")
Back to Journals » The Application of Clinical Genetics » Volume 14
First Two Case Reports of Becker’s Type Myotonia Congenita in Colombia: Clinical and Genetic Features
Authors Olave-Rodriguez JA, Bonilla-Escobar FJ, Candelo E , Rodriguez-Rojas LX
Received 6 June 2021
Accepted for publication 26 October 2021
Published 16 December 2021 Volume 2021:14 Pages 473—479
DOI https://doi.org/10.2147/TACG.S323559
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Jorge Andres Olave-Rodriguez,1 Francisco Javier Bonilla-Escobar,2– 4 Estephania Candelo,5,6 Lisa Ximena Rodriguez-Rojas1,7
1Universidad Icesi, Faculty of Health Sciences, Cali, Colombia; 2Somos Ciencia al Servicio de la Comunidad, Fundación SCISCO/Science to Serve the Community, SCISCO Foundation, Cali, Colombia; 3Universidad del Valle, Cali, Colombia; 4Institute for Clinical Research Education, University of Pittsburgh, Pittsburgh, PA, USA; 5Centro de Investigaciones Clínicas, Fundación Valle del Lili, Cali, Colombia; 6Centro enfermedades raras y malformaciones congenitas (CIACER), Universidad Icesi, Cali, Colombia; 7Human Genetics Department, Fundación Valle del Lili, Cali, Colombia
Correspondence: Lisa Ximena Rodriguez-Rojas Cra. 98 ## 18-49, Cali, Valle del Cauca, Colombia
Email [email protected]; [email protected]
Background: Becker’s type myotonia congenita is an autosomal recessive nondystrophic skeletal muscle disorder characterized by muscle stiffness and the inability of muscle relaxation after voluntary contraction. It is caused by mutations in the CLCN1 gene, which encodes for a chloride channel mainly expressed in the striated muscle. Most cases have been reported in the European population, and only mexiletine has demonstrated a randomized placebo-controlled, double-blinded effectiveness.
Case Presentation: We present two male siblings from Colombia with Latino ancestry, without parental consanguinity, with myotonia during voluntary movements, muscle hypertrophy of lower extremities, transient weakness, and severe muscle fatigue after exercise from three years of age. A genetic panel for dystrophic muscle disorders and a muscle biopsy were both negative. Genetic testing was performed in their second decade of life. Both patients’ exomic sequencing test reported the mutation c.1129C >T (p.Arg377*) affecting exon 10 of the CLCN1, generating a premature stop codon. This mutation was described as pathogenic and observed in only one other patient in the United Kingdom.
Conclusion: To our knowledge, these are the first cases of Becker’s type myotonia congenita reported in Colombia. Increasing awareness of healthcare providers for this type of disease in the region could lead to the identification of undiagnosed patients. Limited availability of medical geneticists as well as genetic testing may be the cause of the lack of previous description of cases, in addition to the delay in the diagnosis of the patients. Further epidemiological studies can reveal underdiagnosed myotonias in the country and in the Latin-American region.
Keywords: Becker type myotonia congenita, myotonia congenita, Colombia, muscular diseases, siblings case reports
Background
Becker’s type Myotonia Congenita (BTMC) (MIM 255700)1 is an autosomal recessive nondystrophic skeletal muscle disorder caused by mutations in the CLCN1 gene.1 Clinically, the disease is characterized by muscle stiffness and the inability to relax after voluntary contraction.2 The CLCN1 gene is located on chromosome 7q34 NC_000007.14 (143316111.143352083) with 23 exons3 and encodes the skeletal muscle chloride channel-1 (CIC-1), mainly expressed in striated muscle.4 This channel is essential for the repolarization of the plasma membrane.5 The protein functions as a homodimer, each monomer has 18 α-helices, 16 of which are transmembrane (Figure 1). During normal muscle physiology, inflow and outflow of ions from muscle cells control contraction and relaxation. In myotonia congenita, too few chloride ions enter the cell, causing hyper excitation of the cell membrane and a prolonged action potential in striated myocytes. This action potential generates a sustained contraction with inability to relax.2
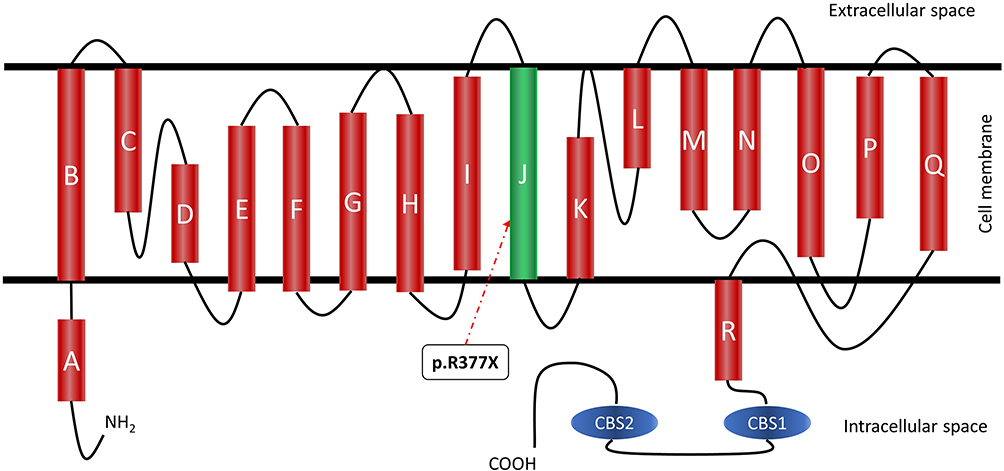 |
Figure 1 Structure of ClC-1 and localization of the mutation (p.R377X) in two Colombian siblings. |
Other symptoms that can be present in BTMC include transient weakness, generalized muscular hypertrophy, and depressed deep tendon reflexes. The reported incidence of myotonia congenita disorders (including Thomsen’s type of autosomal dominant inheritance, MIM: 160800)6 is between 0.3 and 0.6 per 100,000 people in the general population.7
Approximately 290 different mutations are related with myotonia congenita, affecting the chloride channel in different molecular ways from the voltage sensor affecting the fast and slow of the chloride channel, channel conductance, disturbing the ion selectivity and also changing the channel expression by altering the channel cell trafficking2,7 Most cases of BTMC are reported in the European population,8–10 with a higher prevalence in Nordic countries.1,11 There are a few reports of the disease in other populations, limited to Tanzania,12 Costa Rica,13 Morocco,14 and Brazil.15 To our knowledge, there are no reports of BTMC in Colombian individuals.
Several pharmacological approaches have been described and used for congenital myotonia but only mexiletine has demonstrated a randomized placebo-controlled, double-blinded effectiveness, which is a class 1b antiarrhythmic medication with high affinity for muscle sodium channels with previous pre-clinical studies showing a reduction in the muscle fiber excitability produced by the non-dystrophic myotonias (NMD) mutations.14 In the most recent pre-clinical studies, ranolazine has been shown to reduce the clinical symptoms of myotonia on animal model and with a possible superiority compared with mexiletine.15 Based on these findings, a pilot study was developed to demonstrate the effects of ranolazine on signs and symptoms of muscle stiffness and myotonia in MC, showing that this medication might be an alternative for mexiletine in the 25% of the cases with have not demonstrated effectiveness.15 However, blinding and placebo control studies on ranolazine should be performed, as well as comparative studies.
We report two Colombian siblings, of Colombian parents without consanguinity, who were experiencing muscle stiffness and weakness since the age of three years. Multiple studies were inconclusive, including multiple electromyograms, muscle biopsies, and negative results in the study of the myotonic dystrophy protein kinase (DMPK, for myotonic dystrophy type 1, DM1) gene. They were diagnosed in their second decade of life with BTMC which was confirmed genetically. Informed consent was obtained from both patients to publish these cases.
Case Presentation
These two cases are siblings that presented to the Genetics service at the ages of 25 and 24 years old, both with symptoms of proximal muscle weakness, stiffness, and inability to drop objects after they were grabbed. At physical exam, the patients were also found to have myotonia during voluntary movements, muscle hypertrophy of the lower extremities, transient muscle weakness, and severe muscle fatigue after exercise (see Table 1).
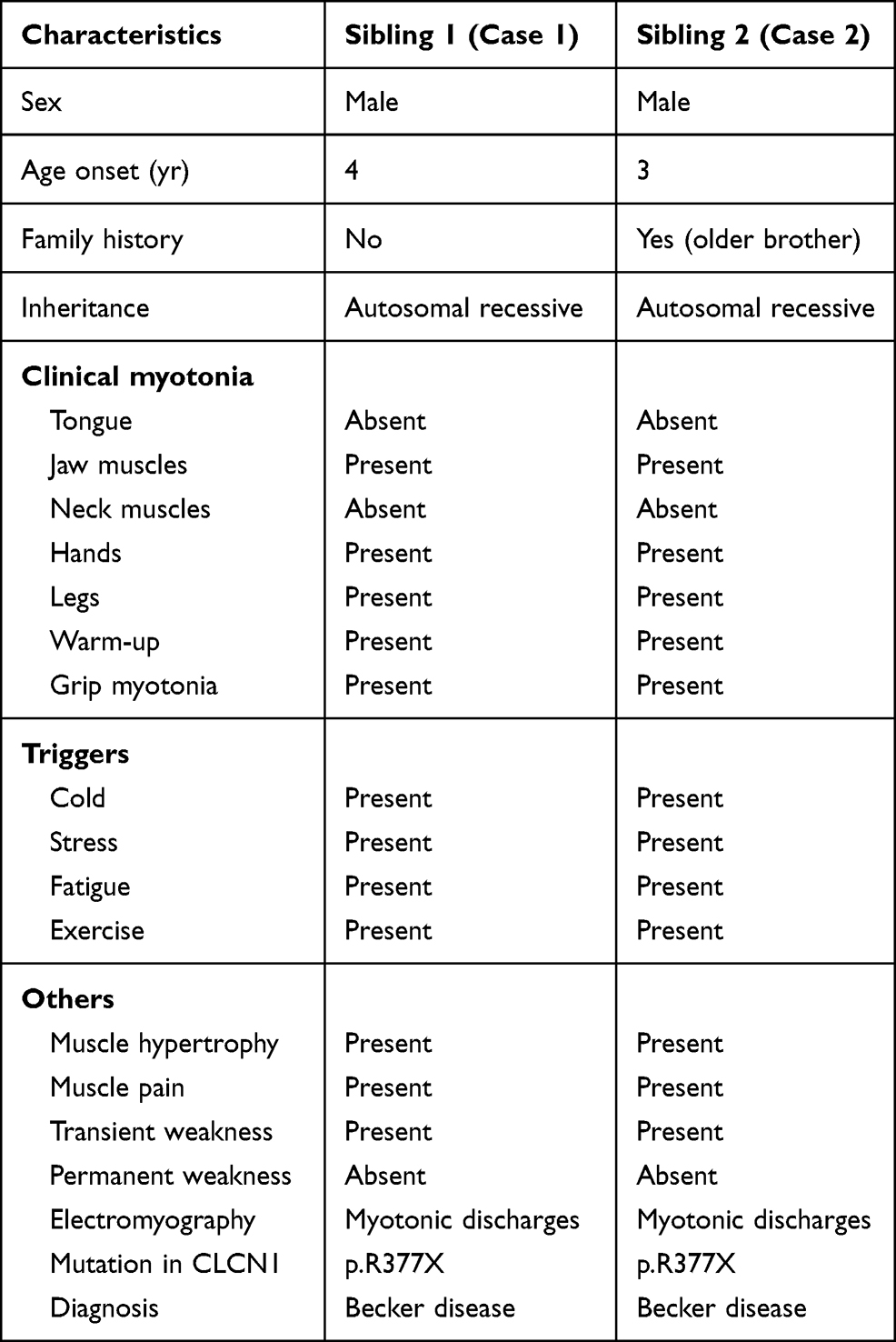 |
Table 1 Clinical Features of Two Patients with the CLCN1 Mutation |
Both patients reported onset of symptoms in infancy, having frequent falls and physical developmental delays at the age of onset for standing up and walking. They were initially managed by an orthopedist with orthotic insoles without success. They refer to the fact that the symptoms have not changed over time.
Case 1
Frequent falls and difficulty walking appeared in the older brother at four years of age. When he was eight years old, he had a physical examination describing calf muscle hypertrophy (gastrocnemius) without Gowers sign, decrease in distal force with thenar atrophy, and inability to perform the walking on heels test. He did not have deficits in relaxation after repetitive muscle contraction, and the walking on toes test was unremarkable.
The patient was born from a G1P1 mother, both parents originating from Cali, Colombia, without consanguinity (Figure 2), with a normal pregnancy and a normal vaginal delivery. The parents deny sharing any relatives in their family tree.
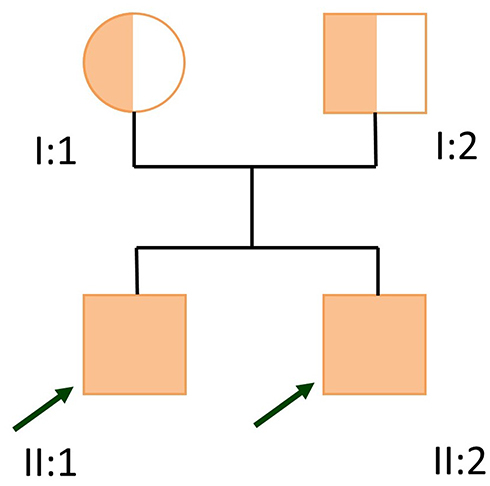 |
Figure 2 Familial pedigree. |
The physical exam was unremarkable, except for decreased strength of upper and lower extremities measured as 3 out of 5 and myotonia in hands with relaxation occurring 10 seconds after squeezing an object with the hands. There were no deformities. A muscle biopsy showed moderately atrophic and angulated fibers, some with internalization of the nucleus and grouping of fibers by reinnervation. It was interpreted as infantile spinal muscular atrophy vs chronic neuropathy. Molecular analysis of the DMPK gene (DM1) was negative. He also had electromyograms at seven, ten, and twelve years of age with findings describing a myopathic pattern.
At the age of twenty-one, with differential diagnoses of progressive spinal muscular atrophy in study vs congenital familial myopathy, and a negative DMPK (DM1), an additional muscle biopsy was performed as well as a test for myotonic dystrophy type 2 (DM2, proximal myotonic myopathy [PROMM]) to evaluate the damage of type 2 muscle fibers.
In the biopsy, there was no suggestion from the morphology of the fibers of a myotonic dystrophy. There were few fibers with variability in their size, and round and occasional fibers of hypercontracted aspect, without the usual dysmorphic pattern. The muscle was described as well preserved for the age of the patient given the chronicity of the case. There was no increase in the endomysial collagen. No findings were suggestive of congenital myopathies or denervation pattern. There was an absence of expression for cytochrome oxidase in occasional fibers. Immunohistochemistry of fast myosin revealed type 1 and 2 fibers with predominance of type 2 fibers. Human leukocyte antigen class I (HLA-I) was not expressed in the muscle fibers. Finally, in the high-resolution and ultrastructure optical microscopy, the sarcomeres were normal, with normal mitochondria. There was no accumulation of lipidic material or other signs of a deposit disease. The cores were normal.
The diagnosis was of a variation in the size of muscle fibers without ruling out dystrophies or inflammatory myopathies. A myotonic dystrophy was ruled out. The treating physician recommended swimming once a week for one and a half hours, practicing yoga, and stretching at home. The patient reported a mild improvement in his symptoms with physical therapy. The patient was seen by cardiology, neurology, ophthalmology, and pneumology with unremarkable findings.
A geneticist ordered an exome sequencing, which reported a mutation in the exon 10 of the gene CLCN1: c.1129c>T (p.Arg377) producing a premature stop codon and was classified as pathogenic. With this result, the patient was diagnosed with autosomal recessive myotonia congenita (Becker myotonia). Phenytoin was prescribed to treat the myotonic symptoms, but was withdrawn shortly because there was no improvement and adverse effects were reported (skin rash and excessive daytime sleepiness).
Case 2
Frequent falls and difficulty walking appeared at three years of age with a similar presentation to the older brother. He was the second son of the same parents, with a normal pregnancy and vaginal delivery.
Physical exam revealed long hands and fingers, myotonic response to movements, hypertrophy of muscles more evident in pectorals and biceps, 3 out of 5 strengths in the upper and lower extremities, and myotonia in hands with relaxation occurring 10 seconds after squeezing an object. There were no deformities. Patient history revealed that the patient does not exercise on a regular basis and stretches at home to counter the symptoms.
At seventeen years of age, an echocardiogram was unremarkable. Two years later, a left ventricular ejection fraction of 65% (normal limits) was reported, and a mild tricuspid insufficiency with similar findings was reported at twenty-one years of age. A control echocardiogram was indicated at an interval of every two years.
Various tests were performed, including a genetic panel for dystrophic muscle disorders (which resulted negative) and a muscle biopsy that did not show signs of a dystrophic muscle disorder. A test for the known mutation of the older brother (mutation c.1129C>T [*p.Arg377*]) was carried out and resulted positive.
Follow-Up and Recommendations for Both Cases
Both patients were genetically counseled about their diagnosis. Counseling included information about the disease (autosomal recessive, orphan disease without treatment, and possibly progressive thus requiring follow-up) and the risks of their descendants. The patients’ siblings will be carriers of the mutation without the disease, or they could have the autosomal dominant form of the disease (Thomsen’s type congenital myotonia).6,16 If the partner of the patient is a carrier of the mutation, their siblings will have a 50% chance of having BTMC and 50% of being healthy carriers or with Thomsen’s type.
Further recommendations were given about anesthetic medications with reported adverse effects in this type of disease such as muscle relaxants, depolarizing drugs, suxamethonium, selective beta-adrenergic agonists, adrenaline, propranolol, fenoterol, and colchicine, among others.
Discussion and Conclusions
These two Colombian young-adult brothers presented with symptoms of proximal muscle weakness, stiffness, inability to drop objects after they were grabbed, myotonia during voluntary movements, lower extremities muscle hypertrophy, transient muscle weakness, severe muscle fatigue, and a history of physical developmental delays (standing up and walking) and frequent falls. At physical exam, the older brother (Case 1) had decreased strength of upper and lower extremities (3/5) and myotonic response to movements like the younger brother (Case 2), who was additionally found to have long hands and fingers and hypertrophy of pectorals and biceps. Only after multiple negative testing that included electromyograms, muscle biopsies, and the analysis of the gene DMPK for DM1, the patients were referred to genetics. Genetic testing reported the same mutation in its homozygous form in both brothers: c.1129C>T (p.Arg377*) in exon 10 of CLCN1 gene, generating a premature stop codon for a diagnosis of BTMC.
A very low total frequency (0.00001414) of the mutation in gnomAD v.2.1.1 has been reported, with additional information from African population (0.0001201) and from European Finns (0.00003980).18 It has not been described before in Latino, Ashkenazi Jews, East Asian, European not Finns, or South-Asian populations. The closest case reported in the literature is in an American male adult diagnosed at 22 years old with a positive familial history, consanguinity and a variant affecting the NM_000083.2 transcript,17 also the Indian population have been described with an occurrence of 8 over 10,012 unrelated population affected with neurological disorders using TrueSight one panel. However, specific data of the localization of the variants are not described.18 The affected amino acid in our patients is in a highly conserved position (GERP 5.11).11 There is a similar report to our knowledge in the literature of a female patient with a similar mutation in the United Kingdom with age of onset of six years, site of onset of the legs with muscle pain, generalized hypertrophy and severe proximal weakness.19 To our knowledge, this is the first report describing two siblings from nonconsanguineous parents with BTMC and the first description of the disease in Colombian patients with Colombian/Latino ancestry.
The effects of the mutations in the CLC-1 channel protein have been described as gating defects. However, an in-vitro study using the mutation of A531V, a myotonic mutation, showed that there were no defects in gating in the channel but instead a disruption in the synthesis/degradation balance of the protein.20 It has been described how the type of mutation and its location differently affects the compound muscle action potential (CMAP). In dominant mutations, the CMAP is decreased.21 Further research is needed to understand the in-vivo effect of the mutation on the action potential. Additionally, the variant provokes an early termination of the protein in the amino at the 337 positions when the protein normally finishes at 989, which generates a loss on the transmembrane, extracellular and cytoplasmic domains (Figure 3). The losses correspond to the cystathionine-beta-synthase (CBS1 and CBS2), which are evolutionarily conserved protein domains that usually present in tandem. Several disorders are caused by mutations in the CBS, in this case affecting channel gating. Despite their clinical relevance, it remains to be established what the precise function of CBS domains is and how they affect the structural and/or functional properties of an enzyme, kinase, or channel.22 Depending on the protein in which they occur, CBS domains have been proposed to affect multimerization and sorting of proteins, channel gating, and ligand binding. However, recent experiments revealing that CBS domains can bind adenosine-containing ligands such ATP, AMP, or S-adenosylmethionine have led to the hypothesis that CBS domains function as sensors of intracellular metabolites.19
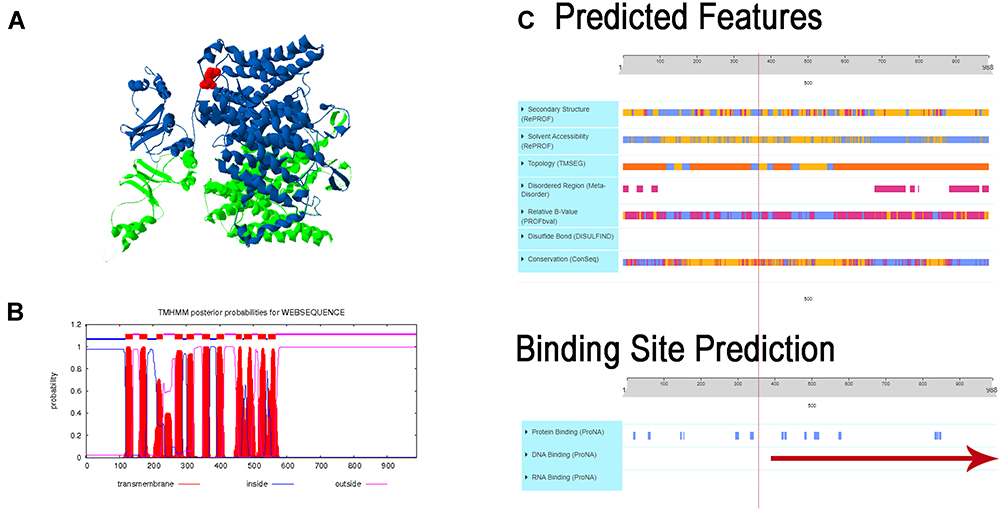 |
Figure 3 Effect of the Arg377* de novo mutation in the CLCN1 protein. (A) Figure shows the tridimensional structure of the CLCN1 protein chain A is in green and chain B in blue in the red dock is marked the c.1129C>T (p.Arg377deletion) CLCN1. (B) Graph B shows the graphical output of the Transmembrane Topology and Signal Peptide Prediction Using Dynamic Bayesian Networks (TMHMM) posterior probabilities sp P35523 CLCN1_Human, demonstrating the posterior probabilities for transmembrane (red), inside (blue), and outside (pink). In the case of the Chloride Voltage-Gated Channel 1 (CLCN1) six transmembrane domains are affected, three and two inside interactions, respectively are disrupted. (C) Predicted features the protein, which are disrupted for the early termination of the protein. The figure shows the predicted changes in the secondary structure, topology, DNA and RNA binding, and the binding site predicted disruption. The red line represents the initiation of the disruption in the protein done by the variant. |
In these patients, the imaging studies or biopsies were not conclusive for a diagnosis. A study analyzing muscle magnetic resonance imaging of patients with BTMC found that there are no skeletal muscle morphologic changes, and the muscle alterations (hypertrophy) are the result of the disease process.23 The time to diagnose and the amount of testing carried out in the patients’ course could be considered excessive. It is important to acknowledge that this is the first time that this disease has been described in Colombia and therefore it was a challenging diagnosis. An early genetic test could expedite the diagnosis and prevent unnecessary testing. This could also prevent exercise recommendations due to their potential to cause exercise-induced damage.24
There are concerns for anesthetic management of patients with BTMC. In a case report in Japan, the authors described the use of non-depolarizing muscle relaxants in a female patient who underwent a right lower lobectomy assisted with thoracoscopy for lung cancer without any complications.25 The patient was given recommendations about anesthetic medications and advised to mention the history of BTMC in case of surgery.
There are medications (sodium channel blockers, mexiletine, phenytoin, carbamazepine) that could improve myotonia, but are indicated only in conditions of low tolerance to the disease and are managed in Colombia by neurologist.26
In conclusion, to the best of our knowledge, these are the first cases of Becker’s type Myotonia Congenita reported in Colombia. Increasing awareness among healthcare providers about this type of disease in Latin-America and specifically in Colombia could lead to the identification of undiagnosed patients. This is relevant given the novelty of the cases and the challenges towards a diagnosis, which include the limited availability of medical geneticists and genetic testing. Further epidemiological studies can reveal underdiagnosed myotonias in Colombia and the Latin-American region.
Abbreviations
BTMC, Becker’s type Myotonia Congenita; DMPK, myotonic dystrophy protein kinase; DM1, myotonic dystrophy type 1; CIC-1, skeletal muscle chloride channel-1; DM2, myotonic dystrophy type 2; PROMM, proximal myotonic myopathy; HLA-I, human leukocyte antigen class I; CBS, cystathionine-beta-synthase; CMAP, compound muscle action potential.
Data Sharing Statement
All data generated or analysed during this study are included in this published article.
Ethics Approval and Consent to Participate
The study was assessed and supported by the biomedical ethics committee at Fundación Valle del Lili. All patients included in this descriptive study filled out the written informed consent. Additionally, written consent to publish this information was obtained from study participants.
Consent for Publication
All patients whose clinical or molecular data are used in this manuscript gave publication consent during their informed consent completion. Written consent for publication was obtained from the study participants.
Acknowledgments
We thank our patients for participating in the study.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval for the version to be published; and agreed to be accountable for all aspects of the work.
Funding
No funding of any kind or funding source was provided during the implementation and execution of this study.
Disclosure
The authors declare that they have no competing interests.
References
1. Sun C, Tranebjaerg L, Torbergsen T, Holmgren G, Van Ghelue M. Spectrum of CLCN1 mutations in patients with myotonia congenita in Northern Scandinavia. Eur J Hum Genet. 2001;9(12):903–909. doi:10.1038/sj.ejhg.5200736
2. Tincheva S, Georgieva B, Todorov T, et al. Myotonia congenita type Becker in Bulgaria: first genetically proven cases and mutation screening of two presumable endemic regions. Neuromuscul Disord. 2016;26(10):675–680. doi:10.1016/j.nmd.2016.08.001
3. Lorenz C, Meyer-Kleine C, Steinmeyer K, Koch MC, Jentsch TJ. Genomic organization of the human muscle chloride channel CIC-1 and analysis of novel mutations leading to Becker-type myotonia. Hum Mol Genet. 1994;3(6):941–946. doi:10.1093/hmg/3.6.941
4. Hudson AJ, Ebers GC, Bulman DE. The skeletal muscle sodium and chloride channel diseases. Brain. 1995;118(Pt 2)):547–563. doi:10.1093/brain/118.2.547
5. Brugnoni R, Kapetis D, Imbrici P, et al. A large cohort of myotonia congenita probands: novel mutations and a high-frequency mutation region in exons 4 and 5 of the CLCN1 gene. J Hum Genet. 2013;58(9):581–587. doi:10.1038/jhg.2013.58
6. Koch M, Harley H, Sarfarazi M, et al. Myotonia congenita (Thomsen’s disease) excluded from the region of the myotonic dystrophy locus on chromosome 19. Hum Genet. 1989;82(2):163–166. doi:10.1007/BF00284051
7. Gutmann L, Phillips LH. Myotonia congenita. Semin Neurol. 1991;11(3):244–248. doi:10.1055/s-2008-1041228
8. de Diego C, Gámez J, Plassart-Schiess E, et al. Novel mutations in the muscle chloride channel CLCN1 gene causing myotonia congenita in Spanish families. J Neurol. 1999;246(9):825–829. doi:10.1007/s004150050462
9. Cassone M, Ferradini V, Longo G, et al. Genotype-phenotype correlation of F484L mutation in three Italian families with Thomsen myotonia. Muscle Nerve. 2017;55(6):E24–5. doi:10.1002/mus.25407
10. Lefter S, Hardiman O, Ryan AM. A population-based epidemiologic study of adult neuromuscular disease in the Republic of Ireland. Neurology. 2017;88(3):304–313. doi:10.1212/WNL.0000000000003504
11. Huber CD, Kim BY, Lohmueller KE. Population genetic models of GERP scores suggest pervasive turnover of constrained sites across mammalian evolution. PLoS Genet. 2020;16(5):e1008827. doi:10.1371/journal.pgen.1008827
12. Dekker MCJ, Tieleman AA, Igogo OJ, van Duyvenvoorde HA, Howlett WP, Hamel BC. First familial Becker muscular dystrophy in Tanzania: clinical and genetic features. Neuromuscular Disorders. 2019;29(4):317–320. doi:10.1016/j.nmd.2019.01.006
13. Morales F, Cuenca P, Del Valle G, et al. Clinical and molecular diagnosis of a Costa Rican family with autosomal recessive myotonia congenita (Becker disease) carrying a new mutation in the CLCN1 gene. Rev Biol Trop. 2008;56(1):1–11. doi:10.15517/rbt.v56i1.5505
14. Ratbi I, Elalaoui SC, Escudero A, Kriouile Y, Molano J, Sefiani A. Moroccan consanguineous family with Becker myotonia and review. Ann Indian Acad Neurol. 2011;14(4):307–309. doi:10.4103/0972-2327.91963
15. Azevedo HC, Mendonça LI, Salum PN, et al. Congenital myotonia. Report of 7 patients. Arq Neuropsiquiatr. 1996;54(4):595–600. doi:10.1590/S0004-282X1996000400008
16. Dunø M, Myotonia Congenita VJ, Adam MP, et al. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 1993. Available from http://www.ncbi.nlm.nih.gov/books/NBK1355/.
17. Trujillano D, Bertoli-Avella AM, Kumar Kandaswamy K, et al. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur J Hum Genet. 2017;25(2):176–182. doi:10.1038/ejhg.2016.146
18. Ganapathy A, Mishra A, Soni MR, et al. Multi-gene testing in neurological disorders showed an improved diagnostic yield: data from over 1000 Indian patients. J Neurol. 2019;266(8):1919–1926. doi:10.1007/s00415-019-09358-1
19. Fialho D, Schorge S, Pucovska U, et al. Chloride channel myotonia: exon 8 hot-spot for dominant-negative interactions. Brain. 2007;130(Pt 12):3265–3274. doi:10.1093/brain/awm248
20. Lee -T-T, Zhang X-D, Chuang -C-C, et al. Myotonia Congenita Mutation Enhances the Degradation of Human CLC-1 Chloride Channels. PLoS One. 2013;8(2):e55930. doi:10.1371/journal.pone.0055930
21. Colding-Jørgensen E, Dunø M, Schwartz M, Vissing J. Decrement of compound muscle action potential is related to mutation type in myotonia congenita. Muscle Nerve. 2003;27(4):449–455. doi:10.1002/mus.10347
22. Ignoul S, Eggermont J. CBS domains: structure, function, and pathology in human proteins. Am J Physiol Cell Physiol. 2005;289(6):C1369–1378. doi:10.1152/ajpcell.00282.2005
23. Kornblum C, Lutterbey GG, Czermin B, et al. Whole-body high-field MRI shows no skeletal muscle degeneration in young patients with recessive myotonia congenita. Acta Neurol Scand. 2010;121(2):131–135. doi:10.1111/j.1600-0404.2009.01228.x
24. Andersen SP, Sveen M-L, Hansen RS, et al. Creatine kinase response to high-intensity aerobic exercise in adult-onset muscular dystrophy. Muscle Nerve. 2013;48(6):897–901. doi:10.1002/mus.23846
25. Hayashida S, Yanagi F, Tashiro M, Terasaki H. [Anesthetic managements of a patient with congenital myotonia (Becker type)]. Masui. 2004;53(11):1293–1296. Japanese.
26. Stunnenberg BC, LoRusso S, Arnold WD, et al. Guidelines on clinical presentation and management of nondystrophic myotonias. Muscle Nerve. 2020;62(4):430–444. doi:10.1002/mus.26887
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.