Back to Journals » Infection and Drug Resistance » Volume 13
First Full-Length Genome Sequence of Dengue Virus Serotype 2 Circulating in Vietnam in 2017
Authors Dang TT ![]() , Pham MH, Bui HV, Le DV
, Pham MH, Bui HV, Le DV ![]()
Received 27 August 2020
Accepted for publication 15 October 2020
Published 10 November 2020 Volume 2020:13 Pages 4061—4068
DOI https://doi.org/10.2147/IDR.S275645
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Suresh Antony
Thuy Thi Dang,1 My Ha Pham,2 Huy Vu Bui,1,3 Duyet Van Le1
1National Hospital for Tropical Diseases, Hanoi, Vietnam; 2Oxford University Clinical Research Unit, Hanoi, Vietnam; 3Hanoi Medical University, Hanoi, Vietnam
Correspondence: Duyet Van Le Tel +84 936647979
Email [email protected]
Introduction: Dengue hemorrhagic fever is caused by four serotypes of dengue viruses transmitted by mosquitoes. In Vietnam, dengue outbreaks occur every year, and all four serotypes have been found circulating with the dominant one varying over time. However, in 2017 an unusual dengue fever outbreak occurred in the North of Vietnam, predominantly caused by DENV1 (92%) and DENV2 (7.3%). The objective of the present study was to obtain and characterize the full-length genome sequence of seven DENV2 strains in 2017 epidemic.
Materials and Methods: Whole-genome sequencing of seven DENV2 isolates from the 2017 outbreak were obtained using the Illumina MiSeq next generation sequencer system. Complete genome sequences were then analyzed to find out genetic variants and genetic relationships between these DENV2 with other strains that circulated in Vietnam previously and other regions of the world.
Results: The complete genome sequence of seven DENV2 isolates in the 2017 dengue outbreak comprised 10,696 nucleotides with an open reading frame coding for 3392 amino acids. The genome analysis showed only a small number of amino acid changes which were obtained in all genes, in which a few amino acids substitutions were distributed over the positions such as G156 (NS1), V106 (NS2A), and L258/T260 (NS5). The phylogenetic analysis revealed that the DENV2 isolates in the 2017 outbreak were most closely related to the dengue virus from India in 2006, suggesting that the causative virus originated from the DENV2 that caused dengue hemorrhagic fever in 2006 in India.
Conclusion: The first complete genome sequences of seven DENV2 isolates in the 2017 dengue outbreak in Northern Vietnam were successfully obtained. The genetic and phylogenetic data indicated that these DENV2 isolates were not causative virus circulating in Vietnam previously but originated from India in 2006. These data are emerging and providing valuable information for the management and surveillance of dengue in Vietnam.
Keywords: dengue virus, DENV, full-length genome, dengue virus type 2, DENV2, genetic variants, dengue epidemic
Introduction
There are four dengue viruses that cause dengue hemorrhagic fever, transmitted by mosquitoes, is the fastest spreading mosquito-borne viral disease in many parts of the world.1–4 In the 1970s, dengue fever was only reported in eight Asian countries, but the incidence of dengue has grown 30 times in the past 50 years and has expanded geographically from Asia to America, Australia, and Africa. Dengue hemorrhagic fever is not limited to tropical climates but has spread to subtropical and temperate climates.5
The genome of the dengue virus is a single-stranded, positive-sense, capped RNA of approximately 10–11 kb in length that encodes a single polyprotein that is then cleaved into seven non-structural and three structural proteins. The function of viral proteins is in reference to the structural proteins which elucidate the pathogenic function of the virus such as the virion particle dynamics, the flexibility of host adhesion, the role of replication, virulence, and mutations in the epitopes.6 Other studies suggested that under the hyperendemic conditions the nonstructural proteins could play important roles in the evolution of the dengue virus. Amino acid variation in nonstructural proteins may associate with increasing severity in epidemiology and transmission.7
Based on the sequence of the E gene, DENV2 is divided into six genotypes, including Asian I (AI), Asian II (AII), Cosmopolitan (C), American (AM), Asian/American (AA), and sylvatic (S). The consequences of dengue fever are caused mainly by a combination of four different serotypes while the human body only has a partial immune mechanism, and not to provide long-term the cross-protective immune system to the remaining serotypes. Therefore, the genetic diversity and evolution of the dengue virus have spurred the worldwide spread of dengue hemorrhagic fever. Understanding DENV evolution offers valuable opportunities to enhance surveillance, and response to DENV epidemics through advances in RNA sequencing, bioinformatics and genealogy, genetics, evolutionary biological methods, and other computational biological methods.
In Vietnam, previous studies showed that all four DENV serotypes were found to be circulating together, but the most common were the DENV1 and DENV2 serotypes, but the prominence of the dengue serotypes vary year by year and seasonally.3,8,9 According to the WHO Representative Office in Vietnam, dengue hemorrhagic fever in Vietnam between 2001 and 2011 was increased rapidly, the southern provinces accounted for more than 85% of cases and 90% of deaths, with more than 90% were children. Dengue epidemic outbreaks occur in a cycle of 3–5 years.10,11 Vietnam has been assessed successfully in mortality control, since 2005 the mortality rate has been less than ratio 1:1000, but it is still limited in reducing the number of cases.10
In the previous outbreak in 2009 and 2014 in Hanoi, there were about 16,262 and 43,000 cases had been recorded that spread to all districts, and Hanoi accounted for 87% of all patients in the Northern region.12 The number of people infected with dengue in 2009 is 6.7 times higher than in 2008 and the dengue epidemic in 2014 spread to 53 provinces with 28 deaths.13 In 2017, there were 183,287 cases of dengue hemorrhagic fever and 154,552 cases were hospitalized within a short period of time, and the patients were recorded for all age groups.14 The dengue outbreak in 2017, there were 59,063 cases in Northern Vietnam, approximately 8 times higher than in 2016.15
Hanoi is one of the two largest cities in the North of Vietnam, with the number of cases, was 70 times higher than in previous times. 2017 epidemic with high incidences of dengue hemorrhagic fever have been linked to primary infection with DENV1 followed by infection with DENV2, DENV3 and DENV4. There are limited studies on DENV genome characteristics in Vietnam, especially DENV2.
In this study, we sequenced the complete genomes of seven DENV2 viruses collected in the outbreak of dengue hemorrhagic fever 2017 in Hanoi. Data analysis of genetic variants, relationships of these viruses with other DENV2 viruses circulating in Vietnam, as well as other regions of the world, will be a valuable dataset to improve knowledge, monitor, and evaluate dengue outbreaks.
Materials and Methods
Patients and Samples
In a total of 300 adult patients with clinical symptoms of dengue and NS-1 positive antigen using the rapid test from Cortez kit (USA) were admitted and treated at the National Hospital of Tropical Diseases in Hanoi between June and August 2017. The patient’s blood samples were then collected (2–3 mL), the plasma was isolated and stored at −80 °C. All patients participating in the study were explained and signed written informed consent forms. This study was approved by the ethical board of the National Hospital of Tropical Diseases (approval number: 23/HĐĐĐ-NĐTƯ) and conducted in accordance with the Declaration of Helsinki.
Dengue Serotyping and Quantification
The serotype and viral load of DENV were done by using one-step multiplex RT-PCR. In which, the plasma of patients was used to isolated viral RNA using Qiagen kits (QIAamp Viral RNA Mini Kit; Qiagen Sciences Germantown, MD, USA) in accordance with the manufacturer’s protocol. Thereafter, the real-time RT PCR was conducted in a 50-µL reaction volume using SuperScript™ III One-Step RT-PCR system with PlatinumTM Taq DNA polymerase (Invitrogen; CA, USA). PCR was made of two reactions; reaction 1 contained 25 µL reaction mix, 1 µL of DENV1 primers (DENV1-F, DENF1-R), 1 µL of DENV3 primers (DENV3-F, DENV3-R), 0.5 µL DENV1 probe (DENV1-Probe), 0.5 µL DENV3 probe (DENV3-Probe), 5 µL template RNA, and the final volume were made up to 50 µL with distilled water. Reaction 2 contained 25 µL reaction mix, 1 µL of DENV2 primers (DENV2-F, DENF2-R), 1 µL of DENV4 primers (DENV4-F, DENV4-R), 0.5 µL DENV2 probe (DENV2-Probe), 0.5 µL DENV4 probe (DENV4-Probe), 5 µL template RNA, and the final volume were made up to 50 µL with distilled water. The primer sequences were listed in the supplementary table 1. For viral load measurement, four standard positive controls with the concentrations are 102, 104, 106, 108 copies/mL, were added to the experiment. PCR reactions were performed in a LightCycler 480 Thermal Cycler (Roche) with a 105 °C heated lid. The cycling parameters were 1 cycle of 50 °C for 30 min, 1 cycle of 95 °C for 2 min, followed by 45 cycles of 95 °C for 15 s, 60 °C for 30 s (reading the fluorescence). The serotypes of the DENV were detected by specific fluorescence and viral load was determined via threshold cycle values (Ct).
Whole-Genome Sequencing
Large scale viral RNA isolation of DENV2 strains was performed using QIAamp Viral RNA Mini Kit (Qiagen Sciences Germantown, MD, USA). The cDNA was created using ProtoScript® II First Strand cDNA Synthesis Kit (New England BioLabs, MA, USA). Thereafter, PCR reactions were performed using two primer pools (Supplementary table 2) to replicate DNA. The DNA products were then checked using agarose gel, the DNA concentrations were standardized. The DNA products were then cut into short fragments, followed by Index1 and Index2 insertion. The excess products were discarded via the purification step, the products were standardized and quantified for library preparation. The samples from the library were used to read the sequences on the Illumina MiSeq machine. The sequences were then analyzed to remove the links and poor quality reads. The CLC software was used to assemble the complete sequence of DENV2. The reference sequence was from a DENV2 from 2008 (GenBank accession number FJ639718.1). The SAM tools were used to calculate the coverage of each nucleotide position on the gene sequence.
Genetic Variation Analysis
The Capsid, PrM, E, NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5 of DENV2 amino acid sequences distributed in Vietnam (2004, 2008), Thailand (2001), Malaysia (2008), Indonesia (1998), India (2006), China (2013), Cambodia (2008), Brunei (2005) and Brazil (2006) were from the GenBank database (NCBI) with accession numbers EU482774, FJ410215, FJ639832, FJ467493, AB189124, FJ898454, MF459663, GU131902, EU179858 and FJ850088, respectively. The amino acid sequences of each gene were analyzed for all seven DENV2 to find substitutions using MEGA (version 10.1.9) and ClustalX 2.2.
Fast Genome Distance Estimation
The amino acid similarities between forty-nine reference sequences and seven DENV2 sequences were conducted using MASH 2.0 software (the reference sequences were selected based on geographical distance). The calculation includes two steps: The aim of the first step was to create a sketch. All sequences were divided into two groups, group 1 includes sequences of seven DENV strains, each sequence was cut into k-mer (short sequences – 20 bp) and assigned as a hash (random identifier). From MASH, each DENV2 selected 1000 k-mer groups (1000 hash groups), the sketch file contained seven sets of k-mer groups. The second group consisted of forty-nine reference sequences and was repeated as above procedure. In the second step, MASH calculated the distance between group 1 and groups 2 sequences. The value <0.5 corresponds to average nucleotide identity (ANI) > 95%, the nucleotide similarity level between two genomes is presented by the ANI indicator.
Phylogenetic Analysis
The sequences of seven DENV2 in this study and forty-nine reference sequences were analyzed using MAFFT software (Multiple Alignment using Fast Fourier Transform; https://mafft.cbrc.jp/alignment/software/). The genome sequences of DENV2 and references were aligned to create single nucleotide polymorphisms (SNPs), the multiple alignment data were separated by SNP-sites methods (https://github.com/sanger-pathogens/snp-sites#usage). A phylogenetic tree was created based on SNPs data using the IQ-tree system (http://www.iqtree.org). The accuracy was increased by selecting the maximum likelihood method and repeatability ultrafast bootstrap 1000.
Results
Serotyping and Quantification
In a total of the 300 blood samples with NS1 positive, realtime RT-PCR results showed that 276 samples were positive for DENV-1, 22 samples for DENV2, one sample for DENV3, and one sample for DENV4. We selected seven DENV2 samples with the highest viral load to perform whole-genome sequencing.
Full-Length Sequence and Genetic Variants
Results of genome sequencing of seven DENV2 samples showed that the average repetition of the whole genome was very high, the smallest value was about 250 x and the maximum value reached 15,000 x. Especially in gene segments coding for structural proteins such as E, C, and M with repeatability >1000 x. With such a high level of repetition, the resulting sequence will increase accuracy when analyzing changes in the genome. The repetition of the gene segments encoding unstructured proteins ranges from 500 to 7000 x.
The amino acid alterations of DENV2 strains are listed in Table 1. The amino acid analysis results of seven DENV2 revealed 13 amino acid substitutions between seven DENV2 strains and 10 reference strains. Of these mutations, the comparison results reported that only 3 gene regions did not appear to have mutations including prM, NS2B, and NS4B. However, one amino acid replacement was found in the five other genes such as C (position 76 −1R/6K), E (120–1I/6T), NS1 (156–1G/6S), NS3 (133–1G/6D), NS4A (71–1G/6S). In addition, the NS2A gene showed two amino acid alterations at position 91 (2T/5S) and 106 (1I/6V). While the majority of amino acid mutations appeared in the NS5 gene such as position 258 (2L/5I), 260 (2T/5M), 639 (2I/5L), 683 (1Q/H), 684 (1M/6I), and 687 (1L/6V). The comparison results also indicated that the amino acid sequence of seven DENV2 strains differed significantly from the DENV2 strain that caused a dengue outbreak in Vietnam in 2004, but was very similar to the DENV2 that caused epidemics in India, China, and Indonesia.
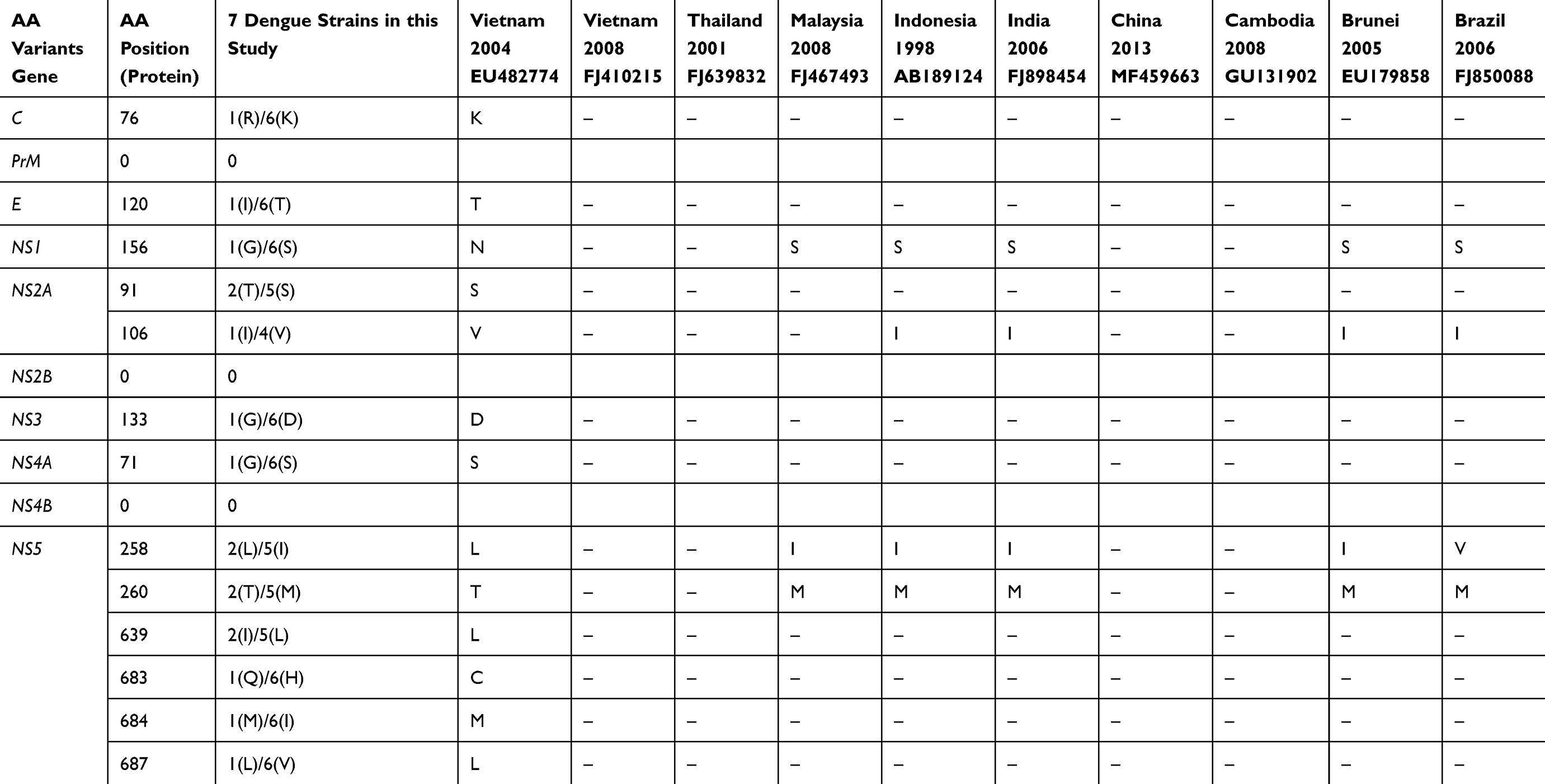 |
Table 1 Genetic Variations in the Dengue Type 2 Genome Circulated in Dengue Epidemic of 2017 in Vietnam. Description of Amino Acid Substitution in the 7 DENV-2 Virus Compared to the 10 Reference Strains. Homologous Amino Acids are Denoted by (-), and the Modified Amino Acids are Denoted by Letters. The Amino Acid Site Including Replacement of Hydrophilic and Hydrophobic are Listed in Each Gene |
Genome Distance Estimation
Results of comparing genome similarity between seven DENV2 with forty-nine reference strains were conducted by MASH 2.0 software and showed in the box chart of Supplementary Figure 1, the lower distance (Mash-distance), the greater level of nucleotide similarity. The data of Mash-distance were presented that the DENV2 viruses in the present study were at the lowest distance from the DENV2 in India in 2006 (distance 0.025), followed by DENV2 strains circulating in 1976 and 1998 (distance 0.053–0.062) and other DENV2 strains in Asia such as China, Taiwan, Singapore and Brunei (Supplementary Figure 2). The DENV2 strain circulating in Malaysia in 2008 exhibited the largest gap with the DENV2 strains in Vietnam in 2017 (distance 0.203). Especially, DENV2 strains causing disease in Vietnam (1988, 2003, 2004, 2006, 2007, 2008) and other Southeast Asia countries such as Thailand (1990, 1995, 2001), Cambodia (2003, 2005), and the Philippines (2015) revealed very high distances with the DENV2 strains in Vietnam in 2017.
The Similarity of DENV-2genome Sequence
Heatmap analysis showed that the DENV2 patterns (denoted D2) almost coincided with the pathogen DENV2 that circulated in India 2006 (dark blue) (Figure 1). Moreover, the DENV2 revealed a lower similarity to the DENV2 strains from Indonesia 1976 (medium blue) and 1998 (light blue), followed by several strains from China (2010, 1987), Brunei (2005, 2006), Indonesia (1975), New-Guinea (1998), Comlumbia (1944), Singapore (2004), USA (2009) and Taiwan (2008). In contrast, there seemed to be a significant difference with the strain circulated in Malaysia 2008 (red), the sequence of DENV2 samples were also exhibited distinct difference with the strain from Vietnam (2003, 2004, 2006, 2007, 2008), Cambodia (2001, 2004, 2007, 2008) and other parts of the world (yellow) although these countries are in Southeast Asia.
 |
Figure 1 Genomic clustering of 7 DENV2 samples and 49 reference sequences based on MASH distances. Heatmap illustrates the pairwise similarity between samples, color scale ranges from 0 (blue) to 0.1 (red). Two large groups are identified and colored with the same key. The MASH clustering also reflects the significant difference between references from Asia and from other continents when comparing with the individual samples in this study. |
Phylogenetic Tree Analysis
The phylogenetic tree of seven DENV2 strains that caused epimedic in Vietnam in 2017 with other DENV2 strains circulating in Vietnam and other regions of the world in previous years is shown in Figure 2. The phylogenetic tree was built based on the full-genome sequence of DENV2, in which seven DENV2 strains were denoted 1–7D2, reference DENV2 were denoted country, GenBank number, and year of finding. The analysis results showed that seven DENV2 strains in Vietnam in 2017 had the highest genetic similarity to the strain in India in 2006. In addition, the genetic relationship from the phylogenetic tree indicated that these strains belonged to the group of DENV2 from Indonesia (1976, 1998), Brunei (2005, 2006), China (2010), Singapore (2004), Philippine (2015) and were not clustered into the same groups with pathogens from Malaysia, Mexico, Columbia, Peru, Fiji, Haiti, Samoa. Although in the same geographical area, the analysis also revealed that the DENV2 viruses of this study had a significant difference as compared with the DENV2 strains in Vietnam (2003, 2004, 2006, 2007, and 2008).
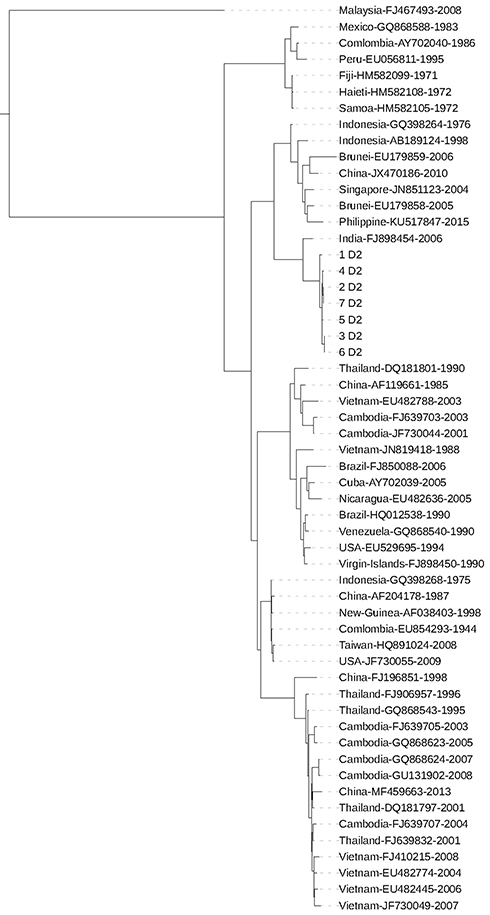 |
Figure 2 Phylogenetic tree based on the level of a similar sequence of 7 DENV2 strains and 49 reference viruses. The viruses from Vietnam in 2017 are denoted by D2, the strains circulated in Vietnam in the past and in the world are denoted by the country name, GenBank accession number and the year causing the disease. |
Discussion
The rate of molecular evolution of the dengue virus, especially the envelope gene, has been estimated in various studies. Costa et al were reported the most accurate rate of evolution of DENV in all serotypes was inferred as 7.6 x 10−4 substitutions/site/year.16 However, substitutions retes were very homogeneous among the serotypes; subsequent studies have shown that four serotypes have uneven rates of evolution. We successfully sequenced the genomes of the seven epidemic DENV2 samples in 2017 in Northern Vietnam and genomic analysis showed that there were different amino acid changes in all structural and unstructured genes. However, we found that the amino acid alterations occurred in the genome of seven DENV2 strains were less than that of DENV-1 strains,17 although these strains both were circulated at the same time in the same geographic region.
In the genome sequences of DENV1 strains, we found many mutations in the E gene,17 the change of amino acids in the envelope protein may enhance the neutralization during pathogenesis. In addition, amino acid variants in several positions were observed in the NS2A, NS2B, NS3, NS4A, NS4B, NS5 genes. These changes may be referred to as the virulence of DENV. However, in the genome sequence of DENV2, there were very few mutations in the E gene, but the mutations were mainly found in the NS5 gene. The difference in mutation rate between DENV1 and DENV2 causing the epidemic in 2017 in Vietnam may be due to a number of reasons such as RNA replication errors of RNA-dependent RNA polymerase enzymes. Several studies show that DENV has an estimated replication error across the entire genome copy of about one mutation/replication due to the lack of proofreading exonuclease activity of the RNA-dependent RNA polymerase.18 Replication errors made by this enzyme lead to a mutation in the genome of the virus, leading to the formation of a genetic variation population. In addition, other reasons such as the recombination of DENV serotypes, selection pressure, adaptive evolution, and ecology may also be factors that contribute to the genetic diversity of DENV during infection in human populations.
Comparison of genetic alterations between DENV2 caused epidemic in Vietnam in 2017 with the DENV2 strains that circulated in Vietnam in 2004 and 2008 as well as DENV2 strains in Thailand (2001), Malaysia (2008), Indonesia (1998), India (2006), China (2013), Cambodia (2008), Brunei (2005) and Brazil (2006) showed differences in several amino acid positions, especially DENV2 strain caused epidemics in 2004 and 2008 in Vietnam, indicating that DENV2 in Vietnam in 2017 did not share an ancestor with DENV2 from 2004, 2008, although they both caused dengue hemorrhagic fever in the same geographic region. Thus, it can be seen that in the same geographical area there exist many different types of DENV; the emergence of a new type of DENV in a geographical area possibly the spread of geographic regions due to globalization.
Previous epidemiology studies indicated that the dengue virus circulating in Vietnam was mainly endemic strains, however, several outbreaks have been recorded as imported strains from other geographical regions. Therefore, analyzing genome sequence as well as other characteristics of the dengue virus gives important information to trace the origins of the dengue virus that caused the outbreak in 2017 and a solution for prevention. Data from boxplot and heatmap draws a significant genetic relationship between seven dengue type 2 circulated in Vietnam in 2017 and forty-nine dengue type 2 in the world. The data obtained in this study suggested that the dengue type2 strains that caused the outbreak in 2017 are very similar to the dengue virus circulated in India in 2006. In addition, other dengue viruses type 2 previously showed a close genetic relationship with the dengue viruses type 2 from Brazil, Cuba, Venezuela, Nicaragua … These results suggest that there was an imported dengue virus type 2 from other geographic regions or the introduction of genetic transfer. Consequently, the fusion of genetic resources between dengue viruses from different origins to create new virus strains.
The genetic relationship via phylogenetic tree showed that the dengue virus type 2 strains that caused an outbreak in Vietnam 2017 likely originated from the strains that circulated in India 2006 (Figure 2). These findings suggest that the dengue virus type 2 in the outbreak in Vietnam in 2017 was likely derived from the dengue virus that was previously circulated in India 2006. Surprisingly, the data from the phylogenetic tree also reported that seven DENV2 in Vietnam 2017 were genetically close to other DENV2 from Indonesia (1976, 1998), Brunei (2006), China (2010), Singapore (2010) and Philipine (2015). The analytical results indicated the genetic resource and origin of the DENV2 that caused the dengue outbreak in Vietnam in 2017. The results also showed the penetration (import) of dengue virus strains from different geographical areas, previous studies in China, Brunei, and Singapore have also reported that the dengue virus strains were imported from different geographical areas.
This is the first study in Vietnam to report the complete genome sequence of DENV2 strains that caused the dengue outbreak in Northern Vietnam in 2017. The genome sequence of seven DENV2 strains with amino acid variations and genetic relationships with other DENV2 strains circulated in different parts of the world have been described and compared. This is an important database to reveal the pathogenetic characteristics of DENV2 and their role in dengue outbreaks in Vietnam.
Funding
This research was supported in part by a grant from the Ministry of Science and Technology of Vietnam (Grant number KC.10.22/16–20).
Disclosure
All authors declare no conflicts of interest.
References
1. World Health Organization. Dengue: Guidelines for Diagnosis, Treatment, Prevention and Control.
2. World Health Organization. Fact sheet dengue and severe dengue; 2019. Available from:https://www.who.int/news-room/fact-sheets/detail/dengue-and-severe-dengue.
3. World Health Organization.Global Strategy for Dengue Prevention and Control 2012-2020. Availabe from: https://www.who.int/immunization/sage/meetings/2013/april/5_Dengue_SAGE_Apr2013_Global_Strategy.pdf.Accessed October 30, 2020.
4. World Health Organization. Dengue control. 2018.
5. Arima Y, Chiew M, Matsui T, Emerging Disease S. Epidemiological update on the dengue situation in the Western Pacific Region, 2012. Western Pac Surveill Response J. 2015;6(2):82–89. doi:10.5365/wpsar.2014.5.4.002
6. Owens RJ, Limn C, Roy P. Role of an arbovirus nonstructural protein in cellular pathogenesis and virus release. J Virol. 2004;78(12):6649–6656. doi:10.1128/JVI.78.12.6649-6656.2004
7. Rodriguez-Roche R, Gould EA. Understanding the dengue viruses and progress towards their control. Biomed Res Int. 2013;2013:690835. doi:10.1155/2013/690835
8. Ha DQ, Huan TQ. Dengue activity in viet nam and its control programme, 19971998. Dengue Bulletin. 1997;21:35–40.
9. Ha DQ, Ninh TU. Virological surveillance of dengue haemorrhagic fever in Viet Nam, 19871999. Dengue Bulletin. 2000;24:18–23.
10. WPRO - WHO Representative Office Viet Nam. Fact sheet Dengue; 2018. Available from: http://www.wpro.who.int/vietnam/vi/.
11. Arima Y, Edelstein ZR, Han HK, Matsui T. Epidemiologic Update on the Dengue Situation in the Western Pacific Region,2011. Western Pac Surveill Response J. 2013;4(2):647–54.
12. Minh An DT, Rocklov J. Epidemiology of dengue fever in Hanoi from 2002 to 2010 and its meteorological determinants. Glob Health Action. 2014;7(1):23074. doi:10.3402/gha.v7.23074
13. Pham LD, Phung NH, Le NT, Vo TQ. Economic report on the cost of dengue fever in Vietnam: case of a provincial hospital. Clinicoecon Outcomes Res. 2017;9:1–8. doi:10.2147/CEOR.S124023
14. Tran BX, Vu G T, Hoang Nguyen L, et al. Cost-of-illness and the health-related quality of life of patients in the dengue fever outbreak in Hanoi in 2017. Int J Environ Res Public Health. 2018;15:6. doi:10.3390/ijerph15061174
15. Huong Van Nguyen PQTT, Nguyen TH, Vu GT, et al. Knowledge, attitude and practice about dengue fever among patients experiencing the 2017 outbreak in Vietnam. Int J Environ Res Public Health. 2019;16:976–988. doi:10.3390/ijerph16060976
16. Costa RL, Voloch CM, Schrago CG. Comparative evolutionary epidemiology of dengue virus serotypes. Infect Genet Evol. 2012;12(2):309–314. doi:10.1016/j.meegid.2011.12.011
17. Dang TT, Pham MH, Bui HV, Van Le D. Whole genome sequencing and genetic variations in several dengue virus type 1 strains from unusual dengue epidemic of 2017 in Vietnam. Virol J. 2020;17(1):7. doi:10.1186/s12985-020-1280-z
18. Venkataraman S, Prasad B, Selvarajan R. RNA dependent RNA polymerases: insights from structure, function and evolution. Viruses. 2018;10:2. doi:10.3390/v10020076
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.