-hour Holter monitoring (in ~100 patients/arm); an assessment of β2-agonist-induced safety effects (maximum reductions in serum potassium and diastolic blood pressure, maximum increments in heart rate, systolic blood pressure and QT interval) (in ~125 patients/arm); and 24-hour urinary cortisol estimation (in ~50 patients/arm without ICS exposure at screening).
Statistics
Assuming a 20% reduction in exacerbation rate with combination therapy, an exacerbation rate of 0.8 exacerbations/patient/year in the formoterol group, 5% of patients being excluded from the full analysis population, and a two-sided alpha of 5%, a sample size of 586 patients per treatment group was required.20
The primary endpoint, the annualized rate of moderate/severe exacerbations was analyzed using a negative binomial regression model with fixed terms for treatment, FEV1% predicted category, number of exacerbations in the previous year category, smoking status, prior ICS use, and country, and the logarithm of time on treatment as an offset variable. A hierarchical gatekeeping procedure was employed to control for multiplicity given the 2 comparisons for the primary endpoint: the secondary comparison FP/FORM 250/10 μg versus FORM was analyzed in a confirmatory manner only if the primary comparison (FP/FORM 500/20 μg versus FORM) was significant at the 5% level. Control for multiplicity for pre-defined key secondary endpoints was done using a Hochberg closed testing procedure. The same negative binomial model was used to analyze EXACT exacerbations (but including baseline EXACT total score as an additional covariate) and post hoc analyses of exacerbations by baseline blood eosinophil count. Time to first moderate/severe exacerbation was analyzed with a Cox proportional hazards model with fixed terms for treatment, FEV1% predicted category, number of exacerbations in the previous year category, smoking status, prior ICS use and country. An identical model was used to analyze, post hoc, time to first CID. Spirometry, SGRQ-C, E-RS total and breathlessness scores were analyzed using repeated measures analysis of covariance with fixed terms for treatment, FEV1% predicted category, number of exacerbations in the previous year category, the respective baseline value, smoking status, prior ICS use, country, time-point, and treatment by time-point interaction. SGRQ-C responder rates were analyzed for each defined timepoint using a logistic regression model with fixed terms for treatment, FEV1% predicted category, number of exacerbations in the previous year category, smoking status, prior ICS use, and country. For details of biomarker analyses please refer to the earlier methodological manuscript.20
Results
A total of 1,765 patients were randomized at 223 sites in 16 countries (Bulgaria, Czech Republic, Germany, Hungary, Latvia, Lithuania, Republic of Macedonia, Poland, Romania, Russian Federation, Slovakia, South Africa, South Korea, Spain, Ukraine, and UK). The full analysis population (FAP: patients receiving ≥1 dose of study treatment and having ≥1 post-baseline exacerbation assessment) and safety population (patients receiving ≥1 dose of study treatment) were identical; and comprised 587, 588 and 590 patients in FP/FORM 500/20 μg, FP/FORM 250/10 μg and FORM arms, respectively. Patient disposition is summarized in Figure 1. There were fewer early discontinuations among patients randomized to FP/FORM 500/20 μg (20.6%) and 250/10 μg (24.0%) compared with those receiving FORM (26.1%; Figure 2). A post hoc analysis of time to discontinuation indicated that FORM-treated patients discontinued earlier than those treated with FP/FORM 500/20 μg (hazard ratio [HR]: 0.77; P=0.029), but not FP/FORM 250/10 μg (HR: 0.90; P=0.348). Baseline demographic and disease characteristics of the randomized subjects were well balanced across treatment groups and are summarized in Table 1. A post hoc analysis showed a similar population distribution of eosinophil counts in each treatment group (Supplementary materials, Table S2).
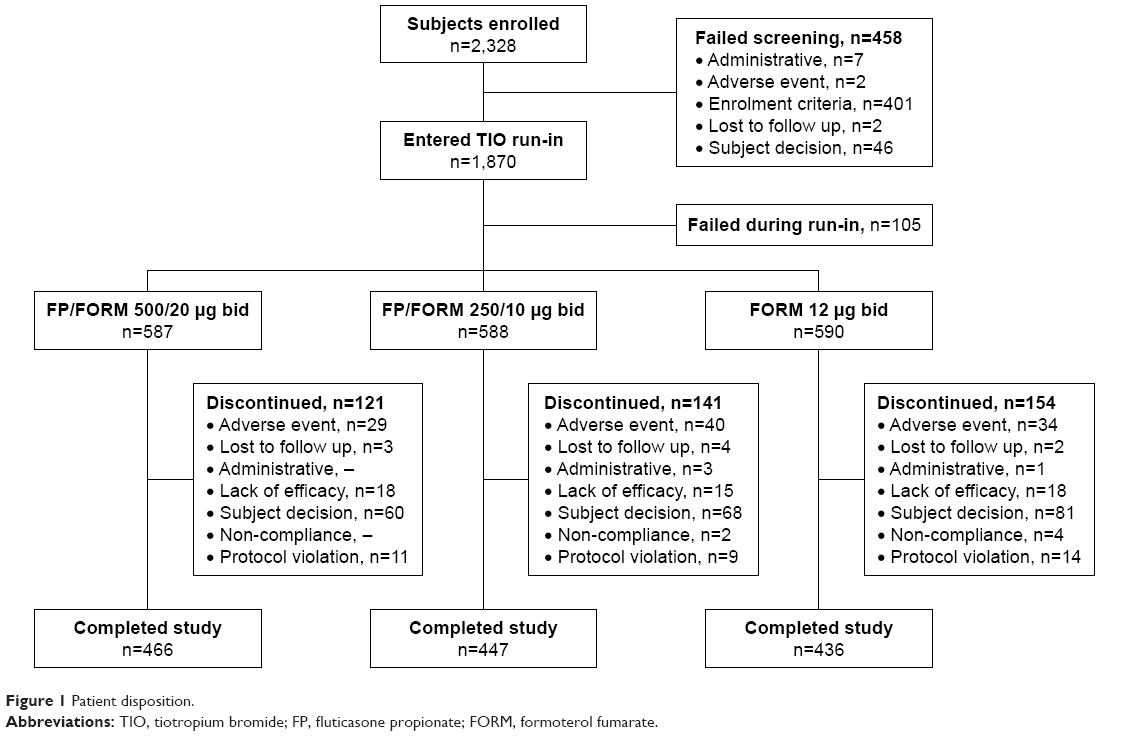 | Figure 1 Patient disposition. |
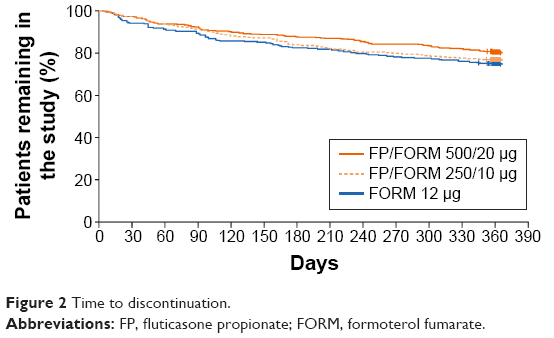 | Figure 2 Time to discontinuation. |
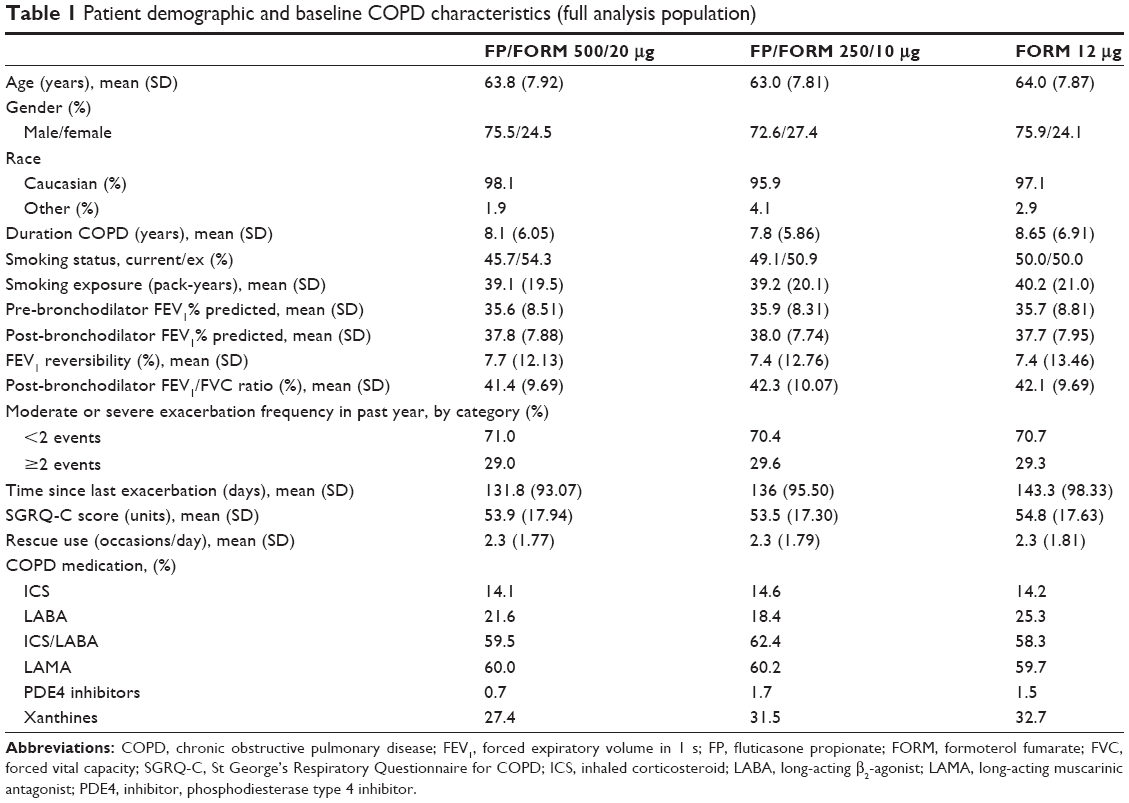 | Table 1 Patient demographic and baseline COPD characteristics (full analysis population) |
Efficacy
Exacerbations
A high proportion of patients (~59%) experienced no exacerbations during the course of the study despite reporting at least 1 moderate/severe event in the prior year. No difference was seen in the annualized rate of moderate/severe COPD exacerbations between either FP/FORM arm compared with FORM (primary endpoint) (Table 2). In view of the sequential testing procedure employed and the non-significant result for the primary endpoint, subsequent inferential analyses should be considered exploratory in nature. No difference was seen in the time to moderate/severe exacerbations (Table 2). In patients with at least 2 moderate/severe COPD exacerbations in the year preceding the study, there was a 21% reduction in the rate of moderate/severe events with FP/FORM 500/20 μg versus FORM that marginally failed to achieve significance at the 5% level (rate ratios [RR]: 0.79 [95% CI: 0.61, 1.03]; P=0.084); whereas exacerbation rates were similar for FP/FORM 250/10 μg and monotherapy (RR: 1.09 [95% CI: 0.85, 1.40]; P=0.484). Of note, even in this subpopulation of reported frequent exacerbators, almost 50% of patients experienced no exacerbation events throughout the entire study.
 | Table 2 Moderate/severe exacerbations analysis (full analysis population) |
In view of the above results, a series of post hoc analyses were conducted in subgroups whereby the overall FAP population was dichotomized using baseline blood eosinophil cut-offs of 2%, 3% and 4% (Supplementary materials, Table S3). Exacerbation risk reduction with FP/FORM versus FORM was found to be no greater in any of the patient subgroups with a higher blood eosinophil count. Additionally, a graded increase in exacerbation rates in FORM-treated patients with increasing eosinophil counts (≥2%, ≥3%, ≥4%) was not seen.
Lung function
Average pre- and 1-hour post-dose FEV1 and FVC over the course of the study were greater with FP/FORM 500/20 μg than FORM. With FP/FORM 250/10 μg, post-dose FEV1 and FVC exceeded that with monotherapy (Table 3). Differences between FP/FORM and FORM in these parameters at week 52 were similar to differences in the average values between treatments over the course of the study, albeit were magnified at week 52 by approximately a further 10 mL for FEV1 and 25 mL for FVC.
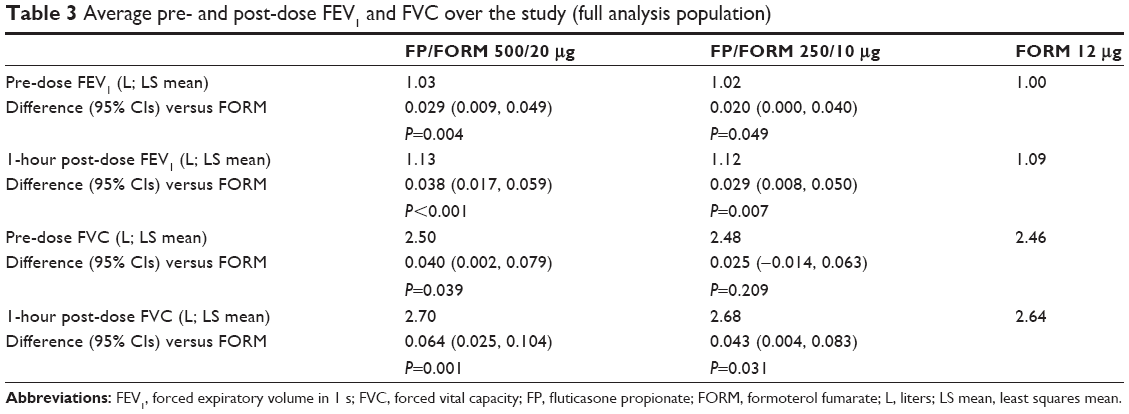 | Table 3 Average pre- and post-dose FEV1 and FVC over the study (full analysis population) |
Other efficacy endpoints
Approximately 48% of patients experienced no EXACT-defined exacerbation events throughout the study duration. There was a 13% reduction in the annualized rate of EXACT exacerbations with FP/FORM 500/20 μg versus FORM that approached significance at the 5% level, whereas EXACT exacerbation rates were similar for FP/FORM 250/10 μg and FORM (Table 4).
 | Table 4 EXACT exacerbations analysis (full analysis population) |
SGRQ-C scores showed modest changes from baseline during the study across all treatment arms (Figure 3). Average SGRQ-C scores over the treatment period were significantly lower (improved) in the FP/FORM 500/20 μg arm versus FORM. The responder analyses at weeks 6, 23 and 52, with response defined per the minimum clinically important improvement threshold of −4 units23 (and with imputation of “non-response” for early discontinuations), indicated a greater likelihood of response with FP/FORM 500/20 μg than FORM (odds ratios [OR] [at weeks 6, 23 and 52] ≥1.28; P≤0.054). Differences between FP/FORM 250/10 μg and FORM were evident only at the week 6 time point (OR =1.31; P=0.036).
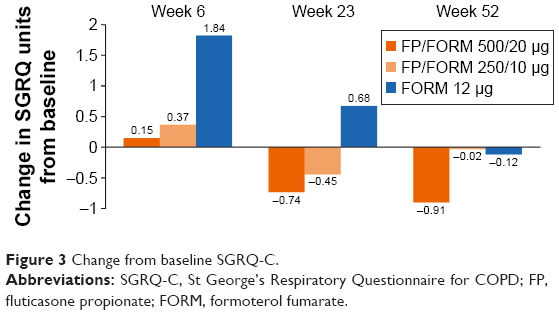 | Figure 3 Change from baseline SGRQ-C. |
The E-RS total score showed greater reductions in symptom scores with both FP/FORM 500/20 μg and 250/10 μg versus FORM (treatment difference −0.47 units [P=0.039] and −0.52 units [P=0.021], respectively). The E-RS breathlessness subscale indicated lower dyspnea scores with combination treatment versus FORM (treatment difference −0.22 [P=0.066] and −0.27 [P=0.024]) for FP/FORM 500/20 μg and 250/10 μg, respectively. Patients with sleep disturbance at baseline gained ~11%–12% additional awakening-free nights with all treatments over the course of the study, although no between-treatment differences were noted.
A post hoc analysis of time to first CID revealed a prolonged time to event occurrence with FP/FORM 500/20 μg and 250/10 μg versus FORM (hazard ratios 0.77 [P<0.001] and 0.88 [P=0.044]), respectively (Figure 4). Similar results were obtained for two other exploratory definitions of CID.
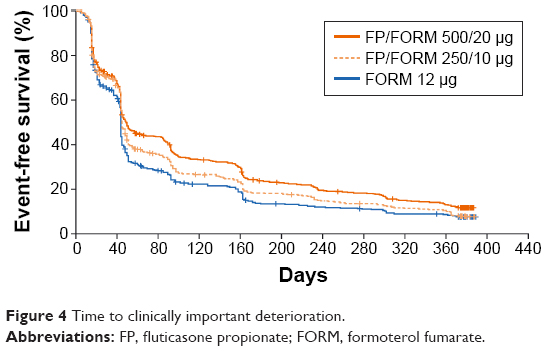 | Figure 4 Time to clinically important deterioration. |
Biomarkers
Changes in SP-D and CCL-18 from baseline to week 6 are summarized in Supplementary materials, Tables S4 and S5, respectively. No between-treatment differences were observed for either biomarker, nor were associations with efficacy (for SP-D) or safety (for CCL-18) outcomes evident.
Safety
An overall summary of adverse events, and a summary of the most commonly affected organ systems, are presented in Tables 5 and 6, respectively, while a summary of common individual adverse events is presented in Supplementary materials, Table S6. Event frequencies were generally similar across treatment groups. Oral fungal infections were reported by 5 to 6 patients in all treatment groups, as were diabetic/hyperglycemic events. None of the latter was considered related to treatment by investigators. There were no reports of skin thinning or bruising. Radiologically confirmed pneumonia in accordance with BTS criteria was reported in 14 (2.4%), 19 (3.2%) and 9 (1.5%) of patients in the FP/FORM 500/20 μg, FP/FORM 250/10 μg and FORM groups, respectively. Applying radiological and/or clinical criteria, again in accordance with BTS standards, 17 (2.9%), 23 (3.9%) and 11 (1.9%) of patients in the corresponding groups were diagnosed with pneumonia. There were no overt differences between-treatments observed in the occurrence of serious cardiovascular events.
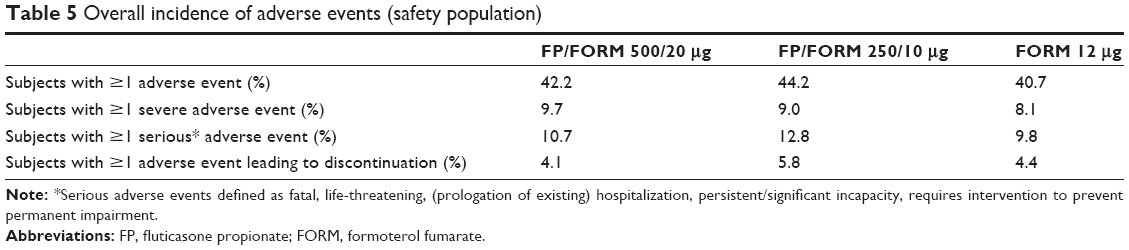 | Table 5 Overall incidence of adverse events (safety population) |
 | Table 6 Incidence (%) of frequent adverse events by system organ class (safety population) |
Assessments of maximal acute β2-agonist-induced effects (decreases in serum potassium and diastolic blood pressure, increases in heart rate, QTc interval and systolic blood pressure) revealed no evidence of a greater effect with FP/FORM 500/20 μg than with the other two treatments incorporating a lower formoterol dose. Furthermore, 24-hour Holter monitoring also revealed no relevant between-treatment differences. Estimation of 24-hour urinary-free cortisol corrected for creatinine (24-hour UFCC) at weeks 6 and 52 in a subgroup of subjects not on inhaled corticosteroids pre-study demonstrated a similar mean reduction from baseline in 24-hour UFCC in both FP/FORM arms but no change from baseline in the FORM group.
Fifty-three patients died (21 [3.6%] on FP/FORM 500/20 μg; 19 [3.2%] on FP/FORM 250/10 μg; 13 [2.2%] on FORM). None of the deaths was reported as related to the study medication by the treating physician. Three patients died following pneumonia: 1 and 2 on FP/FORM 500/20 μg and 250/10 μg, respectively, while further 2 patients, 1 in each FP/FORM group, died following other lower respiratory tract infections (bronchitis and lower respiratory tract infection).
Discussion
The study did not meet its primary endpoint: there was a non-significant 7% reduction in the rate of moderate/severe exacerbations with FP/FORM (both dose levels) compared with FORM. This was a somewhat surprising result since the components/doses within FP/FORM have proven to be effective in previous COPD studies.2–8,24 However, it is also relevant that other trials of ICS/LABA combinations have failed to show exacerbation risk reduction versus LABA monotherapy.25–29 In the light of earlier unsuccessful ICS/LABA trials, several steps were taken to mitigate such an outcome: patients had severe airways obstruction; documentation confirming an exacerbation within the past 12 months was required for enrolment; “recent” exacerbations did not preclude enrolment other than if occurring during the run-in or at screening; a tiotropium run-in was employed to facilitate retention of as many patients as possible during the pre-randomization phase (<6% of subjects discontinued during the run-in); the study was of 12 months duration, thereby mitigating for seasonal variations in exacerbation rates;30 and the EXACT PRO was used to encourage detection of a greater proportion of unreported exacerbation events, as per the recent FORWARD study.8,31 Furthermore, the pooled exacerbation rate was monitored on an ongoing basis and additional subjects were enrolled via a protocol amendment to preserve study power when it became apparent that the observed overall exacerbation rate was lower than initially predicted.20
The patients enrolled had a mean post-bronchodilator FEV1 of 38% predicted, were symptomatic (mean SGRQ-C score of 54 units) and reported 1.4 moderate/severe exacerbations on average over the previous year. Furthermore, over 60% had ≥2% blood eosinophils at baseline. These characteristics are in keeping with recent ICS/LABA exacerbation studies,8,32–34 albeit airways obstruction was particularly severe in our study. Thus, the population enrolled would have been expected to be prone to exacerbations35 and to differentiate the protective effects of ICS/LABA versus LABA in this regard. Interestingly, however, in the recent TRILOGY trial, exacerbation risk reduction with triple therapy versus ICS/LABA was evident only in patients with severe (≥30%–<50% predicted FEV1), but not very severe airways obstruction (<30% predicted).36 This raises the question as to whether the greater severity of airway obstruction in our study in comparison to several previous ICS/LABA trials3,5,7,8,32 was implicated in the observed lack of effect upon exacerbations. To our knowledge, published subgroup analyses of patients with severe and very severe airways obstruction are not available for previous ICS/LABA trials.
Differential withdrawal rates may also be implicated in the failure to show exacerbation risk reduction with FP/FORM as it may have led to a healthy survivor effect. A greater proportion of patients discontinued prematurely in the FORM arm, with discontinuation occurring sooner in these patients than those discontinuing FP/FORM, particularly in the 500/20 μg group. SGRQ-C scores were almost 10 units higher on average in patients who discontinued prematurely (60.3 units) than in those who completed the study (50.7 units). Furthermore, patients discontinuing early reported overall worsening of SGRQ-C scores during the treatment period, unlike patients who remained in the study whose health status improved on average. The loss of patients with markedly impaired and deteriorating health status, prone to exacerbation35 and doing so recurrently,37 may have reduced the likelihood of demonstrating a treatment effect as reported in other studies.38–40 The TORCH study was illuminating in this regard: placebo-treated patients remaining in the study for ≤6 months experienced 6.8 exacerbations/year compared with 0.9 exacerbations/year in those with over 30 months exposure. Placebo-treated patients with exposures between these two extremes showed a stepwise reduction in exacerbation rates.38
A further contributory factor to the observed results may have been under-reporting of exacerbations, an issue, which is well recognized: in cohorts of trained and regularly reviewed British, Canadian and Chinese patients, 50%–70% of exacerbations have been unreported.41–43 It has been postulated that COPD patients may under-report exacerbations given their familiarity with changing symptom levels and acceptance of their disease.41 As previously mentioned, we sought to reduce exacerbation under-reporting by employing the EXACT PRO to trigger patient–physician interactions. Although Wedzicha et al used the diary in a similar manner in their successful beclomethasone/formoterol trial,8,31 a limitation of this approach is the modest concordance between exacerbations defined on the basis of healthcare utilization (HCU) and EXACT exacerbations: 2 separate studies noted that only a third of HCU events fulfilled EXACT exacerbation criteria.44,45
Our post hoc analyses of moderate/severe exacerbation rates in blood eosinophil subgroups also hint at exacerbation under-reporting. Exacerbation rates in FORM-treated patients did not increase with increasing eosinophil levels. Additionally, the exacerbation RR with FP/FORM 500/20 μg versus FORM was ~1 whether in patients with ≥2%, ≥3% or ≥4% eosinophils. These data are inconsistent with a growing body of recent data. The latter have indicated a tendency to increased exacerbation rates in bronchodilator-treated COPD patients with prior exacerbations as blood eosinophil levels rise; and greater exacerbation risk reduction with ICS/LABA versus bronchodilator treatment with increasing eosinophil counts.33,34,46–48 Although a prospectively designed trial to confirm these observations is yet lacking, a recent editorial noted the consistency of such findings across several trials of different design.46
The above observations may in conjunction have contributed to a particularly low exacerbation rate in the FORM arm in our study (0.87 events/patient/year), which was especially notable given the disease severity of the study population. It is recognized that exacerbation rates have diminished over time in randomized trials similar to our own,2,4,5,7,8,25,29,32,39 which may reflect improvements in patient care.32,36 However, the exacerbation rate in FORM-treated patients in our study was ~20%–30% lower than the corresponding rates on LABA treatment in three similar, recent trials.8,32 Indeed, among all comparable ICS/LABA exacerbation studies, only Calverley et al’s study of beclomethasone/formoterol reported lower exacerbation rates in LABA-treated patients than in the present study.29 By contrast, the exacerbation rate with FP/FORM in our study was very consistent with those recently reported for ICS/LABA treatment in the FORWARD trial8 and replicate fluticasone furoate/vilanterol studies.32
A post hoc analysis of time to CID was undertaken given concerns as to the impact of unreported exacerbations, differential drop-out rates and a healthy survivor effect upon the primary endpoint result. The same definition recently proposed by Singh et al49 was employed. The minimum clinically important difference thresholds for lung function50 and SGRQ-C23 incorporated within this definition are well established, and deterioration of lung function and health status is associated with poorer long-term outcomes in COPD51,52 as are moderate/severe exacerbations,53 the third component of this composite measure. It is thus a coherent endpoint to assess treatment benefit in COPD. A significantly slower time to CID was observed for both FP/FORM dose levels versus FORM. Further studies to validate this composite measure, to establish its capacity to differentiate treatments, and define its relationship to prognosis will be required. An initial post hoc analysis of the ECLIPSE and TORCH trials suggests that CID “positivity” is indeed linked to mortality.54
Pneumonia was reported more frequently with FP/FORM compared with FORM monotherapy (approximately a 2-fold difference), albeit the absolute difference in incidence was small (~1%). These findings, and the incidence of pneumonia with FP/FORM, are consistent with previous reports.55 A strength of our study is that pneumonia was identified in accordance with BTS criteria, including radiographic confirmation wherever feasible. There were 5 deaths following pneumonia or other lower respiratory tract infections in this study (2 on FP/FORM 500/20 μg and 3 on FP/FORM 250/10 μg), and overall a slightly increased number of all cause deaths on combination versus monotherapy. Given these findings, albeit in patient numbers too small to permit definitive conclusions, the findings of a recent, large National Institutes of Health-sponsored review are of interest: Festic and Scanlon reviewed randomized controlled trials (RCTs) including ~15,000 ICS-treated patients and observational studies involving ~50,000 ICS-treated patients. Pneumonia risk was increased 2- to 3-fold in RCTs on ICS versus non-ICS treatments, and to a lesser extent, in observational studies. However, pneumonia-related mortality and total mortality were unchanged on ICS in RCTs and were decreased in the majority of observational studies.56 The apparent contradiction between increased pneumonia risk and unchanged/decreased mortality led the authors to speculate that pneumonia may result from the local immunosuppressive effects of ICS, which may, however, modulate the severity of pneumonia via their anti-inflammatory effects. Two recent independent observational studies provide support for this notion.57,58
As with pneumonia, other adverse events, biochemical changes and acute pharmacodynamic effects showed no evidence of a dose response between the 500/20 μg and 250/10 μg FP/FORM dose levels. Similarly, from an efficacy perspective, and as anticipated, few overt between-dose level differences were seen. Nonetheless, although the study did not confirm the efficacy of FP/FORM, it did suggest the higher FP/FORM dose might be more appropriate in COPD patients, subject to confirmation in future studies. In comparison to FORM, there was an apparent trend in favor of the higher FP/FORM dose in terms of risk reduction in frequent exacerbators, lung function, EXACT exacerbations, SGRQ-C responders and time to CID. Whether these incremental benefits are due to the increased fluticasone propionate or formoterol dose in FP/FORM 500/20 μg versus the 250/10 μg dose cannot be definitively ascertained. However, the significant increases in pre-morning dose FEV1 and FVC with FP/FORM 500/20 μg, but not 250/10 μg, versus FORM may suggest a relevant contribution from its ICS component.
Conclusion
FP/FORM did not reduce exacerbation rates in comparison to FORM in patients with COPD and a history of prior exacerbations. Numerical benefits were, however, observed in favor of FP/FORM 500/20 μg versus FORM for a number of secondary variables including pre- and post-dose lung function, EXACT exacerbations, SGRQ-C, and ER-S total and breathlessness scores. Few efficacy differences were evident between FP/FORM 250/10 μg and FORM. Pneumonia was more frequent in FP/FORM-treated patients, although the absolute difference in incidence was small. Adverse event profiles were otherwise similar between treatments.
Acknowledgments
The authors wish to thank all patients, investigators, sites, contract research organization staff and sponsor staff for their involvement in the study. Flutiform is a registered trade mark of Jagotec AG. Atimos and Modulite are registered trademarks of Chiesi Farmaceutici S.p.A. Spiriva is a registered trademark of Boehringer Ingelheim Pharma GmbH & Co. KG.
The study was funded by Mundipharma Research Limited. Editorial assistance to prepare this manuscript was provided by MD Medical Communications Limited, and was funded by Mundipharma Research Limited.
Author contributions
All authors reviewed this manuscript, take responsibility for the integrity of the data herein, drafting and revising the paper, meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship and have given final approval to the version to be published.
Disclosure
Professor Papi reports grants, personal fees, non-financial support and other from Chiesi, AstraZeneca, GlaxoSmithKline, Boehringer Ingelheim, Merck Sharp & Dohme, Pfizer, Takeda, Mundipharma, TEVA; personal fees and non-financial support from Menarini, Novartis, Zambon; and grants from Sanofi, outside the submitted work. Dr Koroknai is an employee of PAREXEL, the contract research organization contracted to perform the study. Ms McAulay, Dr Dalvi and Dr Overend are employees of Mundipharma Research Limited, Cambridge, UK. Dr Mersmann is an employee of Mundipharma Research GmbH & Co. KG, Germany. Professor Dokic, Dr Tzimas, Dr Mészáros, and Dr Olech-Cudzik have no conflicts of interest to declare. The authors report no other conflicts of interest in this work.
References
Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD); 2017. Available from: http://goldcopd.org. Accessed January 30, 2017. | ||
Szafranski W, Cukier A, Ramirez A, et al. Efficacy and safety of budesonide/formoterol in the management of chronic obstructive pulmonary disease. Eur Respir J. 2003;21(1):74–81. | ||
Rennard SI, Tashkin DP, McElhattan J, et al. Efficacy and tolerability of budesonide/formoterol in one hydrofluoroalkane pressurized metered-dose inhaler in patients with chronic obstructive pulmonary disease: results from a 1-year randomized controlled clinical trial. Drugs. 2009;69(5):549–565. | ||
Sharafkhaneh A, Southard JG, Goldman M, Uryniak T, Martin UJ. Effect of budesonide/formoterol pMDI on COPD exacerbations: a double-blind, randomized study. Respir Med. 2012;106(2):257–268. | ||
Anzueto A, Ferguson GT, Feldman G, et al. Effect of fluticasone propionate/salmeterol (250/50) on COPD exacerbations and impact on patient outcomes. COPD. 2009;6(5):320–329. | ||
Kardos P, Wencker M, Glaab T, Vogelmeier C. Impact of salmeterol/fluticasone propionate versus salmeterol on exacerbations in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175(2):144–149. | ||
Ferguson GT, Anzueto A, Fei R, Emmett A, Knobil K, Kalberg C. Effect of fluticasone propionate/salmeterol (250/50 microg) or salmeterol (50 microg) on COPD exacerbations. Respir Med. 2008;102(8):1099–1108. | ||
Wedzicha JA, Singh D, Vestbo J, et al; FORWARD Investigators. Extrafine beclomethasone/formoterol in severe COPD patients with history of exacerbations. Respir Med. 2014;108(8):1153–1162. | ||
Pascoe SJ, Lipson DA, Locantore N, et al. A phase III randomized controlled trial of single-dose triple therapy in COPD: the IMPACT protocol. Eur Respir J. 2016;48(2):320–330. | ||
Bodzenta-Lukaszyk A, Buhl R, Balint B, Lomax M, Spooner K, Dissanayake S. Fluticasone/formoterol combination therapy versus budesonide/formoterol for the treatment of asthma: A randomized, controlled, non-inferiority trial of efficacy and safety. J Asthma. 2012;49:1060–1070. | ||
Aalbers R, Brusselle G, McIver T, Grothe B, Bodzenta-Lukaszyk A. Onset of bronchodilation with fluticasone/formoterol combination versus fluticasone/salmeterol in an open-label, randomized study. Adv Ther. 2012;29:958–969. | ||
Bodzenta-Lukaszyk A, Dymek A, McAulay K, Mansikka H. Fluticasone/formoterol combination therapy is as effective as fluticasone/salmeterol in the treatment of asthma, but has a more rapid onset of action: An open-label, randomized study. BMC Pulm Med. 2011;11:28. | ||
Bodzenta-Lukaszyk A, Pulka G, Dymek A, et al. Efficacy and safety of fluticasone and formoterol in a single pressurized metered dose inhaler. Respir Med. 2011;105:674–682. | ||
Corren J, Mansfield LE, Pertseva T, Blahzko V, Kaiser K. Efficacy and safety of fluticasone/formoterol combination therapy in patients with moderate-to-severe asthma. Respir Med. 2013;107:180–195. | ||
Nathan RA, D’Urzo A, Blazhko V, Kaiser K. Safety and efficacy of fluticasone/formoterol combination therapy in adolescent and adult patients with mild-to-moderate asthma: a randomized controlled trial. BMC Pulm Med. 2012;12:67. | ||
Bodzenta-Lukaszyk A, van Noord J, Schroder-Babo W, McAulay K, McIver T. Efficacy and safety profile of fluticasone/formoterol combination therapy compared to its individual components administered concurrently in asthma: a randomized controlled trial. Curr Med Res Opin. 2013;29:579–588. | ||
Kaiser K, Pertseva T. Long-term safety and efficacy of fluticasone propionate/formoterol fumarate combination therapy in patients with asthma. Prim Care Respir J. 2013;22:A1–A18. | ||
Mansur AH, Kaiser K. Long-term safety and efficacy of fluticasone/formoterol combination therapy in asthma. J Aerosol Med Pulm Drug Deliv. 2013;26:190–199. | ||
Papi A, Mansur AH, Pertseva T, et al. Long-term fluticasone propionate/formoterol fumarate combination therapy is associated with a low incidence of severe asthma exacerbations. J Aerosol Med Pulm Drug Deliv. 2016;29(4):346–361. | ||
Papi A, Jones PW, Dalvi PS, McAulay K, McIver T, Dissanayake S. The EFFECT trial: evaluating exacerbations, biomarkers, and safety outcomes with two dose levels of fluticasone propionate/formoterol in COPD. Int J Chron Obstruct Pulmon Dis. 2015;10:2431–2438. | ||
EXACT-PRO Initiative. The Exacerbations of Chronic Pulmonary Disease Tool (EXACT) User Manual Version 5. Bethesda: Evidera; 2013. | ||
Singh D, Maleki-Yazdi MR, Tombs L, Iqbal A, Fahy WA, Naya I. Prevention of clinically important deteriorations in COPD with umeclidinium/vilanterol. Int J Chron Obstruct Pulmon Dis. 2016;11:1413–1424. | ||
Jones PW. St George’s Respiratory Questionnaire: MCID. COPD. 2005;2(1):75–79. | ||
Burge PS, Calverley PM, Jones PW, Spencer S, Anderson JA, Maslen TK. Randomized, double blind, placebo controlled study of fluticasone propionate in patients with moderate to severe chronic obstructive pulmonary disease: the ISOLDE trial. BMJ. 2000;320(7245):1297–1303. | ||
Calverley P, Pauwels R, Vestbo J, et al; for the TRISTAN study group. Combined salmeterol and fluticasone in the treatment of chronic obstructive pulmonary disease: a randomized controlled trial. Lancet. 2003;361(9356):449–456. | ||
Hanania NA, Darken P, Horstman D, et al. The efficacy and safety of fluticasone propionate (250 microg)/salmeterol (50 microg) combined in the Diskus inhaler for the treatment of COPD. Chest. 2003;124(3):834–843. | ||
Mahler DA, Wire P, Horstman D, et al. Effectiveness of fluticasone propionate and salmeterol combination delivered via the Diskus device in the treatment of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;166(8):1084–1091. | ||
Tashkin DP, Rennard SI, Martin P, et al. Efficacy and safety of budesonide and formoterol in one pressurized metered-dose inhaler in patients with moderate to very severe chronic obstructive pulmonary disease: results of a 6-month randomized clinical trial. Drugs. 2008;68(14):1975–2000. | ||
Calverley PM, Kuna P, Monsó E, et al. Beclomethasone/formoterol in the management of COPD: a randomized controlled trial. Respir Med. 2010;104(12):1858–1868. | ||
Jenkins CR, Celli B, Anderson JA, et al. Seasonality and determinants of moderate and severe COPD exacerbations in the TORCH study. Eur Respir J. 2012;39(1):38–45. | ||
Singh D, Kampschulte J, Wedzicha JA, et al. A trial of beclomethasone/formoterol in COPD using EXACT-PRO to measure exacerbations. Eur Respir J. 2013;41(1):12–17. | ||
Dransfield MT, Bourbeau J, Jones PW, et al. Once-daily inhaled fluticasone furoate and vilanterol versus vilanterol only for prevention of exacerbations of COPD: two replicate double-blind, parallel-group, randomized controlled trials. Lancet Respir Med. 2013;1(3):210–223. | ||
Pascoe S, Locantore N, Dransfield MT, Barnes NC, Pavord ID. Blood eosinophil counts, exacerbations, and response to the addition of inhaled fluticasone furoate to vilanterol in patients with chronic obstructive pulmonary disease: a secondary analysis of data from two parallel randomized controlled trials. Lancet Respir Med. 2015;3(6):435–442. | ||
Siddiqui SH, Guasconi A, Vestbo J, et al. Blood Eosinophils: A Biomarker of Response to Extrafine Beclomethasone/Formoterol in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2015;192(4):523–525. | ||
Hurst JR, Vestbo J, Anzueto A, et al; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med. 2010;363(12):1128–1138. | ||
Singh D, Papi A, Corradi M, et al. Single inhaler triple therapy versus inhaled corticosteroid plus long-acting β2-agonist therapy for chronic obstructive pulmonary disease (TRILOGY): a double-blind, parallel group, randomized controlled trial. Lancet. 2016;388(10048):963–973. | ||
Spencer S, Jones PW. Time course of recovery of health status following an infective exacerbation of chronic bronchitis. Thorax. 2003;58(7):589–593. | ||
Vestbo J, Anderson JA, Calverley PM, et al. Bias due to withdrawal in long-term randomized trials in COPD: evidence from the TORCH study. Clin Respir J. 2011;5(1):44–49. | ||
Calverley PM, Boonsawat W, Cseke Z, Zhong N, Peterson S, Olsson H. Maintenance therapy with budesonide and formoterol in chronic obstructive pulmonary disease. Eur Respir J. 2003;22(6):912–919. | ||
Wedzicha JA, Calverley PM, Seemungal TA, Hagan G, Ansari Z, Stockley RA; INSPIRE Investigators. The prevention of chronic obstructive pulmonary disease exacerbations by salmeterol/fluticasone propionate or tiotropium bromide. Am J Respir Crit Care Med. 2008;177(1):19–26. | ||
Seemungal TA, Donaldson GC, Paul EA, Bestall JC, Jeffries DJ, Wedzicha JA. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157(5 Pt 1):1418–1422. | ||
Xu W, Collet JP, Shapiro S, et al. Negative impacts of unreported COPD exacerbations on health-related quality of life at 1 year. Eur Respir J. 2010;35(5):1022–1030. | ||
Langsetmo L, Platt RW, Ernst P, Bourbeau J. Underreporting exacerbation of chronic obstructive pulmonary disease in a longitudinal cohort. Am J Respir Crit Care Med. 2008;177(4):396–401. | ||
Jones PW, Lamarca R, Chuecos F, et al. Characterisation and impact of reported and unreported exacerbations: results from ATTAIN. Eur Respir J. 2014;44(5):1156–1165. | ||
Mackay AJ, Donaldson GC, Patel AR, Singh R, Kowlessar B, Wedzicha JA. Detection and severity grading of COPD exacerbations using the exacerbations of chronic pulmonary disease tool (EXACT). Eur Respir J. 2014;43(3):735–744. | ||
Iyer AS, Dransfield MT. Serum eosinophils as a COPD biomarker: ready for prime time? Lancet Respir Med. 2016;4(5):341–343. | ||
Pavord ID, Lettis S, Locantore N, et al. Blood eosinophils and inhaled corticosteroid/long-acting β-2 agonist efficacy in COPD. Thorax. 2016;71(2):118–125. | ||
Watz H, Tetzlaff K, Wouters EF, et al. Blood eosinophil count and exacerbations in severe chronic obstructive pulmonary disease after withdrawal of inhaled corticosteroids: a post-hoc analysis of the WISDOM trial. Lancet Respir Med. 2016;4(5):390–398. | ||
Singh D, Maleki-Yazdi MR, Tombs L, Iqbal A, Fahy WA, Naya I. Prevention of clinically important deteriorations in COPD with umeclidinium/vilanterol. Int J Chron Obstruct Pulmon Dis. 2016;11:1413–1424. | ||
Donohue JF. Minimal clinically important differences in COPD lung function. COPD. 2005;2(1):111–124. | ||
Sin DD, Wu L, Man SF. The relationship between reduced lung function and cardiovascular mortality: a population-based study and a systematic review of the literature. Chest. 2005;127(6):1952–1959. | ||
Wilke S, Jones PW, Mullerova H, et al. One-year change in health status and subsequent outcomes in COPD. Thorax. 2015;70(5):420–425. | ||
Schmidt SA, Johansen MB, Olsen M, et al. The impact of exacerbation frequency on mortality following acute exacerbations of COPD: a registry-based cohort study. BMJ Open. 2014;4(12):e006720. | ||
Naya I, Tombs L, Mullerova H, Compton C, Jones P. Long-term outcome following first clinically important deterioration in COPD. Eur Respir J. 2016;48(Suppl 60):PA304. | ||
Singh D, Corradi M, Spinola M, Petruzzelli S, Papi A. Extrafine beclometasone diproprionate/formoterol fumarate: a review of its effects in chronic obstructive pulmonary disease. NPJ Prim Care Respir Med. 2016;26:16030. | ||
Festic E, Scanlon PD. Incident pneumonia and mortality in patients with chronic obstructive pulmonary disease. A double effect of inhaled corticosteroids? Am J Respir Crit Care Med. 2015;191(2):141–148. | ||
Yamauchi Y, Yasunaga H, Hasegawa W, et al. Effect of outpatient therapy with inhaled corticosteroids on decreasing in-hospital mortality from pneumonia in patients with COPD. Int J Chron Obstruct Pulmon Dis. 2016;11:1403–1411. | ||
Di Martino M, Agabiti N, Cascini S, et al; OUTPUL Study Group. The effect on total mortality of adding inhaled corticosteroids to long-acting bronchodilators for COPD: a real practice analysis in Italy. COPD. 2016;13(3):293–302. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.