Back to Journals » Drug Design, Development and Therapy » Volume 15
Fixed Dose Single Tablet Formulation with Differential Release of Amlodipine Besylate and Simvastatin and Its Pharmacokinetic Profile: QbD and Risk Assessment Approach
Authors Kanwal U ![]() , Mukhtar S, Waheed M, Mehreen A, Abbas N
, Mukhtar S, Waheed M, Mehreen A, Abbas N ![]() , Shamim R
, Shamim R ![]() , Hussain K
, Hussain K ![]() , Rasool F, Hussain A, Bukhari NI
, Rasool F, Hussain A, Bukhari NI ![]()
Received 18 December 2020
Accepted for publication 12 March 2021
Published 24 May 2021 Volume 2021:15 Pages 2193—2210
DOI https://doi.org/10.2147/DDDT.S240506
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Ummarah Kanwal,1 Shahid Mukhtar,1 Muzzamil Waheed,2 Arifa Mehreen,3 Nasir Abbas,1 Rahat Shamim,1 Khalid Hussain,1 Fatima Rasool,1 Amjad Hussain,1 Nadeem Irfan Bukhari1
1Punjab University College of Pharmacy, University of the Punjab, Lahore, Pakistan; 2Government College University, Faisalabad, 38850, Pakistan; 3University of Lahore, Lahore, Pakistan
Correspondence: Nadeem Irfan Bukhari
Punjab University College of Pharmacy, University of the Punjab, Allama Iqbal Campus, Lahore, 54000, Pakistan
Tel +923004259738
Email [email protected]
Purpose: A differential release fixed dose matrix tablet of amlodipine besylate (AML-B) and simvastatin (SIM) was formulated to enhance patient compliance.
Material and Method: In the first phase, release controlling parameters of AML-B and SIM granules were identified and in the second phase a fixed dose AML-B and SIM tablet formulation was prepared and optimized for a differential release of the drugs using a quality by design (QbD) and risk assessment approach. A validated HPLC method was employed for simultaneous determination of AML-B and SIM for FDC formulation. A pharmacokinetics of the above drugs was studied in healthy dogs in the third phase.
Results: In QbD-based optimized formulation, Eudragit® RSPO-dicalcium phosphate (DCP) blend controlled the release of AML-B over 8 h, though this diffusion-controlled release assumed first order kinetics. DCP and Eudragit® RS 100 also retarded release of SIM causing SIM release over 8 h after AML-B release from the optimized FDC tablet formulation. The HPLC retention times of AML-B and SIM were 2.10 and 15.52 min, respectively. Linearity for AML-B was 5.0– 50 ng/mL and 0.01– 2.0 μg/mL for SIM with percent recoveries of 92.85– 101.53% and 94.51– 117.75% for AML-B and SIM. AUC0-∞ of AML-B was increased 3 fold, while AUC0-∞ of SIM was decreased 2 fold. The tmax values for AML-B and SIM were 12 and 6 h, respectively. AML-B was absorbed without any lag time (tlag) while tlag was 6.33 ± 0.81 h for SIM, thus met the study objective.
Conclusion: The pharmacokinetic study showed an immediate absorption of AML-B while that of SIM was withheld for 6 h, close to the desired delay time of 8 h.
Keywords: differential release, Eudragit S100, Eudragit RSPO, DCP-Eudragit blend, quality by design, polymers, amlodipine besylate, simvastatin
Introduction
Hypertension is the major risk factor for cardiovascular diseases (CVDs), which are the leading causes of death globally. Co-occurrence of hypertension and dyslipidemia is high.1 According to the third report of the National Cholesterol Education Program guidelines, the treatment of hypertension requires an aggressive management,2 often warranting the prescription of more than one drug. But prescription of numerous drugs along with some other factors reduces the patient compliance.3 Drug compliance can be improved with fixed dose combination (FDC) as compared to a free drugs' combination regimen.4 Currently, amlodipine besylate (AML-B) and simvastatin (SIM) are often prescribed together to reduce CVD risk by managing blood pressure and lowering low-density lipoprotein cholesterol. Thus, FDC of AML-B and SIM is logical. AML-B inhibits cytochrome P450 3A4 enzyme which metabolizes SIM. Thus, simultaneous administration of AML-B and SIM results in a 30% increase in systemic SIM concentration, which may exacerbate the side effects of SIM, including liver damage and rhabdomyolysis.5 Furthermore, the SIM metabolism starts during the evening,6 thus, a non-concurrent administration of both drugs is preferred.7 Keeping the above in view, this work focused on developing a single tablet-based sustained release delivery system with a differential release of AML-B and SIM with a gap of 8 h. Such a fixed dose combined delivery system for both drugs is expected to enhance patient compliance, minimize potential peak trough fluctuations of SIM in plasma by continuous release of AML-B and 8 h-lagged release of SIM, and the AML-led inhibition of cytochrome P450 3A4. The lower fluctuation of plasma SIM concentrations is also expected to reduce SIM’s side effects and an effective cholesterol reduction during the night, due to its delayed release at pH above 7, ie, in the colon. A prolonged colon transit time of a dosage form enhances the time available for drug absorption. Among several approaches for prolonging the colon transit time,8 use of the pH-dependent polymer approach is preferred.9 Both Eudragit RS, pH-dependent and Eudragit S100, pH-independent polymers are effectively used to control drug release through matrix tablets.10
Devising a sustained release delivery system requires the controlling of several factors, which could be best achieved by a combined quality by design (QbD) and risk assessment approach. Formulation by QbD approach initially estimates the quality target product profile (QTPP), a scheduled summary of the end product’s features required essentially for its desired safety and efficacy. The critical quality attributes (CQAs) of a finished product can also be determined by QTPP. The initial risk assessment estimates possible critical factors that can significantly affect the final product quality. Accordingly, optimization of process parameters and material attributes reduces the quality risk of a finished product. In line with the above, in this study, the formulation variables were systematically investigated in a screening experiment11 to establish the relationships between CQAs (factors) and QTPP (responses) using a simplex design with the help of Design Expert®, version 12.
Materials
AML-B (FYNK Pharmaceuticals, Lahore), SIM (Getz Pharma Karachi, Pakistan), Eudragit® RSPO, Eudragit® RLPO and Eudragit® S-100 (Evonik, Germany), Dibasic calcium phosphate anhydrous (EMCOMPRESS®), Magnesium stearate and talcum (Elite Chemicals Karachi), PVP K-30®, sodium phosphate monobasic and tribasic, sodium dodecyl sulfate (SDF), methanol, acetonitrile (Sigma-Aldrich Co. USA).
Methods
Study Design
The study was conducted in three phases as shown in Figure 1. In the first phase, the individual granule formulations for each of AML-B and SIM were prepared and optimized by using QbD through design of experiment (DOE) through Design Expert®, version 12. In the second phase, the optimized granules of both drugs were physically combined for compression into a matrix tablet formulation which was studied for in-vitro release of the respective drugs. A HPLC-UV based simple, accurate, precise, and robust analytical method was developed and validated for simultaneous determination of AML-B and SIM FDC in the samples derived from in-vitro release media and dog plasma. In the third phase, non-compartmental pharmacokinetics of the fixed dose AML-B and SIM in dogs was studied for its suitability in humans.
 |
Figure 1 Schematics for study design. |
Preparation of AML-B Tablet Formulation
Preliminary Screening for Preparation Method
A screening study was carried out to select an appropriate manufacturing method for tablets of just AML-B from direct compression, water- or isopropyl alcohol (IPA)-based granulation techniques. The AML-B tablets were prepared by direct compression (AB-1), water based wet granulation (AB-2) and IPA-based granulation (AB-3), according to the composition given in Table S1 . For the direct compression, all the materials were mixed for 10 min and compressed by using 8 mm biconcave punches on a rotary compression machine (ZP-19, China). For the water-based wet granulation method we added PVP K-30® aqueous solution to the premixed AML-B and dibasic calcium phosphate (DCP). The resulting wet mass was dried in a hot air oven at 60 °C for 20 min, passed through 18# mesh and dried again at 60 °C for 10 min followed by assessment of moisture content using Sartorius® moisture analyzer. Eudragit® RSPO was added to the dried grains and mixed for 5 min. Finally, magnesium stearate was added followed by mixing for 2 min. The granules were compressed into tablets as above. The IPA-based AML-B granules were prepared by mixing and granulating a half quantity of required Eudragit® RSPO, AML-B and DCP with PVP K-30® dissolved in 35 mL IPA while the remaining quantity of Eudragit RSPO and magnesium stearate was dry mixed with the resultant granules. The granules were dried at 45 °C for 25 min, sieved through mesh #30 and compressed into tablets as above.
The IPA-based granulation method was found appropriate, thus five AML-B tablet formulations, AB-4 to AB-8, were prepared, where the amounts of drug, PVP K-30® and magnesium stearate was fixed as 7, 12 and 2 mg, respectively. Only, the amounts of Eudragit® RSPO and DCP were varied (Table 1). Tablets were compressed as above with a final weight of 200 mg. The DoE analyzing software, Design Expert®, generates a statistically-designed template for different levels of factors. However, entry of the user-defined data is also possible in its historical data option. The cause-and-effect data of AB-4 to AB-8 (Table 8) were entered in the historical (happenstance) node in Design Expert® for finding a composition for the desired AML-B release at different time intervals. The response surface plot (Figure 2a) shows the combined effects of the above parameters on the drug release at different times. The Eudragit® RSPO, 20 mg and of DCP, 159 mg were predicted to be the best for the desired properties of an AML-B tablet. The composition of AB-4 formulation was similar to that predicted by the Design Expert for a formulation meeting the desirability for drug release. Thus, in optimization study, the composition was further maneuvered, using QbD approach around the levels of the above factors (detailed in the proceeding text).
 |
Table 1 Composition of AML-B and SIM Tablets for Pilot Study |
 |
Table 2 Design Matrix for Optimization of Amlodipine Tablet |
 |
Table 3 Quality Target Product Profile and Identification of Critical Quality Attributes |
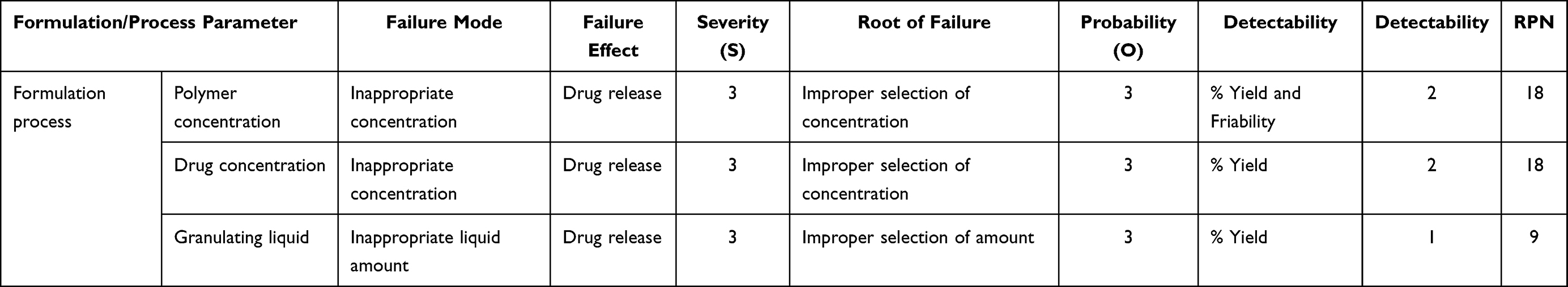 |
Table 4 FMEA Risk Assessment of Fixed Dose Single Tablet Formulation of Amlodipine Besylate and Simvastatin |
 |
Table 5 Composition of Fixed Dose Tablet Formulation of AML-B and SIM |
 |
Table 6 Physical Characteristics of AML-B and SIM Tablets |
 |
Table 7 Physical Characteristics of Pilot Batches of AML-B and Simvastatin (Mean n = 10) |
 |
Table 8 In-vitro Release at Different Time Intervals of AML-B Tablets with R2 Values of Release Models |
 |
Figure 2 Combined effect of Eudragit and DCP on the release of AML-B at different time intervals in: (A) screening study and (B) optimization study. |
Formulation of Amlodipine Besylate by Quality by Design
The simplex lattice design for two factors, Eudragit® RSPO and DCP was employed to generate a matrix for experimentation under the QbD approach after deliberately varied amounts of Eudragit® RSPO and DCP, respectively from 15 to 25 mg and 154 to 164 mg (Table 2). Since a total of the two factors was 179, thus, this value was given as the constraint in the lattice design, comprising 3 simplex, 2 augment and 3 replicate points to fit in the quadratic model. The appropriateness of the model was determined based on the goodness-of-fit statistical criteria. Lack of fit F value should reflect the predictive capability of model, Adeq precision (measure of the signal to noise ratio) should be > 4 and Pred R2 must be in close agreement (difference not more than 0.2) with Adj. R2. AML-B releasedata at 1, 2, 4, 6 and 8 h only in acidic media (0.1 N HCl) were taken as responses. Based on the experimental design, tablet formulations of AML-B were prepared.
Determination of Quality Target Product Profile and Critical Quality Attributes
The quality target product profile (QTPP) begins by determining required dosage form and critical quality attributes (CQAs) of product that should be controlled for quality product as shown in Table 3.12,13
Risk Assessment
Risk assessment was calculated by using the failure mode and effect analysis (FMEA) at the preliminary stage rather than quality checking at the last stage. Risk score matrix was employed to determine a factor's priority and ranked accordingly to probability, severity and detectability. Risk priority number (RPN) was calculated by multiplying three factors and score range was set at 15 and a factor above 15 was taken as high-risk (Table 4).12,14
Preparation of the SIM Tablet Formulation
SIM tablets by wet granulation (S-1) and IPA-based granulation (S-2) were prepared using compositions shown in Table S2. SIM wet granules were prepared and compressed into tablets using the same wet granulation method as employed for the preparation of AML-B, except the following few modifications. DCP was mixed with Eudragit® S-100 and passed the semi-dried material through 30# mesh prior to drying again and no Eudragit® RSPO was added. In the IPA-based granulation (S-2), we mixed SIM, DCP and Eudragit® S-100 using mortar and pestle and granulated with IPA, dried the wet mass at 45 °C for 25 min and compressed into tablets as above. On finding the appropriateness of SIM tablets, formulations, S-3, S-4. S-5 and S-6 were prepared using an IPA-based granulation method. Amount of SIM and magnesium stearate was kept constant as 10 mg and 2 mg, respectively while Eudragit® S-100 and DCP were used in different ratios (Table 3).
Preparation of a Fixed Dose Tablet of AML-B and SIM
The fixed dose tablet formulation was prepared by mixing equal weights of granules AB-4 (of AML-B) and S-3 (of SIM) (Table 5) and compressed into 200 mg per tablet as stated above.
Characterization of Tablets
Solely AML-B formulations AB-1, AB-2 and AB-3 and SIM tablet formulations S-1 and S-2 were evaluated for physical characteristics including thickness, hardness and diameter. While fixed dose tablets were separately evaluated for physical characteristics (thickness, hardness, diameter and friability).
In-Vitro Release Study for AML-B and SIM Tablet Formulations
Drug release from AML-B tablets (AB-4 to AB-8) was studied using USP Type-II apparatus in 500 mL, 0.01 N HCl (pH 1.2) at 37 °C. Samples were drawn at 1, 2, 4, 6 and 8 h and AML-B was determined using UV spectrophotometer (1601 PC Shimadzu, Japan). A granule composition of the AML-B tablet showing release of 25, 40, 55, 65% and not less than 80%, respectively at 1, 2, 4, 6 and 8 h were carried on for further incorporation into a fixed dose tablet. For SIM release from tablet formulations (S-3 to S-6) in acid media, 0.01 N HCl with 0.5% SDF, samples were drawn after 2 h. For assessment of dissolution of drug in basic media, phosphate buffer (pH 7.0), samples were drawn after 2, 4, 6 and 8 h and the respective drugs were determined at 247 nm and 257 nm using UV spectrophotometer (1601 PC Shimadzu, Japan). Formulation with minimal SIM release in acidic media and delayed release in phosphate buffer was selected for development of the FDC tablet.
In-vitro Release Study from FDC of AML-B and SIM
The FDC tablet formulation was evaluated for in-vitro drug release in acid and basic phosphate buffer, pH 6.8 using USP Type II apparatus at 50 rpm. After 2 h of stirring in acid media, the pH of the medium was changed in-situ to 6.86 ± 0.05 by adding 250 mL of 0.146 molar trisodium phosphate in 0.5% SDS. Samples were drawn after 2.5, 5.0 and 8.0 h. Since AML-B and SIM show absorbance at 237 nm, their quantitative analysis of the FDC tablet was carried out using a validated HPLC (Shimadzu Model LC-20AT Japan) method with C18 column coupled diode array detector SPD-M20A. Chromatographic analysis was carried out at ambient temperature (22-25 °C). Isocratic separation of compounds was carried out with mobile phase comprising phosphate buffer, pH 3.5, acetonitrile and methanol (3:5:2). Mobile phase was filtered through 0.45 µm membrane filter (Millipore, Bradford, MA) followed by ultra-sonication for 15 min. The mobile phase was run at a flow rate of 1.0 mL/min and finally 20 µL was injected into the column. Effluent was monitored spectrophotometrically at wavelength 237 nm. FDC tablet formulation was carried forward for pharmacokinetic study.
Pharmacokinetic Study
Animals
For the pharmacokinetics study, dogs of 1.5–5.0 years age, and 30–40 kg body weight were purchased from University of Veterinary and Animal Sciences, Lahore, Pakistan. Dogs were not included in study if they had any disease, recruited for any investigational drug study within 4 weeks of screening. Dogs were excluded from study if found to be allergic to any of the study drugs. The study was approved from the Animal Ethics Committee, Punjab University College of Pharmacy, University of Punjab (Protocol No, AEC/PUCP/1039A-dated 28-5-12). All the experiments in animals in this work were undertaken in accordance with the International Conference on Harmonization ICH guidelines.25
Dose Administration and Blood Sampling
Each dog received FDC differential release single tablet formulation orally with 200 mL of water. A blood sample (3 mL) was collected at pre-scheduled intervals from the cephalic vein and plasma samples were stored at −80 °C until assay. Plasma samples were treated with acetonitrile and methanol to precipitate plasma proteins, vortexed for 1 min, centrifuged at 4000 rpm for 15 min and supernatant was flushed with nitrogen gas. Dried residue was reconstituted with the mobile phase, injected to column and peak areas were recorded.
Pharmacokinetic Data Analysis
A non-compartmental approach was used to compute pharmacokinetic parameters by using Kinetica 4.4.1 software. Pharmacokinetic parameters computed were peak plasma concentration (Cmax), time to Cmax (tmax), (time for first appearance of drug (tlag), total area under the curve (AUC0-∞), extrapolated AUC (AUCt-∞), volume of distribution (Vd), Vd at steady state (Vss), rate of elimination (Kelim), and half-life (t1/2) to correspond to the different pharmacokinetic processes.
Results and Discussion
The optimized fixed dose tablet formulation with continuous release of AML-B and delayed release of SIM, using different polymers was developed. First, the individual tablet dosage forms with desired release characteristics were developed separately for both drugs. Dry compression and granulation methods using different granulating solvents and amounts of internal and external polymers were employed. DCP was used as filler due to potential incompatibility of AML-B with others, such as lactose in the presence of magnesium stearate and water.15
Quality by Design
QTPPs and CQAs for the tablet are listed in Table 3. Risk assessment, to identify various factors affecting drug quality is presented in Table 4. According to ICH Q8 R2 guideline, FMEA was used to identify the potential risk factors, which could affect the tablets (Table 4). The rank for risk quantification whose Risk Point Number (RPN) score was found higher than 15 was evaluated. Then preliminary study was done to select the method for formulation factors for the FDC.
Characteristics of AML-B Tablet Formulations
For AML-B, preliminary study was conducted to find the best granulation technique. AML-B tablets prepared by direct compression technique (AB-1) showed immediate uptake of water, resulting in deformation and transformation to a soft palpable mass, thus further characterization of tablets was not carried out. Granules prepared by wet granulation using water (AB-2) were soft. When sieved through mesh #18, the maximum amount of material passed through with slight hand pressure, as required, dusty fines were at a minimum.16 Moisture content was 2.66% which was slightly above the ideal limit of 1–2%.17 Variation in thickness of tablets was low, probably due to balanced grains to fine ratio, as dry Eudragit® RSPO was added during the final mixing. However, this resulted in a varied hardness from 4.2 to 5.1 (average 4.6) kp. To improve tablet hardness, additional binding impact was required and was achieved by granulating Eudragit® RSPO, AML-B and DCP with PVP K-30®, dissolved in IPA.18 The granules prepared with IPA-dissolved PVP K-30® (AB-3), exhibited an improved binding impact yet the wet mass was difficult to handle, probably due to an additional binding effect of Eudragit® RSPO secondary to its solubility in IPA as Eudragits® also act as binding agents.19 This necessitated using Eudragit® RSPO, to act as an internal binder at granulation stage to improve binding and also incorporating dry Eudragit® RSPO (externally) during the final mixing stage to avoid a sticky thick mass in granulation and allowing a higher ratio of fines in the formulation. Granules were prepared using inter- and intra-granular Eudragit® RSPO 1:1 and were found to be moderately hard, with little fines and having a moisture content of 2.6%. The ratio of fines in granules was improved by adding half Eudragit® RSPO in dry form after granulation and the tablets produced were with the desired characteristics. Physical characteristics of the tablets were satisfactory with improved hardness (Table 6). Variation in tablet thickness was probably due to improper mixing of Eudragit® RSPO with grains due to the difference in size of granules. Hence, to achieve uniform distribution of Eudragit® RSPO with the rest of mass, it was decided to sieve the dried material through 30# mesh instead of 18# mesh in further study. Granulation with IPA gave better hardness as compared to that produced by granulation with water. Thus, IPA-based granulation method was adopted to prepare AML-B tablets.
Characteristics of SIM Tablets
The SIM tablet formulation showing the closest release characteristics to that of delayed release system was selected for incorporation in the FDC formulation and characterization. SIM tablets prepared with wet granulation using water were evaluated for physical characteristics (Table 7) and behavior in 0.01 N HCl and phosphate buffer, pH 7. SIM tablets showed minimal variation in thickness probably due to uniform compression force. However, there was a variation in hardness attributable to the insufficient binding of granules. One tablet was exposed to each 0.01N HCl and phosphate buffer, pH 7 in a petri dish and observed for physical appearance under stagnant condition. The SIM tablet showed signs of penetration of dissolution media as observed by slow erosion of the tablet’s surface within 30 min, a similar behavior in both media. This behavior was contrary to the predicted behavior of Eudragit® S-100 which was resistant to acidic environment and dissolved in basic pH. This reflected a need to granulate material using IPA to improve polymer behavior in acidic and basic media. The physical characteristics of the tablets prepared with IPA-based granulation are given in Table 7 and release behavior in 0.01 N HCl, pH 1.2 and Phosphate buffer, pH 7 in Figure 3. SIM tablets prepared by using IPA as granulating solvent were exposed separately, to 0.01N HCl and phosphate buffer taken in a petri dish and observed for physical appearance under stagnant condition. After 2 h, tablets in 0.01 N HCl remained intact with no sign of erosion (Figure 3A), while that in phosphate buffer, showed an evidence of tablet dissolution (Figure 3B). Tablets were also dislocated from their original place, a clouding ring in Figure 3B indicated. This behavior demonstrated that SIM tablets prepared by the above mentioned process withstood the acidic environment as desired and released drug in the basic pH. This was further investigated by performing an in-vitro release study of SIM tablets in both acidic and basic environments. IPA-based granules were used for preparation of the SIM tablet formulation.
 |
Figure 3 Behavior of simvastatin tablets after 2 h exposure (A) 0.01 N HCl (B) phosphate buffer. |
In the preliminary study, the IPA-based granulation method with inter- and intra-granular polymer at ratio 1:1 was found to produce the better tablet formulation for AML-B. Using granulation with the IPA method, selected composition of AML-B was employed to find range of polymer to be carried out further. For this purpose, amount of polymer was deliberately changed to prepare five formulations. To maintain total weight of tablet, varied amount of polymer was adjusted with quantity of DCP, as bulking agent. Tablets were evaluated for the physical characteristics and in-vitro drug release, given in Tables 7 and 8 (Figure 4).
 |
Figure 4 In-vitro release of tablets at different time intervals (A) AML-B tablets in 0.1 N HCL (B) SIM tablets in 0.1 N HCL + 0.5% sodium dodecyl sulfate and SIM tablets in phosphate buffer containing 0.5% sodium dodecyl sulfate. |
Drug release from all formulations, except formulation AB-4, followed non-Fickian diffusion release mechanism (n>0.5). In formulation AB-4, n<0.5 indicated a Fickian diffusion release mechanism. The slightly higher R2 value suggested that release followed first order kinetics. R2 value of >0.95 for the Higuchi model suggesting that drug release was diffusion based (Table 8).
Tablets demonstrated appropriate hardness (>10 kp) for all five AML-B formulations. In-vitro release profile followed near zero order. AB-4 was with appropriate hardness and demonstrated close to the desired in-vitro release profile, ie, 24.3, 35.3, 49.39, 59.10 and 68.80% after 1, 2, 4, 6 and 8 h, respectively. Range of polymer and DCP around 20 mg (ie, 15–25 mg) and 159 mg (ie, 154–164 mg), respectively were manipulated to achieve the optimized formulation. Formulation AB-4 was further optimized by 8-runs plan (design matrix), through DOE. Based on an experimental design matrix, eight tablet formulations of AML-B were prepared using different ranges of Eudragit® RSPO and DCP while the amount of drug, PVP K-30® and magnesium stearate were fixed. Release profiles of the tablets at different time intervals was measured and given in Table 8.
Release data at different time points were fitted to a quadratic mixture model. Release data of AML-B at 1 h revealed a significant interaction between Eudragit® RSPO (Polymer, A) and DCP (B). All numerical and goodness of fit statistical criteria, indicated appropriateness of the selected model which could capture the effects of the changing levels of Eudragit® RSPO and DCP without noise, despite lesser experimental runs. Only the pred R2 was not close to Adj R2, which might be due to the lesser number of experiments. The equation for AML-B release from tablet formulation at 1 h (Rel_at_1h = +27.41 × A + 28.97 × B + 7.66 A × B) approximated the contribution of Eudragit® RSPO and DCP or their interaction (Eudragit® RSPO × DCP) to its release at 1 h. The equation reflected that Eudragit® RSPO (A), DCP (B) and their interaction (A×B) have a positive effect on release, DCP effect being the most prominent (indicated by value 28.97). These findings were in accordance with a previous study.20 The goodness of fit statistics criteria for release data of AML-B at 2 h were appropriate. The equation of release at 2 h (Rel_at_2h = +37.70 × A + 45.39 × B) indicated a more prominent effect of DCP (B) for 2 h release as compared to Eudragit® RSPO (A). For release, only the Pred R2 was not close to Adj R2 as normally expected, which might be indicative of a possible problem with the model and/or data and could be due to the lesser number of experiments. Nevertheless, other model parameters were appropriate, thus, the selected model could be proceeded as it could be used to navigate design space. Rel_at_4h resulted in the equation, +52.09 × A +72.14 × B indicating that Eudragit® RSPO (A) and DCP (B) positively affected the release, DCP effect being most strong, indicated by value 72.14 as compared to that of Eudragit® RSPO (52.09 for A). The Adeq precision ratio for Rel_at_6h indicated an adequate signal, thus the model was appropriate. The equation, Rel_at_6h = +60.40 × A + 82.32 × B + 61.07 × A × B indicated the positive (but insignificant) effect of Eudragit® RSPO, DCP and their interaction on release, DCP effect being most intense, indicated by value 82.32. Adeq precision ratio for Rel_at_8h indicated the adequate signal, thus the model was appropriate. The equation for Rel_at_8h, + 75.49 × A + 90.15 × B + 49.48 ×A × B showed that all the terms (parameters and their interaction) have a positive effect, though non-significant on release at this time point, DCP effect being most prominent, indicated by the value of 90.15. Individually or in blend, Eudragit and DCP affected the overall properties of granules-based tablet formulation and also modulated the drug release at different time intervals (Table 9). The combined effect of Eudragit and DCP on the AML-B release, as a response surface plot has been given in Figure 2A and B.
 |
Table 9 Release of Different Formulation of AML-B at Specified Time Intervals (Mean, n = 3) |
Since one of the emphases of this study was to find the CQAs, under QbD approach, the DCP was also a focus. DCP possesses good flowability and is used as a bulking agent. It improves the compression properties, and causes an increased erosion rate of the matrix, by reducing the Fickian drug diffusion in a matrix-based uncoated sustained release tablet formulation.21,22 DCP may retard drug release due to its hydrophobic properties.22 However, the DPC’s potential as a binder and release retardant have not been well reported.23 In line with the previous findings,24 DCP exhibited a hybrid functionality as a binder and release retardant. Furthermore, QbD supported, a synergistic interaction of DCP with Eudragit RSPO as binder and release retardant for AML-B, a breakthrough of QbD approach.25 The concentrations of both, in a blend, also critically affected the hardness. All the responses were optimized using desired release characteristics, stated under “desirability” in Table S3.
Figure 5 reflects the most desirable conditions, as shown by blue dot for AML-B release at different time intervals. A flag shows predicted levels of factors and desired release for all time intervals. Based, on the predicted levels of factors, polymer and DCP for optimized AML-B tablet formulations (Table S4), a composition using Eudragit® RSPO 24.0 mg and DCP 155.0 mg, PVP K-30 12 mg and magnesium stearate 2 mg was selected for validation. The hardness of tablets was appropriate and content was 99.76%. The in-vitro release of formulation (Table 10) was comparable to the DoE-predicted values of release at different time intervals with only slight variations in drug release at 8 h.
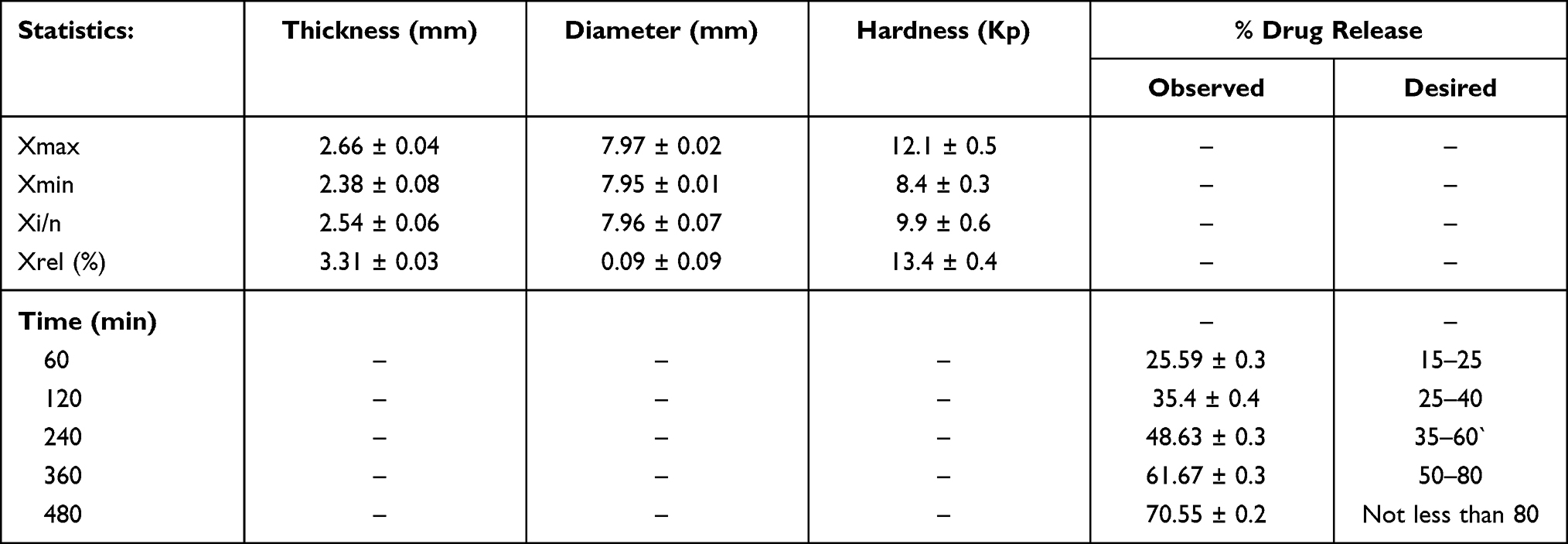 |
Table 10 Physical Characteristics and In-vitro Release of Validation Formulation of AML-B Tablet |
 |
Figure 5 Factor levels adjustment to achieve predicted release at (A) 1 h (B) 2 h (C) 4 h (D) 6 h (E) 8 h. |
Characteristics of SIM Tablet Formulations
For SIM, initially four formulations were prepared with varying concentrations of polymer (Eudragit® S100) and evaluated for physical characteristics and in-vitro drug release. As the amount of polymer in formulation was increased, the hardness increased, probably due to the additional binding impact of Eudragit® S100, when mass was granulated with IPA.19 Table 8 shows release data of tablets in acid and phosphate buffer. Eudragit® S-100, being a pH-sensitive polymer was insoluble at acidic pH, while soluble at basic pH, thus, SIM formulations with a greater amount of polymer exhibited less release in acidic media. Inverse behavior was observed for tablets tested in basic phosphate buffer. Increased polymer concentration in the formulation raised the drug release in phosphate buffer due to higher solubility and dissolution of polymer at alkaline pH, creating a porous matrix structure. These findings were in accordance with the previous observations.26 However, as reported when enteric polymers were only used as binders in pellets, some drug released in the stomach, showing that such simple matrices could not be used to carry drugs to the lower parts of GIT.27 However, due to selectivity in pH, such systems behaved as slow release formulations. As we required SIM formulation exhibiting resistance in acidic media and delayed release at basic pH, formulation, S-3 with 1% polymer was selected for incorporation in FDC. Since the SIM tablet formulation, S-3, met the desired characteristics without optimization study, thus for simplicity, a formal optimization study for SIM was not carried out.
Physical characteristics of fixed dose tablet (200 mg) included thickness (2.36 mm), diameter (7.92 mm) and hardness (7.9 Kp). The assay of the tablet was satisfactory for AML-B, ie, 95.18% and SIM 100.81%. Release profile of SIM tablet showed better release, closer to the desired release profile of delayed release system of AML-B at 2.5 and 5 h and of SIM in basic media at 5 and 8 h. Thus, this formulation was selected for further in-vitro drug release study up to 24 h. The SIM tablets in dissolution media for 24 h, resulted in a slightly better release profile (Figure 6). The comparative release of AML-B from all FDC tablets in the first 2.5 h was as expected, but after changing pH of media, release was slightly reduced, which may be due to the changed amount of bulking agent in combined tablet formulation or alkaline pH of media. SDS in the media was added as a compendial requirement to facilitate SIM release.28 Individual release profiles of FDC tablet in acidic media for AML-B or release of both AML-B and SIM by using a different surfactant, like polysorbate 80 may be sought in future studies. Release of SIM in acidic media without using surfactant can be confirmed in an in-vivo or pharmacokinetic study. Also, in future the SIM can be directly treated with a pH-selective polymer like Eudragit® S-100 and compressed with a zero order or immediate release formulation of AML-B.
 |
Figure 6 (A) Release profile of validated formulation (B) In-vitro drug release of fixed dose tablet of AML-B and SIM. |
Drugs in Unknown Plasma Sample Using HPLC Method
For simultaneous analysis of AML-B and SIM, calibration curves in dog plasma were observed to be linear for AML-B and SIM in concentration ranges 5.0–50 ng/mL and 0.01–2.0 ng/mL, respectively. AML-B and SIM were eluted at retention times, 2.10 min and 15.52 min, respectively. LOQ and LOD of AML-B were 0.10 and 0.03 ng/mL, respectively and the same parameters for SIM were 0.024 and 0.008 ng/mL, respectively. Method was validated in dog plasma to determine AML-B and SIM simultaneously. Recoveries for AML-B and SIM were 93–101% and 94–117%, respectively with RSD < 5% for both drugs (Table 11). Reproducibility and repeatability of method was supported by the results ensuring that it could be applied for pharmacokinetic studies of AML-B and SIM.
 |
Table 11 Percentage Recovery, Intraday and Inter-Day Accuracy, Precision of AML-B and SIM |
Pharmacokinetic Parameters
Pharmacokinetic study of fixed dose tablet was conducted to find whether the aim of formulation was met, ie, continuous release of AML-B and withholding of SIM release up to 8 h. After administration of FDC of AML-B and SIM, the plasma level time curve of AML-B in all six dogs (Figure 6) showed appearance of AML-B at 0.5 h indicating rapid absorption of drug, consistent with the earlier reports.29 In all dogs, AML-B concentration could be noted up to 48 h except in Dog 3 where drug concentration was below LOD at this time interval and thus, taken as zero for calculation of pharmacokinetic parameters. Among all, in Dog 6, the AML-B concentration was consistently higher at all-time intervals, contrarily in Dog 3, drug concentration was lower than all others (Figure 7).
 |
Figure 7 Plasma concentration vs time of FDC AML-B (5 mg) and SIM (10 mg) after a single oral administration in dogs (n=6). |
SIM appeared in dog 1 and dog 3 after 6 h while in the rest of the animals, it was absorbed a bit later, ie, at 8 h. In Dog 1, post dosing at 8 h, showed maximum SIM concentration, ie, 5.22 µg/mL. As compared to all dogs, Dog 1 showed higher SIM concentration at 16–48 h. At 12 h highest drug concentration of 3.5 µg/mL was observed in Dog 4. At the last time interval, ie, 48 h, SIM concentration was observable in Dog 1 and Dog 6, while in Dog 3, Dog 4 and Dog 5 the drug was below the LOD and thus shown as 0 µg/mL.
In the absorption phase, the drug did not incline continuously, yet after reaching a peak, concentration declined, which again was raised to peak concentration, creating what has been termed as a shoulder in plasma level time curve. Shoulders of varying heights could be noted at a time around 3 h in plasma level time profile of AML-B before reaching Cmax in all dogs (Figure 7). This justifies the provision of the plasma profile of AML-B of all dogs (Figure 8). In the present study appearance of plasma level time profile with a shoulder was not unusual, more than one peak in plasma level time curve of AML-B has reported earlier.29 The possible reasons for the multiple peaks phenomena could be due to irregular blood partition of drugs between plasma and blood cells, enterohepatic recycling of drugs, rapid uptake of the drugs in the tissues and then late release from the tissues to blood, and/or the gastric emptying pattern of drugs.30 Nevertheless, maximum peak of AML-B in plasma level time curve was sharp enough to estimate Cmax of AML-B.
 |
Figure 8 Mean plasma concentration vs time of fixed dose combination AML-B (5 mg) and SIM (10 mg) after a single oral administration in dogs (n = 6). |
A double peak phenomenon was clearly observed in plasma level time profile of SIM in all animals except in Dog 6 and Dog 2. Double peaks phenomenon was completely absent in Dog 6 however, though in Dog 2, plasma level time curve did not reveal second peak, yet a small rise in elimination curve could be seen (Figure 8), necessitating showing plasma level time curves of all dogs. In Dog 6, a rise in concentration of SIM might be shifted to some other time interval where drug concentration had not measured. The appearance of multi-peak plasma level time profiles has been hypothesized by several mechanisms. Enterohepatic circulation is one of the well-established causes of multi-peak in plasma level time profile.31 Dosage forms may influence the biphasic release of a drug and thus, may cause two peaks in plasma level time curve. Being a matrix tablet, this could also be the cause of an appearance of a double peak. However, biphasic release was not evident in in-vitro drug dissolution profile of the drug. The regional differences in drug absorption could be a possible reason for the appearance of a double peak of SIM in its plasma level time curve. However, this could not be supported from the available literature.
The appearance of multiple peaks in the plasma concentration time curve of both drugs, complicated the determination of elimination rate constant (Kelim) and, thereby, half-life (t1/2) might not be reliable. For such drugs, a prolonged sampling time is suggested for the precise determination of Kelim.32 In this study, on one hand the samples had been taken for a reasonable length of time, particularly for AML-B and on the other hand, sufficient time points after second peak were available in the terminal portions for the reliable calculation of the above parameters. The adequacy of sampling time intervals could be assessed from the percent contribution of extrapolated area under curve (AUCt-∞) in the total AUC (AUC0-∞). In this study, AUCt-∞ contributed less than 20% in AUC0-∞, thus the number of sample points was sufficient for both drugs. Furthermore, three points in terminal portions are usually used to calculate Kelim. Thus, computation of the above parameters was likely to be reliable.
Pharmacokinetic parameters of AML-B and SIM after oral administration of fixed dose showed in Table 12. AUC0-48 of AML-B was 3-fold higher and Cmax was slightly above the reported Cmax values. AML-B tmax was 12 h, higher as compared to the reported 4 h thus, reflecting a plasma level of a sustained release formulation. The prolonged plasma half-life of AML-B, 13.03 h as compared to 7 h could be attributed to a lower clearance (ClT) of 5.11 L/h. No lag time (tlag) was observed for AML-B and demonstrated its continuous release post administration. An early release and appearance of AML-B is the requirement of antihypertensive therapy for early hypertensive coverage in patients.29,33
 |
Table 12 Pharmacokinetic Parameters of AML-B and SIM After Oral Administration of Fixed Dose |
If an interaction of AML-B had occurred, the concentration of SIM should have increased, secondary to inhibition of metabolism by CYP 3A4, with simultaneous administration of AML-B and SIM.5 However, in this study, the Cmax of SIM was reduced 5-fold compared to the reported Cmax value.34 The current formulation was designed so that it could release AML-B but withhold release of SIM after about 8 h. Thus, reduction in Cmax was likely due to its delayed release till 8 h, though further proof is required to substantiate the hypothesis. The in-vitro release data revealed successful achievement of this aim of dosage form development which was supported by the pharmacokinetic study as well. AML-B appeared within 0.5 h of its administration yet SIM appeared in blood after 8 h in the majority of dogs, post administration of dosage form. Appearance of blood concentration of SIM at 6 h in Dog 1 and 3 was slightly earlier than the expected 8 h. However, it seems that SIM release from dosage form after this time interval will work since SIM metabolism is started in the evening. At 8 h after the single administration of the test formulation, the concentration of SIM was at its peak. At the same time interval, the concentration of AML-B was rising to achieve its maximum at 12 h and ranged from 28.12 ng/mL in Dog 1 to 60.4 ng/mL in Dog 6.
SIM tmax was 8.28 h, delayed from the reported 4 h which was a feature of controlled release formulation. The tlag of SIM was 6.33±0.81 h which was a required objective of the present study. AUC0-∞ of SIM in the current study was reduced 2-fold compared to reported values.34 Immediate release formulations can potentially lead to the saturation of intestinal metabolic enzymes, subsequently leading to an enhanced bioavailability.35,36 SIM is subjected to pre-systemic metabolism mainly by CYP 3A4 located in the enterocytes. It was possible that the intestinal metabolic enzymes became saturated by an instantaneously released high dose of SIM, facilitating the rapid absorption of SIM.35,37 In this study, the pharmacokinetics of drugs was compared to that of the reported values in normal dogs. It would be worth studying the comparative pharmacokinetics after the administration of AML-B and SIM to dogs as separate tablets also.
The Vd for AML-B and SIM was 82.90 L and 2.3 L, respectively. Vss is used to calculate drug amount in body under equilibrium conditions and it is a clearance-independent Vd. Vss is used to compute loading dose and also predict fluctuation of plasma concentrations during a dosage interval for an intermittent dosage regimen. Therefore, larger Vss, shows smaller fluctuations and vice versa. Thus, dose and dosing interval can be appropriately selected for drugs with narrow therapeutic windows to minimize plasma concentration fluctuations. For the Vss of AML-B, the maximum value of 172.30 L was seen in Dog 3 while the lowest, 80.54 L, was noticed in Dog 6. For SIM, Vss was found to be 111.07± 34.76 L. Regardless of the frequency of drug administration, time required to reach steady-state conditions is approximately 4–5 times the terminal half-life after which Vss is achieved.38 Therefore, in the present study, although the dose was administered once, the study drugs, AML-B and SIM, had completed 4 and 7 half-lives, respectively achieving steady state conditions. The Kelim of AML-B was 0.05 h−1 and for SIM, it was found to be 0.12 h−1. The values of plasma half-life of AML-B and SIM were 13.03 h and 7.12 h. respectively. The total body clearance (ClT) of AML-B was 2.97 L/h and that of SIM was 0.27 L/h.
Conclusion
AML-B and SIM in separate tablet formulations, for continuous and delayed release of drug, respectively were successfully developed and optimized. DCP-Eudragit blend played a paramount role in the release of the drugs, showing that DCP, in combination with Eudragit could be manipulated in formulation to control the release of drugs. Differential release of the two drugs from the FDC single tablet was also achieved successfully. AML-B showed continuous release and SIM was withheld for close to the desired time of 8 h after administration of FDC. HPLC method offered simultaneous quantitation of AML-B and SIM in FDC with a shorter run time and high resolution of analytes’ peaks. HPLC method was simple, sensitive and specific for objective drugs, accurate, and precise over a wide range of analytes concentration. Pharmacokinetic parameters, such as area under curve, peak plasma concentration and time to reach peak concentration demonstrated that drugs could be suitable for administration to human after a pharmacokinetic study in humans, however, with this FDC, liver damage and rhabdomyolysis must be evaluated with acute and chronic use of this system. Furthermore, multidose pharmacokinetic studies are necessary to find the accumulation factors for both drugs.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Selby JV, Peng T, Karter AJ, et al. High rates of co-occurrence of hypertension, elevated low-density lipoprotein cholesterol, and diabetes mellitus in a large managed care population. Am J Manag Care. 2004;10(2 Pt 2):163–170.
2. Patra CN, Kumar AB, Pandit HK, Singh SP, Devi MV. Design and evaluation of sustained release bilayer tablets of propranolol hydrochloride. Acta Pharm. 2007;57(4):479–489. doi:10.2478/v10007-007-0038-0
3. Kiortsis DN, Giral P, Bruckert E, Turpin G. Factors associated with low compliance with lipid-lowering drugs in hyperlipidemic patients. J Clin Pharm Ther. 2000;25(6):445–451. doi:10.1046/j.1365-2710.2000.00315.x
4. Bangalore S, Kamalakkannan G, Parkar S, Messerli FH. Fixed-dose combinations improve medication compliance: a meta-analysis. Am J Med. 2007;120(8):713–719. doi:10.1016/j.amjmed.2006.08.033
5. Nishio S, Watanabe H, Kosuge K, Uchida S, Hayashi H, Ohashi K. Interaction between amlodipine and simvastatin in patients with hypercholesterolemia and hypertension. Hypertens Res. 2005;28(3):223–227. doi:10.1291/hypres.28.223
6. Lee JW, Morris JK, Wald NJ. Grapefruit juice and statins. Am J Med. 2016;129(1):26–29. doi:10.1016/j.amjmed.2015.07.036
7. Park CG, Lee H, Choi JW, Lee SJ, Kim SH, Lim HE. Non-concurrent dosing attenuates the pharmacokinetic interaction between amlodipine and simvastatin. Int J Clin Pharmacol Ther. 2010;48(8):497–503. doi:10.5414/CPP48497
8. Sinha V, Kumria R. Colonic drug delivery: prodrug approach. Pharm Res. 2001;18(5):557–564. doi:10.1023/A:1011033121528
9. Leopold CS. Coated dosage forms for colon-specific drug delivery. Pharmaceut Sci Tech Today. 1999;2(5):197–204. doi:10.1016/S1461-5347(99)00151-0
10. Kim C. Advanced Pharmaceutics: Physicochemical Principles. CRC; 2004.
11. Shah HP. Quality by design enabled development and optimization of gastroretentive floating matrix tablets of dipyridamole. Asian J Pharm. 2017;11(02).
12. Rapalli VK, Khosa A, Singhvi G, Girdhar V, Jain R, Dubey SK. Application of QbD principles in nanocarrier-based drug delivery systems. In: Pharmaceutical Quality by Design. Academic Press; 2019:255–296.
13. Theismann E-M, Keppler K, Owen M, Schwarz K, Schlindwein WJP. Modelling the effect of process parameters on the wet extrusion and spheronisation of high-loaded nicotinamide pellets using a quality by design approach. Pharmaceutics. 2019;11(4):154. doi:10.3390/pharmaceutics11040154
14. Nayak AK, Ahmed SA, Beg S, Tabish M, Hasnain MS. Application of Quality by design for the development of biopharmaceuticals. In: Pharmaceutical Quality by Design. Elsevier. 2019:399–411.
15. Abdoh A, Al-Omari M, Badwan A, Jaber A. Amlodipine besylate-excipients interaction in solid dosage form. Pharm Dev Technol. 2004;9(1):15–24. doi:10.1081/PDT-120027414
16. Bandelin FJ. Compressed tablets by wet granulation. Pharma Dosage Forms. 1989;2:131–193.
17. Solanki HK, Basuri T, Thakkar JH, Patel A. Recent advances in granulation technology. Int J Pharm Sci Rev Res. 2010;5(3):48–54.
18. Becker D, Rigassi T, Bauer-Brandl A. Effectiveness of binders in wet granulation: a comparison using model formulations of different tabletability. Drug Dev Ind Pharm. 1997;23(8):791–808. doi:10.3109/03639049709150550
19. Rowe RC, Sheskey PJ, Owen SC, Association AP, Library R. Handbook of Pharmaceutical Excipients. Vol. 4. Pharmaceutical press London; 2003.
20. Lin SY, Cheng CL, Lin PC. Preparation and evaluation of sodium diclofenac controlled-release tablets. Pharm World Sci. 1995;17(2):42–47. doi:10.1007/BF01875053
21. Bharate S, Bharate S, Bajaj A. Interactions and incompatibilities of pharmaceutical excipients with active pharmaceutical ingredients: a comprehensive review. J Excip Food Chem. 2016;1(3):1131.
22. Rao MS, Madhuri K, Gv K. Formulation and evaluation of controlled release floating tablets of lamivudine employing HPMC K4M and sodium alginate.Pharm Sci Res 2013;4(1):396.
23. Rowe R, Sheskey P, Quinn M. Handbook of Pharmaceutical Excipients. Vol. 18.
24. Khan AM, Hanif M, Bukhari NI, et al. Artificial Neural Network (ANN) approach to predict an optimized pH-dependent mesalamine matrix tablet. Drug Des Devel Ther. 2020;14:2434–2449. doi:10.2147/DDDT.S244016
25. Bhoop BSJPT. Quality by Design (QbD) for holistic pharma excellence and regulatory compliance. Pharm Times. 2014;46(8):26–33.
26. Asghar LFA, Chandran S. Design and evaluation of matrices of Eudragit with polycarbophil and carbopol for colon-specific delivery. J Drug Target. 2008;16(10):741–757. doi:10.1080/10611860802473345
27. Marvola M, Nykänen P, Rautio S, Isonen N, Autere AM. Enteric polymers as binders and coating materials in multiple-unit site-specific drug delivery systems. Eur J Pharm Sci. 1999;7(3):259–267. doi:10.1016/S0928-0987(98)00032-3
28. Rockville M.The United States Pharmacopeial Convection.
29. Kwak HH, Kim JO, Chung HK, et al. Pharmacokinetics of oral amlodipine orotate in vagotomized dogs. Biopharm Drug Dispos. 2006;27(3):141–145. doi:10.1002/bdd.495
30. Gao Y, Shao J, Jiang Z, et al. Drug enterohepatic circulation and disposition: constituents of systems pharmacokinetics. Drug Discov Today. 2014;19(3):326–340. doi:10.1016/j.drudis.2013.11.020
31. Ezzet F, Krishna G, Wexler DB, Statkevich P, Kosoglou T, Batra VK. A population pharmacokinetic model that describes multiple peaks due to enterohepatic recirculation of ezetimibe. Clin Ther. 2001;23(6):871–885. doi:10.1016/S0149-2918(01)80075-8
32. Suttle AB, Pollack GM, Brouwer KL. Use of a pharmacokinetic model incorporating discontinuous gastrointestinal absorption to examine the occurrence of double peaks in oral concentration–time profiles. Pharm Res. 1992;9(3):350–356. doi:10.1023/A:1015890918883
33. Wang B, Sheng L, Li Y. Simultaneous determination of telmisartan and amlodipine in dog plasma by LC–MS-MS. J Chromatogr Sci. 2015;53(10):1708–1713. doi:10.1093/chromsci/bmv078
34. Shanmugam S, Ryu J-K, Yoo S-D, Choi H-G, Woo J-S. Evaluation of pharmacokinetics of simvastatin and its pharmacologically active metabolite from controlled-release tablets of simvastatin in rodent and canine animal models. Biomol Ther. 2011;19(2):248–254. doi:10.4062/biomolther.2011.19.2.248
35. Shitara Y, Sugiyama Y. Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors: drug–drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacol Ther. 2006;112(1):71–105. doi:10.1016/j.pharmthera.2006.03.003
36. Sawada T, Sako K, Yoshihara K, Nakamura K, Yokohama S, Hayashi M. Timed‐release formulation to avoid drug–drug interaction between diltiazem and midazolam. J Pharm Sci. 2003;92(4):790–797. doi:10.1002/jps.10336
37. Tubic-Grozdanis M, Hilfinger JM, Amidon GL, et al. Pharmacokinetics of the CYP 3A substrate simvastatin following administration of delayed versus immediate release oral dosage forms. Pharm Res. 2008;25(7):1591–1600. doi:10.1007/s11095-007-9519-6
38. Toutain P-L, Bousquet‐Mélou A. Plasma terminal half‐life. J Vet Pharmacol Ther. 2004;27(6):427–439. doi:10.1111/j.1365-2885.2004.00600.x
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.