Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 14
Fine-particulate matter aggravates cigarette smoke extract–induced airway inflammation via Wnt5a–ERK pathway in COPD
Authors Wang Z, Zhao J, Wang T, Du X, Xie J ![]()
Received 23 November 2018
Accepted for publication 11 February 2019
Published 9 May 2019 Volume 2019:14 Pages 979—994
DOI https://doi.org/10.2147/COPD.S195794
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Chunxue Bai
Zhihua Wang, Junling Zhao, Ting Wang, Xiaohui Du, Jungang Xie
Department of Respiratory and Critical Care Medicine, National Clinical Research Center of Respiratory Disease, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, People’s Republic of China
Background: Exposure to environmental particulate matter (PM) ≤2.5 µm in diameter (PM2.5) and smoking are common contributors to COPD, and pertinent research implicates both factors in pulmonary inflammation. Using in vivo mouse and in vitro human cellular models, we investigated the joint impact of PM2.5 pollution, and cigarette smoke (CS) in mice or cigarette-smoke extract (CSE) in cells on COPD inflammation, and explored potential mechanisms.
Methods: Tissue changes in lungs of C57BL/6 mice exposed to PM2.5 and CS were studied by light microscopy, H&E, immunochemistry, and immunofluorescence-stained sections. Levels of inflammatory factors induced by PM2.5/CS in mice and PM2.5/CSE in 16HBE cells were also monitored by quantitative reverse-transcription (qRT)-PCR and ELISA. Expression of genes related to the Wnt5a-signaling pathway was assessed at transcriptional and protein levels using immunofluorescence, qRT-PCR, and Western blotting.
Results: Inflammatory response to combined exposure of PM2.5 and CS or CSE in mouse and 16HBE cells surpassed responses incited separately. Although separate PM2.5 and CS/CSE exposure upregulated the expression of Wnt5a (a member of the Wnt-secreted glycoprotein family), combined PM2.5 and CS/CSE exposure produced a steeper rise in Wnt5a levels. Use of a Wnt5a antagonist (BOX5) successfully blocked related inflammatory effects. ERK phosphorylation appeared to mediate the effects of Wnt5a in the COPD model, promoting PM2.5 aggravation of CS/CSE-induced airway inflammation.
Conclusion: Our findings suggest that combined PM2.5 and CS/CSE exposure induce airway inflammation and Wnt5a expression in vivo in mice and in vitro in 16HBE cells. Furthermore, PM2.5 seems to aggravate CS/CSE-induced inflammation via the Wnt5a–ERK pathway in the context of COPD.
Keywords: COPD, airway inflammation, PM2.5, Wnt5a
Introduction
COPD is a common disease characterized by airway obstruction due to structural alterations of small airways and accompanied by systemic inflammation.1,2 This disease is creating immense fiscal demand and fueling individual/societal health care burdens.3 Epidemiologic and genetic risk factors for COPD include long-term smoking, chronic environmental exposure to air pollution and chemical substances, and respiratory infections.4 As the primary risk factor, cigarette smoking is the chief cause for concern in COPD.5–7 Another strong risk factor for COPD is exposure to toxic atmospheric pollutants, especially fine-particulate matter ≤2.5 μm in diameter (PM2.5).8
Extended exposure to PM2.5 may hasten deterioration of lung function and heighten the risk of developing COPD.9 Numerous epidemiological studies have confirmed that in patients with COPD, PM2.5 may cause genetic and epigenetic alterations, as well as immunodysfunction.10–13 Results of a large epidemiological survey suggested that smokers are more susceptible than nonsmokers to PM2.5 pollution.14 Furthermore, recent COPD studies have determined that PM2.5 exposure may incite immune disorders, promote pulmonary inflammation, or aggravate airway remodeling in animal or cell models,15–18 although the precise mechanisms have yet to be fully clarified.
Wnt proteins have critical roles in many organisms related to carcinogenesis and embryonic development. Several Wnt ligands expressed in the developing and adult lung include Wnt2, Wnt3a, Wnt5a, Wnt5b, Wnt7b, Wnt10a, Wnt11, and Wnt13.19 Among them, Wnt5a and Wnt7b are expressed at high levels, particularly in the developing airway epithelium during lung development. A new study has revealed the essential role of secreted Wnt4 in respiratory system development.20 Three pathways are implicated in Wnt signaling: the canonical Wnt–β-actin pathway, the Wnt–Ca2+ pathway, and planar cell polarity.21 Downstream effects of Wnt signaling are achieved through various intracellular components, according to which pathway is activated. In fact, dysregulated Wnt signaling is an important aspect of chronic pulmonary diseases, such as COPD and idiopathic pulmonary fibrosis.22 Wnt5a, one of the well-studied Wnt family members, regulates cellular responses by activating or inhibiting canonical Wnt signaling, depending on the type of receptor involved.23
Studies examining the molecular mechanism of Wnt5a in COPD have been factually underweighted, and the role of Wnt5a with respect to interplay between COPD and airway inflammation induced by PM2.5/cigarette-smoke extract (CSE) still needs to be defined. Using animal and cellular models, we sought to investigate the joint impact of PM2.5 pollution and CS on COPD inflammation and explore the mechanisms potentially involved.
Methods
Cell culture and treatment
A purchased human airway epithelial cell line transfected with human papillomavirus type 16 (HPV16) E6/E7 oncogenes (16HBE; American Type Culture Collection, Manassas, VA, USA) was cultured in RPMI 1640 (KeyGen Biotech, Nanjing, China) supplemented with 10% heat-inactivated FBS (Scientan, Beijing, China) and 1% penicillin–streptomycin (Scientan). The cells were incubated at 37°C in 5% CO2 with humidification, then exposed (or not) to PM2.5 (100 μg/mL) or 10% CSE. To investigate effects of Wnt5a on inflammation induced by PM2.5 and CSE, 16HBE cells were pretreated with the Wnt5a antagonist BOX5 (200 μM; Merck Millipore, Burlington, MA, USA) for 1 hour before adding 100 μg/mL PM2.5 or 10% CSE.
Cell-viability assay
The viability of 16HBE cells subjected to various treatments was gauged using a WST8 assay kit (CCK8; Promoter Biotechnology, Wuhan, China), according to the manufacturer’s instructions. Cells were exposed to PM2.5 (25, 50, 100, or 200 μg/mL) plus 10% CSE for 24 hours or BOX5 (100, 200, or 300 μM) in RPMI 1640 culture medium (100 μL). Each group occupied five replicate wells. Data were drawn from three independent experiments. OD at 450 nm was determined with a microplate reader (Multiskan MK3; Thermo Fisher Scientific, Waltham, MA, USA).
RNA extraction and quantitative reverse-transcription PCR
Total RNA extracts of mouse lung tissue and cells were obtained using RNAiso Plus (Takara Bio, Kusatsu, Japan) as instructed, and their concentrations were measured by microvolume spectrophotometry (NanoDrop; Thermo Fisher Scientific). A reverse-transcription kit (PrimeScript RT; Takara Bio) was employed for cDNA synthesis, and thereafter using an ABI Fast 7900 HT real-time PCR system (Applied Biosystems, Foster City, CA, USA) for quantification. Analysis utilized the 2−ΔΔCt method, with β-actin as the reference gene. Primer sequences are listed in Table 1.
 | Table 1 Primer sequences |
Western blot analysis
Total proteins of mouse lung tissue and cells were extracted in lysis buffer containing phosphatase inhibitors using a BCA protein-assay kit (Bioyear Biotechnology, Wuhan, China) to measure concentrations. Once separated by 10% SDS-PAGE, the proteins were transferred to polyvinylidene difluoride membranes (Merck Millipore) and blocked (5% evaporated milk in Tris-buffered saline containing 0.05% Tween 20 [TBST]) for 1–2 hours, followed by overnight incubation with primary antibodies at 4°C. Corresponding primary antibodies were applied: anti-Wnt5a (1:1,000; ABclonal Biotechnology, Wuhan, China), anti-P-ERK1/2 (1:2,000; Cell Signaling Technology, Danvers, MA, USA), anti-T-ERK1/2 (1:1,000, Cell Signaling Technology), and anti-β-actin antibody (1:4,000; ABclonal Biotechnology). Membranes were then washed with TBST and incubated for 1 hour at room temperature with secondary antibodies conjugated to HRP (1:4,000; Bioyear Biotechnology). Protein bands were imaged (ChemiDoc XRS + system; Bio-Rad, Hercules, CA, USA) and quantitatively analyzed (Image Lab; Bio-Rad).
ELISA for IL6 and IL8 levels
Cell cultures were collected and centrifuged at 3,000 rpm for 5 minutes at 4°C, and then stored at −80°C for later analysis. IL6 and IL8 concentrations in culture supernatants were detected by corresponding DuoSet ELISA kits (R&D Systems, Minneapolis, MN, USA) following the manufacturer’s instructions (limits of detection: IL6 9.38 pg/mL, IL8 31.3 pg/mL).
Routine, immunohistochemical, and immunofluorescence staining of mouse lung tissue
The right lung of each mouse was sampled, fixed in 4% paraformaldehyde, and processed using standard techniques by staining sections of paraffin-embedded tissue for H&E, immunohistochemistry, and immunofluorescence. For immunohistochemistry, the primary antibody rabbit polyclonal anti-PCNA (Proteintech, Rosemont, IL, USA) was used. For Wnt5a immunofluorescence staining, sections of mouse lungs were deparaffinized, rehydrated, and subjected to heat-induced epitope retrieval. After washing and blocking, the slides were incubated overnight with mouse anti-Wnt5a primary antibody at 4°C. On the following day, incubation with fluorescein isothiocyanate–conjugated antimouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) took place for 1 hour in the dark at room temperature. Sections were subsequently washed three times, and DAPI nuclear stain was applied for fluorescence microscopy. For dual immunofluorescence staining, primary antibodies, including antineutrophils (mouse Ly6G; Abcam, Cambridge, UK) and T cells (rabbit CD3; Proteintech), were used to assess inflammatory cell populations.
Animal care and PM2.5/smoking-exposure protocols
Six-week-old male C57BL/6 mice were obtained from the Animal Experiment Center of Wuhan University and housed in the Laboratory Animal Center, Tongji Medical College. All animal experiments were approved by the animal experiment ethics committee of Tongji Medical College, Huazhong University of Science and Technology and adhered to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, Bethesda, MD, USA).
Once adapted to the experimental environment, the mice were randomly assigned to one of four groups: controls, PM2.5, smoking, and PM2.5 + smoking (n=7 per group). Healthy control mice were exposed to room air without PM2.5, mice in the PM2.5 group inhaled PM2.5 (110 μg/m3) delivered by ultrasonic atomizer, and mice in the smoking group inhaled CS (ten cigarettes over 2.5 hours) delivered in a PAB-S200 animal passive smoking–exposure system (Biolab Technology, Beijing, China). Mice in the PM2.5 + smoking group were exposed simultaneously to both. All mice were treated twice daily 5 days per week for 10 months. At completion, the mice were killed and lung tissue collected for analysis.
PM2.5-sample preparation
PM2.5 was prepared as described previously with slight modifications.24 Between November 2016 and May 2017, daily PM2.5 samples were collected on Teflon filters (Ryder Keith Technology, Beijing, China) using a high-volume PM2.5 sampler (UAS-310). Samples were gathered on a building rooftop 24 hours per day. Used filters were carefully packed and stored at −20°C until extraction, at which time they were cut into pieces (4×4 cm), soaked in sterilized water, and sonicated for 45 minutes. Water-extracted samples were then collected and frozen at −80°C. Frozen PM2.5 suspensions were vacuum freeze-dried (Gydevang 17-19 DK-3450; Heto-Holten, Allerød, Denmark), collected, and stored at −20°C. Prior to delivery to mice or cells, they were resuspended in sterile PBS or RPMI 1640 culture medium and sonicated for 30 minutes.
Cigarette smoke–extract preparation
CSE was obtained as previously described with few modifications.25 In brief, CSE was prepared by bubbling smoke from two filtered cigarettes (3R4F) into a 50 mL centrifuge tube containing RPMI 1640 culture medium (20 mL), aided by vacuum extraction. The extracts obtained were sterilized by filtration (0.22 μm) and considered full concentrations of CSE.
Statistical analysis
All data are expressed as mean ± SD. Groupwise differences were first tested by one-way ANOVA and then Student’s t-test. The relationship between Wnt5a mRNA expression and proinflammatory cytokines was assessed using Pearson or Spearman analysis. Standard software (Prism version 5.0; GraphPad Software, San Diego, CA, USA) was used for all computations, setting significance at P<0.05.
Results
PM2.5 aggravated smoking-induced changes in lungs of mice
To study histological changes produced by PM2.5 and smoking separately and together, experimental mice were grouped as controls, PM2.5, smoking, and PM2.5 + smoking. H&E-stained tissue sections from each group were then examined, and the results indicated that separate exposure to PM2.5 and smoking showed marked tracheal influx of inflammatory cells and hyperplasia of airway epithelial cells. Such effects were substantially more pronounced in the PM2.5 + smoking group (Figure 1A–D).
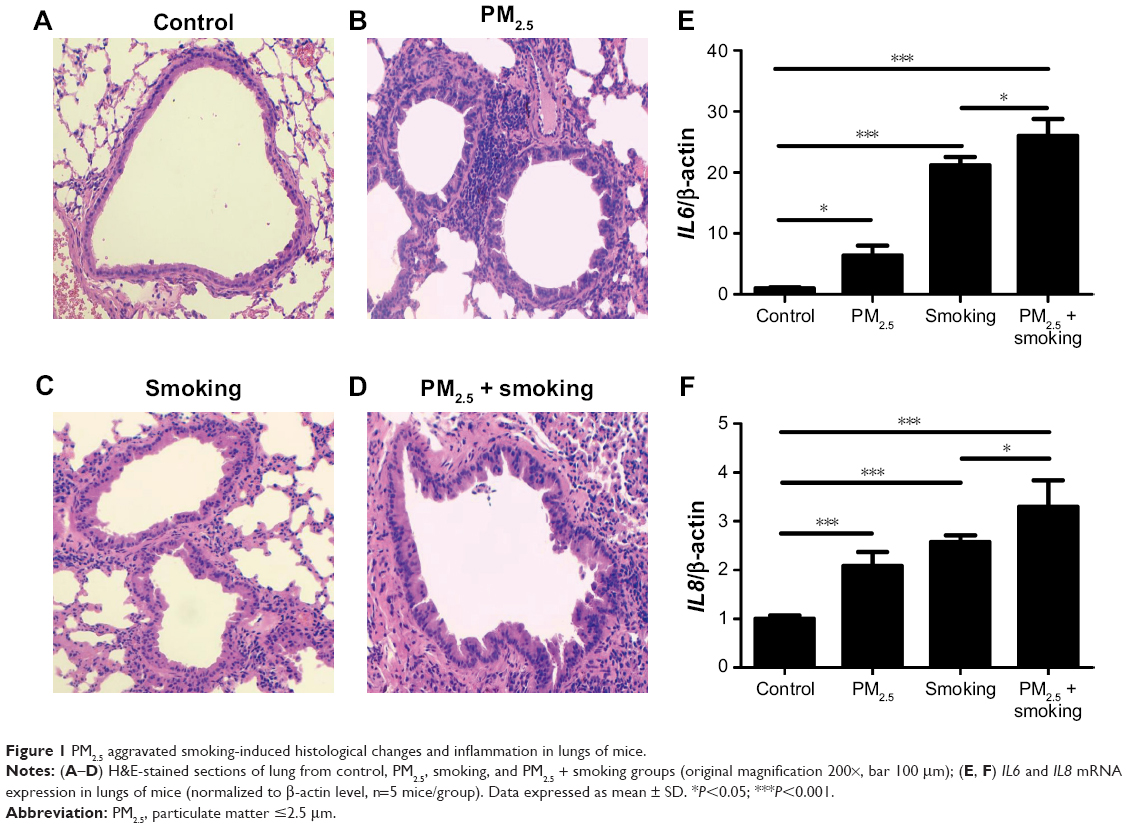 | Figure 1 PM2.5 aggravated smoking-induced histological changes and inflammation in lungs of mice. |
In addition, immunofluorescence results revealed that both PM2.5 and smoking significantly increased the number of Ly6G+ neutrophils and CD3+ lymphocytes that infiltrated the trachea, respectively, compared with the control group, and the populations of these inflammatory cells increased more significantly in the PM2.5 + smoking group, suggesting that PM2.5 promotes smoking-induced airway inflammation in mice (Figure S1). While the immunoreactivity of the proliferation marker PCNA was present in alveolar epithelial cells and small-airway epithelia of the control group, it was significantly increased in both the PM2.5 and smoking groups and increased to an even higher degree in the PM2.5 + smoking group (Figure S2).
PM2.5 promoted smoking-induced inflammation in lungs of mice
As is well known, the release of inflammatory mediators by inflammatory cells is crucial for COPD development. To investigate the influence of PM2.5 on smoking-induced inflammation in mice further, we measured expression levels of proinflammatory factors (IL6 and IL8) in lungs of mice. Likewise, levels of IL6 and IL8 mRNA expression (both elevated in the PM2.5 and smoking groups individually) were upregulated even more so in mice of the PM2.5 + smoking group (Figure 1E and F). Furthermore, in addition to the mRNA levels of inflammatory factors in mouse lung tissue, we also collected mouse bronchoalveolar lavage fluid to measure the levels of these cytokines. In fact, changes in levels of IL6 and IL8 in mouse bronchoalveolar lavage fluid were consistent with observed differences in their mRNA levels (data to be shown in forthcoming article).
PM2.5 and CSE individually upregulated inflammation in 16HBE cells
Prior to addressing the added effect of PM2.5 on CSE-induced inflammation in vitro, we first showed that PM2.5 and CSE were separately capable of triggering cellular inflammation by exposing 16HBE cells to various concentrations of CSE (2.5%, 5%, 10%, or 20%) for 24 hours or to 10% CSE for various periods (6 hours, 12 hours, 24 hours, or 48 hours). Levels of IL6 and IL8 in supernatants of cell cultures were determined thereafter by ELISA. As shown in Figure 2A and B, both cytokines displayed dose- and time-dependent upregulation. Similarly, in 16HBE cells at variable PM2.5 exposure (25 μg/mL, 50 μg/mL, 100 μg/mL, or 200 μg/mL) for 24 hours or at PM2.5 exposure (100 μg/mL) for various periods (6 hours, 12 hours, 24 hours, or 48 hours), levels of these cytokines surged (Figure 2C and D).
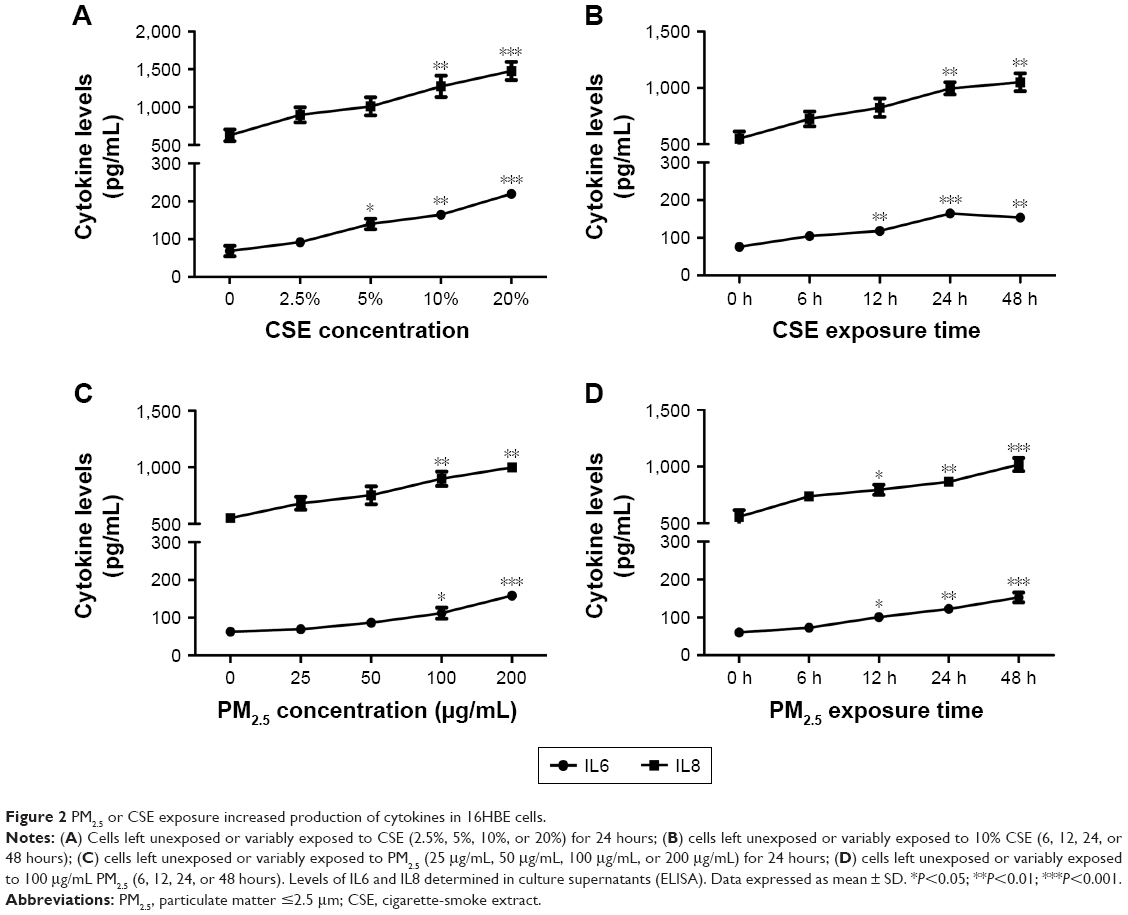 | Figure 2 PM2.5 or CSE exposure increased production of cytokines in 16HBE cells. |
Cytotoxicity induced by exposing 16HBE cells to both PM2.5 and CSE
To assess the presumptive cytotoxicity of a combination of PM2.5 and CSE, the viability of 16HBE cells was examined after exposures to 25 μg/mL, 50 μg/mL, 100 μg/mL, or 200 μg/mL PM2.5 in combination with 10% CSE. As indicated in Figure 3A, a dose-dependent decline in cell viability was observed. At fixed exposure of PM2.5 (100 μg/mL) and CSE (10%), cell viability declined >10% compared with controls, so this level of exposure was deemed optimal for the following experiment.
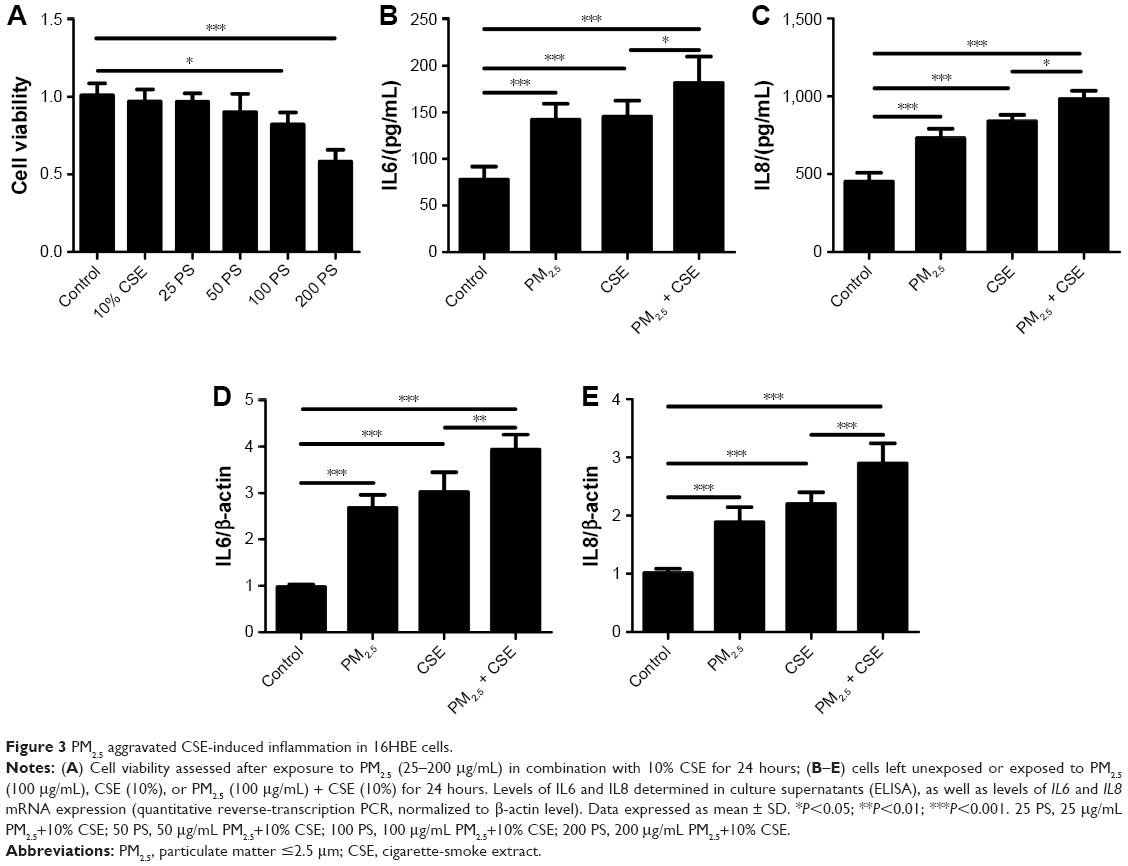 | Figure 3 PM2.5 aggravated CSE-induced inflammation in 16HBE cells. |
PM2.5 exposure enhanced CSE-induced inflammation in 16HBE cells
To determine the effects of PM2.5 and CSE, 16HBE cells were exposed to PM2.5 (100 μg/mL), CSE (10%), or PM2.5 (100 μg/mL) + CSE (10%) for 24 hours separately, measuring IL6 and IL8 expression levels by ELISA. Compared with distinct cytokine increases observed after separate exposure to PM2.5/CSE, significantly greater upregulation of IL6 and IL8 followed combined PM2.5 and CSE exposure (Figure 3B and C). To confirm these findings, we also measured transcriptional levels of these cytokines at various exposure by quantitative reverse-transcription (qRT)-PCR, all proving similar to ELISA results (Figure 3D and E).
Wnt5a expression increased in lungs of mice in PM2.5/smoking group
Immunofluorescence imaging indicated that levels of Wnt5a observed in lungs of mice exposed to a combination of PM2.5 and smoking increased compared with controls and clearly surpassed levels found after PM2.5 or smoking exposure alone (Figure 4A–D). Furthermore, shifts in Wnt5a at the transcriptional level corresponded with protein fluctuations in mouse lungs (Figure 4E).
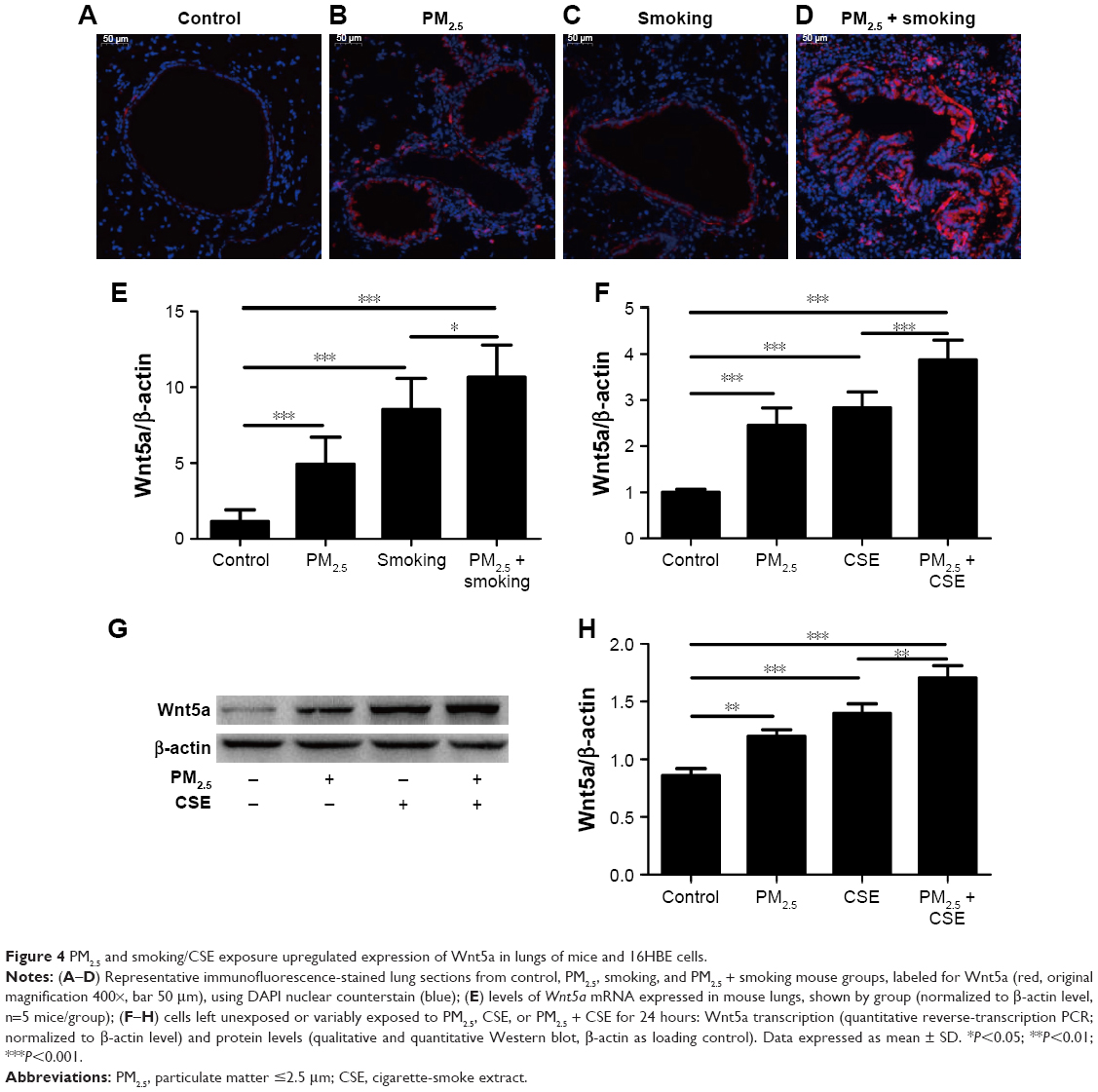 | Figure 4 PM2.5 and smoking/CSE exposure upregulated expression of Wnt5a in lungs of mice and 16HBE cells. |
PM2.5/CSE exposure induced upregulation of Wnt5a in 16HBE cells
To study the effect of PM2.5/CSE on Wnt5a expression and its potential function in airway inflammation, 16HBE cells were subjected to various exposure for 24 hours: PM2.5 (100 μg/mL), CSE (10%), or PM2.5 (100 μg/mL) + CSE (10%). As shown in Figure 4F, transcriptional levels of Wnt5a were upregulated markedly in the separate PM2.5 and CSE groups compared with controls; moreover, expression of Wnt5a peaked in the PM2.5 + CSE group. Wnt5a protein-expression levels assessed by Western blot were consistent with these results (Figure 4G and H).
Correlations between Wnt5a and inflammatory cytokines in both mice and 16HBE cells
We evaluated the correlation between Wnt5a mRNA levels and airway inflammation in mouse lung tissue, and found that a significant and positive correlation was present between Wnt5a mRNA and levels of inflammatory genes (Figure S3A and B). In addition, a similar correlation was found in 16HBE cells (Figure S3C and D). These results suggest that the inflammation observed can be attributed to the high expression levels of Wnt5a in both mice exposed to PM2.5/smoking and 16HBE cells exposed to PM2.5/CSE.
Wnt5a antagonist downregulated expression levels of proinflammatory factors in 16HBE cells
To evaluate the role of Wnt5a in airway inflammation, we used BOX5, a Wnt5a-derived antagonistic peptide, to block endogenous Wnt5a signaling, initially testing its toxicity in 16HBE cells. Various doses of BOX5 (100–300 μM) showed no obvious toxicity (Figure 5A), so 16HBE cells were preincubated with BOX5 (200 μM) or vehicle (PBS) for 1 hour and with exposure to PM2.5/CSE for 24 hours. Supernatants of cell-culture medium were collected to determine levels of inflammatory cytokines by ELISA, whereas total RNA was extracted from 16HBE cells for qRT-PCR. As depicted in Figure 5B–E, administration of BOX5 appeared to attenuate the upregulation of inflammatory cytokines induced by PM2.5/CSE in 16HBE cells, whether at protein or transcriptional level. These results indicated that Wnt5a at least partly mediated the upregulation of inflammatory cytokines induced by PM2.5/CSE.
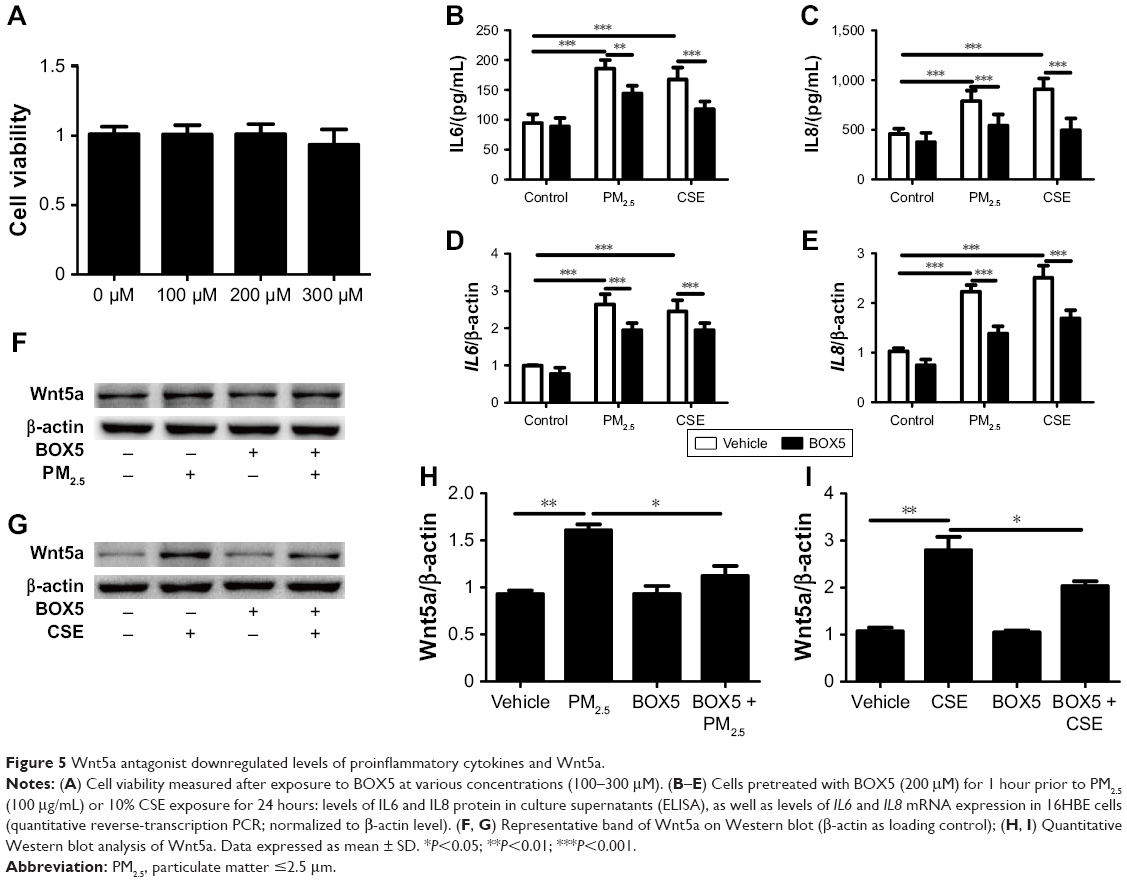 | Figure 5 Wnt5a antagonist downregulated levels of proinflammatory cytokines and Wnt5a. |
BOX5 treatment also significantly reduced Wnt5a expression in 16HBE cells (Figure 5F–I), implicating Wnt5a as a major factor in airway inflammation induced by PM2.5/CSE. In addition, the level of Wnt5a in BOX5 + PM2.5 + CSE group was downregulated in comparison with that in PM2.5 + CSE group, which further illustrated the role of Wnt5a in the joint effect of PM2.5 in combination with CSE (Figure S4).
Downregulation of Wnt5a attenuated proinflammatory cytokine secretion via ERK pathway in 16HBE cells
To investigate mechanisms further by which Wnt5a modulates airway inflammation related to PM2.5/CSE exposure, we examined the potential involvement of ERK signaling in Wnt5a-regulated inflammation, initially observing ERK-pathway activity in mice and in 16HBE cells. Expression levels of P-ERK1/2 and T-ERK1/2 were determined by Western blot. As is illustrated in Figure 6A, the level of P-ERK1/2 and the ratio of P-ERK1/2:T-ERK1/2 increased significantly in mice after PM2.5 and smoking exposure, respectively, becoming even more pronounced after joint exposure to PM2.5 and smoking. In 16HBE cells, PM2.5 and CSE exerted similar effects on the ERK pathway (Figure 6B). Thereafter, 16HBE cells were preincubated with 200 μM BOX5 or vehicle (PBS) for 1 hour, followed by 24 hours’ incubation with or without exposure to PM2.5/CSE. As shown in Figure 6C and D, upregulated phosphorylation of ERK1/2 (P-ERK1/2) induced by PM2.5/CSE was inhibited partly by BOX5. These data suggest that in the context of COPD, the Wnt5a–ERK pathway takes part in airway inflammation triggered by PM2.5/CSE. We also observed protein levels of other inflammation-related signaling pathways, such as JNK and p38MAPK (members of the MAPK family); however, preincubation of 16HBE cells with BOX5 alone with or without PM2.5/CSE did not alter the expressions of these genes significantly (data not shown).
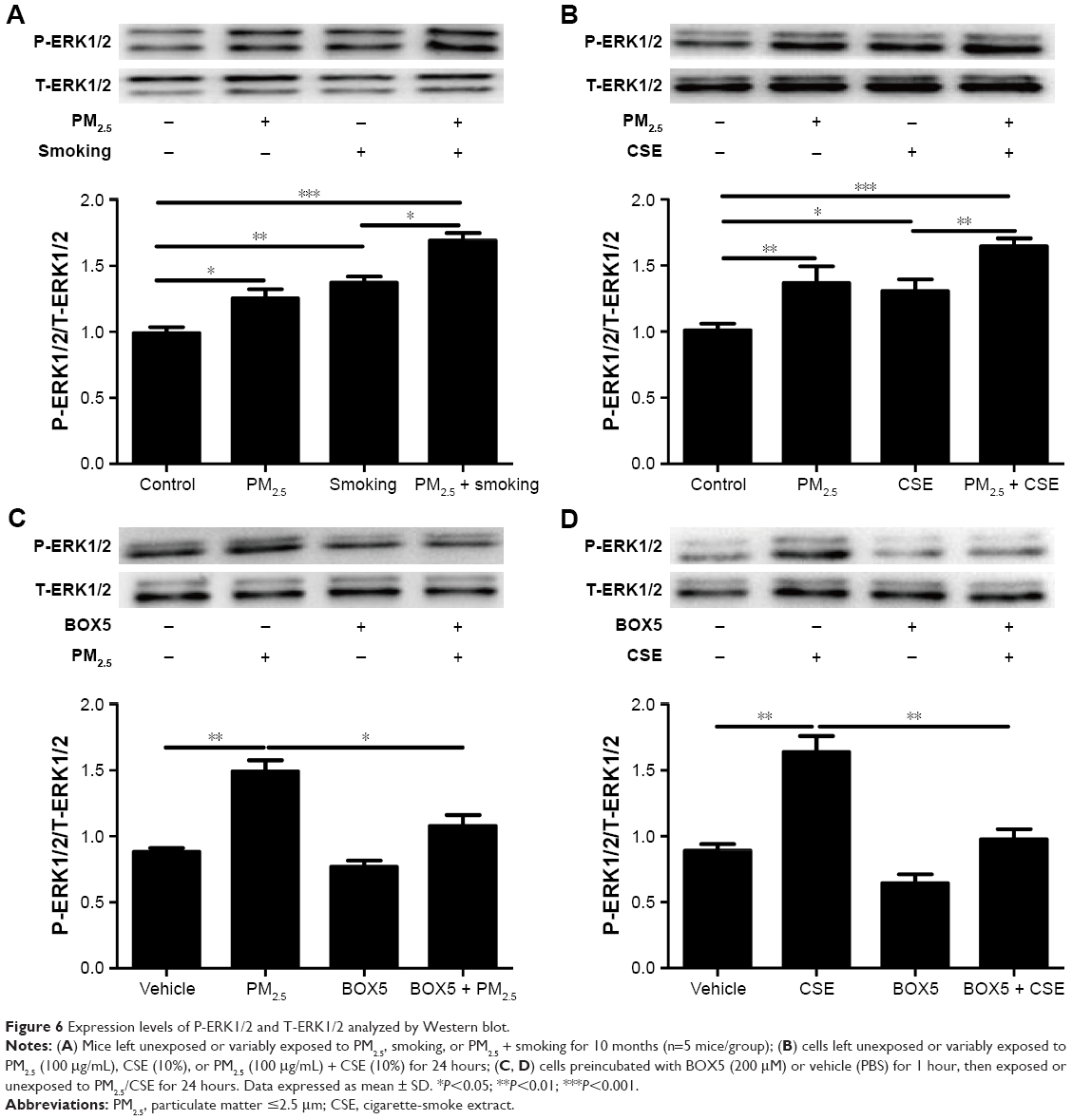 | Figure 6 Expression levels of P-ERK1/2 and T-ERK1/2 analyzed by Western blot. |
Discussion
Our research has shown that exposure to PM2.5 or CSE causes aberrant upregulation of Wnt5a in both in vivo mouse and in vitro human cell models. Upregulated Wnt5a then promotes inflammatory cytokine production via ERK-pathway activation. After combined exposure to PM2.5 and CSE in vivo and in vitro, such effects were more pronounced. It is thus evident that PM2.5 inhalation promotes and aggravates CSE-induced airway inflammation via the Wnt5a–ERK pathway, making Wnt5a a potential therapeutic target for combating airway inflammation in the context of COPD.
A key characteristic of COPD is the presence of chronic inflammation.26 Consequently, IL6 has become a prognostic biomarker in patients with this disease.27 As a member of the CXC chemokine family, IL8 is also linked to the development of COPD, high levels materializing in bronchoalveolar lavage fluid and sputum samples of affected patients.2,27 Reports have already indicated that expression levels of IL6 and IL8 may be upregulated in patients with COPD.28,29 Therefore, we selected both as inflammatory markers for the present study. In our experiments, IL6 and IL8 levels were upregulated in the lungs of mice subjected to long-term smoking exposure. Protein- and mRNA-expression levels of these cytokines were also heightened in 16HBE cells, showing dose- and time-dependent elevations in response to CSE. This evidence further supports the concept that chronic airway and systemic inflammation is ongoing in COPD related to CSE.30,31
Epidemiological studies have also demonstrated that patients with COPD are more susceptible to ambient air pollutants, such as PM2.5, although the basis has never been fully explained.32 The relatively small diameter yet substantial density of PM2.5 enables breaching of the blood–gas barrier and lung parenchyma/airway deposition, causing great damage to bronchial lining cells and alveolar epithelium.33 As in other reports, which noted that PM2.5 exposure boosts levels of inflammatory cytokines to induce airway inflammation,34–37 we found that PM2.5 exposure increased proinflammatory cytokine levels of 16HBE cells in dose- and time-dependent manners, and experimental mice with COPD showed more intense pulmonary inflammation after PM2.5 inhalation. Similarly, both protein and mRNA proinflammatory cytokine levels in cells showed greater gains following combined exposure compared to exposure to PM2.5 or CSE alone. As such, our COPD models induced by CSE were more vulnerable to PM2.5 exposure, which served to aggravate ongoing inflammation.
Wnt5a belongs to the Wnt-secreted glycoprotein family and is a prototypic ligand that activates β-catenin-dependent or -independent Wnt signaling to regulate cell migration, polarity, proliferation, and survival.38 In addition, Wnt5a is known to participate in distal lung morphogenesis, with homozygous Wnt5a-knockout mice displaying lethal pulmonary dysplasia.39 According to previous reports, Wnt5a serves as an activator in lung diseases, particularly lung cancer,40 and acts as an important proinflammatory mediator in chronic inflammatory diseases, such as metabolic disorders, atherosclerosis, rheumatoid arthritis, and other conditions.41–43 It has been reported that Wnt5a regulates fundamental mechanisms that mediate airway smooth-muscle contraction, and thus may be relevant to airway hyperresponsiveness in asthma.44 Furthermore, Wnt5a impairs Wnt/β-catenin mediated alveolar epithelial cell repair, and inhibition of Wnt5a in vivo attenuates lung-tissue destruction and improves lung function in models of COPD.45 A recent study has shown that Wnt5a is upregulated by CSE in tandem with inflammatory factors in a model of COPD.46 Our data on the impact of smoking/CSE in Wnt5a expression are aligned with the aforementioned findings.
The present study is the first to demonstrate that PM2.5 may also induce Wnt5a expression at both transcriptional and translational levels in mice and 16HBE cells. Furthermore, we found that expression of Wnt5a was significantly enhanced by combined PM2.5 and CSE exposure, offering further proof that Wnt5a is positively regulated by PM2.5/CSE. Meanwhile, the relationship between Wnt5a and inflammation due to expression of inflammatory cytokines was also established in our study. The obvious correlation between Wnt5a and IL6/IL8 in both mice and 16HBE cells proves that overexpressed Wnt5a is closely related to airway inflammation in COPD. Finally, BOX5 (a specific Wnt5a antagonist) clearly attenuated the upsurge in proinflammatory cytokines induced by PM2.5/CSE, suggesting a Wnt5a-mediated mechanism by which PM2.5/CSE exposure upregulates inflammatory cytokines in the context of COPD.
As a canonical inflammatory signaling pathway, phosphorylation of ERK is critical in inflammation and innate immunity.47 Earlier studies have documented ERK phosphorylation in mouse lungs and in human airway epithelial cells.48,49 PM2.5 likely promotes amphiregulin expression through the ERK pathway to sustain proinflammatory responses in bronchial epithelial cells.50 In our experiments, we observed that ERK phosphorylation was separately upregulated by PM2.5 and smoking in mice, and likewise separately regulated by PM2.5 and CSE in 16HBE cells, consistent with previous data, and again, we noted more robust activation of ERK by combined exposure.
Some researchers have found that ERK may serve as a Wnt5a repressor in gastric cancer cells,51 whereas other sources have cited Wnt5a activation of ERK signaling in mouse cardiac fibroblasts and in human dental pulp cells.52,53 At this juncture, there have been no reports on the relationship between Wnt5a and ERK-pathway activation in the setting of airway inflammation. The present efforts are the first to document that BOX5 administration downregulates phosphorylation of ERK in 16HBE cells, implicating Wnt5a as a modulator of airway inflammation that is induced by PM2.5/CSE exposure in COPD, at least in part in an ERK-dependent manner.
Limitations
Our study had certain limitations, the first being that in our time-consuming animal model, BOX5 (a Wnt5a-derived N-butyloxycarbonyl hexapeptide [Met-Asp-Gly-Cys-Glu-Leu]) was not administered to confirm the effects of Wnt5a in animals.54 However, this is precisely our next pursuit. In addition, primary bronchial epithelial cells should ideally be used for experimental in vitro studies. Because of technical limitations and the reality of growth retardation for primary cells in culture, we used the human bronchial epithelial cell line 16HBE, and cannot exclude the possibility of differences due to altered sensitivity.
Conclusion
In the course of this study, we used mice and human cells to confirm the role of the Wnt5a–ERK pathway in CSE-induced airway inflammation aggravated by PM2.5 exposure. PM2.5 appears to enhance Wnt5a expression, which positively regulates secretion of inflammatory cytokines via ERK-pathway activation. Moreover, BOX5 (a Wnt5a antagonist) efficiently attenuated airway inflammation due to PM2.5/CSE through Wnt5a–ERK signaling blockade (Figure S5). As such, the present study has fully elaborated the relationship between PM2.5, smoking, and airway inflammation in the course of COPD. By exploring the regulatory potential of BOX5 in terms of the Wnt5a–ERK pathway, novel therapeutic strategies may emerge to combat the inflammation of COPD at a molecular level.
Abbreviations
PM2.5, environmental particulate matter ≤2.5 μm in diameter; CSE, cigarette smoke extract; 16HBE cells, 16 human bronchial epithelial cells; CCK-8, cell counting kit; PCNA, proliferating cell nuclear antigen; qRT-PCR, quantitative reverse transcription-PCR; P-ERK1/2, phosphorylation of ERK1/2; BALF, bronchoalveolar lavage fluid; HRP, horseradish peroxidase.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (81570033, 81570047, 81370145, and 81370156), National Key Basic Research and Development Program (973 Program, 20l5CB553403), Chinese Medical Association Research Project (2013BAI09B00), National Key Technologies R&D Program (2016YFC1303900 and 2016YFC1304700), and National Key Research and Development Program in China (2016YFC0903600).
Disclosure
The authors report no conflicts of interest in this work.
References
Cazzola M, Donner CF, Hanania NA. One hundred years of chronic obstructive pulmonary disease (COPD). Respir Med. 2007;101(6):1049–1065. doi:10.1016/j.rmed.2007.01.015 | ||
Wang Y, Xu J, Meng Y, et al. Role of inflammatory cells in airway remodeling in COPD. Int J Chron Obstruct Pulmon Dis. 2018;13:3341–3348. doi:10.2147/COPD.S176122 | ||
Zhu B, Wang Y, Ming J, et al. Disease burden of COPD in China: a systematic review. Int J Chron Obstruct Pulmon Dis. 2018;13:1353–1364. doi:10.2147/COPD.S161555 | ||
Lerong C, Yuan X, Zou L, et al. Effects of 1,25-Dihydroxyvitamin D3 on the prevention of chronic obstructive pulmonary disease (COPD) in rats exposed to air pollutant particles less than 2.5 micrometers in diameter (PM2.5). Med Sci Monit. 2018;24:356–362. | ||
Woodruff PG, Agusti A, Roche N, et al. Current concepts in targeting chronic obstructive pulmonary disease pharmacotherapy: making progress towards personalised management. Lancet. 2015;385(9979):1789–1798. doi:10.1016/S0140-6736(15)60693-6 | ||
Kankaanranta H, Harju T, Kilpeläinen M, et al. Diagnosis and pharmacotherapy of stable chronic obstructive pulmonary disease: the finnish guidelines. Basic Clin Pharmacol Toxicol. 2015;116(4):291–307. doi:10.1111/bcpt.12366 | ||
Kammerl IE, Dann A, Mossina A, et al. Impairment of immunoproteasome function by cigarette smoke and in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2016;193(11):1230–1241. doi:10.1164/rccm.201506-1122OC | ||
DeVries R, Kriebel D, Sama S. Outdoor air pollution and COPD-related emergency department visits, hospital admissions, and mortality: a meta-analysis. COPD. 2017;14(1):113–121. doi:10.1080/15412555.2016.1216956 | ||
Cui G, Zhang Z, Lau AKH, et al. Effect of long-term exposure to fine particulate matter on lung function decline and risk of chronic obstructive pulmonary disease in Taiwan: a longitudinal, cohort study. Lancet Planet Health. 2018;2(3):e114-e125. doi:10.1016/S2542-5196(18)30028-7 | ||
Audi C, Baïz N, Maesano CN, et al. Serum cytokine levels related to exposure to volatile organic compounds and PM2.5 in dwellings and workplaces in French farmers – a mechanism to explain nonsmoking COPD. Int J Chron Obstruct Pulmon Dis. 2017;12:1363–1374. doi:10.2147/COPD.S117866 | ||
Cortez-Lugo M, Ramirez-Aguilar M, Perez-Padilla R, et al. Effect of Personal Exposure to PM2.5 on Respiratory Health in a Mexican Panel of Patients with COPD. Int J Environ Res Public Health. 2015;12(9):10635–10647. doi:10.3390/ijerph120910635` | ||
Leclercq B, Platel A, Antherieu S, et al. Genetic and epigenetic alterations in normal and sensitive COPD-diseased human bronchial epithelial cells repeatedly exposed to air pollution-derived PM2.5. Environ Pollut. 2017;230:163–177. doi:10.1016/j.envpol.2017.06.028 | ||
Montoya-Estrada A, Torres-Ramos YD, Flores-Pliego A, et al. Urban PM2.5 activates GAPDH and induces RBC damage in COPD patients. Front Biosci. 2013;5:638–649. doi:10.2741/S396 | ||
Strak M, Janssen N, Beelen R, et al. Associations between lifestyle and air pollution exposure: potential for confounding in large administrative data cohorts. Environ Res. 2017;156:364–373. doi:10.1016/j.envres.2017.03.050 | ||
Gu XY, Chu X, Zeng XL, et al. Effects of PM2.5 exposure on the Notch signaling pathway and immune imbalance in chronic obstructive pulmonary disease. Environ Pollut. 2017;226:163–173. doi:10.1016/j.envpol.2017.03.070 | ||
Zhang K, Guo L, Wei Q, et al. COPD rat model is more susceptible to cold stress and PM2.5 exposure and the underlying mechanism. Environ Pollut. 2018;241:26–34. doi:10.1016/j.envpol.2018.05.034 | ||
Liu J, Guo L, Zhang K, et al. The probable roles of valsartan in alleviating chronic obstructive pulmonary disease following co-exposure to cold stress and fine particulate matter. Environ Toxicol Pharmacol. 2018;60:230–236. doi:10.1016/j.etap.2018.05.002 | ||
Ye X, Hong W, Hao B, et al. PM2.5 promotes human bronchial smooth muscle cell migration via the sonic hedgehog signaling pathway. Respir Res. 2018;19(1):37. doi:10.1186/s12931-017-0702-y | ||
Morrisey EE. Wnt signaling and pulmonary fibrosis. Am J Pathol. 2003;162(5):1393–1397. doi:10.1016/S0002-9440(10)64271-X | ||
Caprioli A, Villasenor A, Wylie LA, et al. Wnt4 is essential to normal mammalian lung development. Dev Biol. 2015;406(2):222–234. doi:10.1016/j.ydbio.2015.08.017 | ||
Pashirzad M, Shafiee M, Rahmani F, et al. Role of Wnt5a in the pathogenesis of inflammatory diseases. J Cell Physiol. 2017;232(7):1611–1616. doi:10.1002/jcp.25687 | ||
Shi J, Li F, Luo M, et al. Distinct roles of Wnt/beta-catenin signaling in the pathogenesis of chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Mediators Inflamm. 2017;2017:3520581. doi:10.1155/2017/3520581 | ||
Liao HD, Mao Y, Ying YG. The involvement of the laminin-integrin alpha7 beta1 signaling pathway in mechanical ventilation-induced pulmonary fibrosis. J Thorac Dis. 2017;9(10):3961–3972. doi:10.21037/jtd.2017.09.60 | ||
Shang Y, Zhu T, Lenz AG, et al. Reduced in vitro toxicity of fine particulate matter collected during the 2008 Summer Olympic Games in Beijing: the roles of chemical and biological components. Toxicol in Vitro. 2013;27(7):2084–2093. doi:10.1016/j.tiv.2013.08.004 | ||
Wang J, Li Q, Xie J, et al. Cigarette smoke inhibits BAFF expression and mucosal immunoglobulin A responses in the lung during influenza virus infection. Respir Res. 2015;16:37. doi:10.1186/s12931-015-0201-y | ||
Ezegbunam W, Foronjy R. Posttranscriptional control of airway inflammation. Wiley Interdiscip Rev RNA. 2018;9(1):e1455. doi:10.1002/wrna.1455 | ||
Wu YP, Cao C, Wu YF, et al. Activating transcription factor 3 represses cigarette smoke-induced IL6 and IL8 expression via suppressing NF-kappaB activation. Toxicol Lett. 2017;270:17–24. doi:10.1016/j.toxlet.2017.02.002 | ||
de Moraes MR, Da Costa AC, Correa Kde S, et al. Interleukin-6 and interleukin-8 blood levels’ poor association with the severity and clinical profile of ex-smokers with COPD. Int J Chron Obstruct Pulmon Dis. 2014;9:735–743. doi:10.2147/COPD.S64135 | ||
Cordoba-Lanus E, Baz-Davila R, Espinoza-Jimenez A, et al. IL-8 gene variants are associated with lung function decline and multidimensional BODE index in COPD patients but not with disease susceptibility: a validation study. COPD. 2015;12(1):55–61. doi:10.3109/15412555.2014.908831 | ||
Bucher H, Duechs MJ, Tilp C, et al. Tiotropium attenuates virus-induced pulmonary inflammation in cigarette smoke-exposed mice. J Pharmacol Exp Ther. 2016;357(3):606–618. doi:10.1124/jpet.116.232009 | ||
Lee JS, Park SJ, Cho YS, et al. Role of AMP-Activated Protein Kinase (AMPK) in smoking-induced lung inflammation and emphysema. Tuberc Respir Dis (Seoul). 2015;78(1):8–17. doi:10.4046/trd.2015.78.1.8 | ||
Heinrich J, Schikowski T. COPD patients as vulnerable subpopulation for exposure to ambient air pollution. Curr Environ Health Rep. 2018;5(1):70–76. doi:10.1007/s40572-018-0178-z | ||
Gilmour PS, Schladweiler MC, Richards JH, et al. Hypertensive rats are susceptible to TLR4-mediated signaling following exposure to combustion source particulate matter. Inhal Toxicol. 2004;16(Suppl 1):5–18. doi:10.1080/08958370490442827 | ||
Luo B, Shi H, Wang L, et al. Rat lung response to PM2.5 exposure under different cold stresses. Int J Environ Res Public Health. 2014;11(12):12915–12926. doi:10.3390/ijerph111212915 | ||
van Berlo D, Hullmann M, Schins RP. Toxicology of ambient particulate matter. Exp Suppl. 2012;101:165–217. doi:10.1007/978-3-7643-8340-4_7 | ||
Li R, Kou X, Xie L, et al. Effects of ambient PM2.5 on pathological injury, inflammation, oxidative stress, metabolic enzyme activity, and expression of c-fos and c-jun in lungs of rats. Environ Sci Pollut Res Int. 2015;22(24):20167–20176. doi:10.1007/s11356-015-5222-z | ||
Riva DR, Magalhaes CB, Lopes AA, et al. Low dose of fine particulate matter (PM2.5) can induce acute oxidative stress, inflammation and pulmonary impairment in healthy mice. Inhal Toxicol. 2011;23(5):257–267. doi:10.3109/08958378.2011.566290 | ||
Kumawat K, Gosens R. WNT-5A: signaling and functions in health and disease. Cell Mol Life Sci. 2016;73(3):567–587. doi:10.1007/s00018-015-2076-y | ||
Li C, Xiao J, Hormi K, et al. Wnt5a participates in distal lung morphogenesis. Dev Biol. 2002;248(1):68–81. | ||
Yang J, Zhang K, Wu J, et al. Wnt5a increases properties of lung cancer stem cells and resistance to cisplatin through activation of Wnt5a/PKC signaling pathway. Stem Cells Int. 2016;2016:1690896. doi:10.1155/2016/1243659 | ||
Zhu Z, Yin S, Wu K, et al. Downregulation of Sfrp5 in insulin resistant rats promotes macrophage-mediated pulmonary inflammation through activation of Wnt5a/JNK1 signaling. Biochem Biophys Res Commun. 2018;505(2):498–504. doi:10.1016/j.bbrc.2018.09.070 | ||
Malgor R, Bhatt PM, Connolly BA, et al. Wnt5a, TLR2 and TLR4 are elevated in advanced human atherosclerotic lesions. Inflamm Res. 2014;63(4):277–285. doi:10.1007/s00011-013-0697-x | ||
MacLauchlan S, Zuriaga MA, Fuster JJ, et al. Genetic deficiency of Wnt5a diminishes disease severity in a murine model of rheumatoid arthritis. Arthritis Res Ther. 2017;19(1):166. doi:10.1186/s13075-017-1375-0 | ||
Koopmans T, Kumawat K, Halayko AJ, Gosens R. Regulation of actin dynamics by WNT-5A: implications for human airway smooth muscle contraction. Sci Rep. 2016;6:30676. doi:10.1038/srep30676 | ||
Baarsma HA, Skronska-Wasek W, Mutze K, et al. Noncanonical WNT-5A signaling impairs endogenous lung repair in COPD. J Exp Med. 2017;214(1):143–163. doi:10.1084/jem.20160675 | ||
Feller D, Kun J, Ruzsics I, et al. Cigarette smoke-induced pulmonary inflammation becomes systemic by circulating extracellular vesicles containing Wnt5a and inflammatory cytokines. Front Immunol. 2018;9:1724. doi:10.3389/fimmu.2018.01724 | ||
Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92(2):689–737. doi:10.1152/physrev.00028.2011 | ||
Wang H, Yang T, Wang T, et al. Phloretin attenuates mucus hypersecretion and airway inflammation induced by cigarette smoke. Int Immunopharmacol. 2018;55:112–119. doi:10.1016/j.intimp.2017.12.009 | ||
Zeng Z, Li M, Chen J, et al. Reduced MBD2 expression enhances airway inflammation in bronchial epithelium in COPD. Int J Chron Obstruct Pulmon Dis. 2018;13:703–715. doi:10.2147/COPD.S148595 | ||
Blanchet S, Ramgolam K, Baulig A, Marano F, Baeza-Squiban A. Fine particulate matter induces amphiregulin secretion by bronchial epithelial cells. Am J Respir Cell Mol Biol. 2004;30(4):421–427. doi:10.1165/rcmb.2003-0281RC | ||
Zhang Y, Du J, Zheng J, et al. EGF-reduced Wnt5a transcription induces epithelial-mesenchymal transition via Arf6-ERK signaling in gastric cancer cells. Oncotarget. 2015;6(9):7244–7261. doi:10.18632/oncotarget.3133 | ||
Abraityte A, Vinge LE, Askevold ET, et al. Wnt5a is elevated in heart failure and affects cardiac fibroblast function. J Mol Med. 2017;95(7):767–777. doi:10.1007/s00109-017-1529-1 | ||
Zhao Y, Wang CL, Li RM, et al. Wnt5a promotes inflammatory responses via nuclear factor kappaB (NF-kappaB) and mitogen-activated protein kinase (MAPK) pathways in human dental pulp cells. J Biol Chem. 2014;289(30):21028–21039. doi:10.1074/jbc.M113.546523 | ||
Jenei V, Sherwood V, Howlin J, et al. A t-butyloxycarbonyl-modified Wnt5a-derived hexapeptide functions as a potent antagonist of Wnt5a-dependent melanoma cell invasion. Proc Natl Acad Sci U S A. 2009;106(46):19473–19478. doi:10.1073/pnas.0909409106. |
Supplementary materials
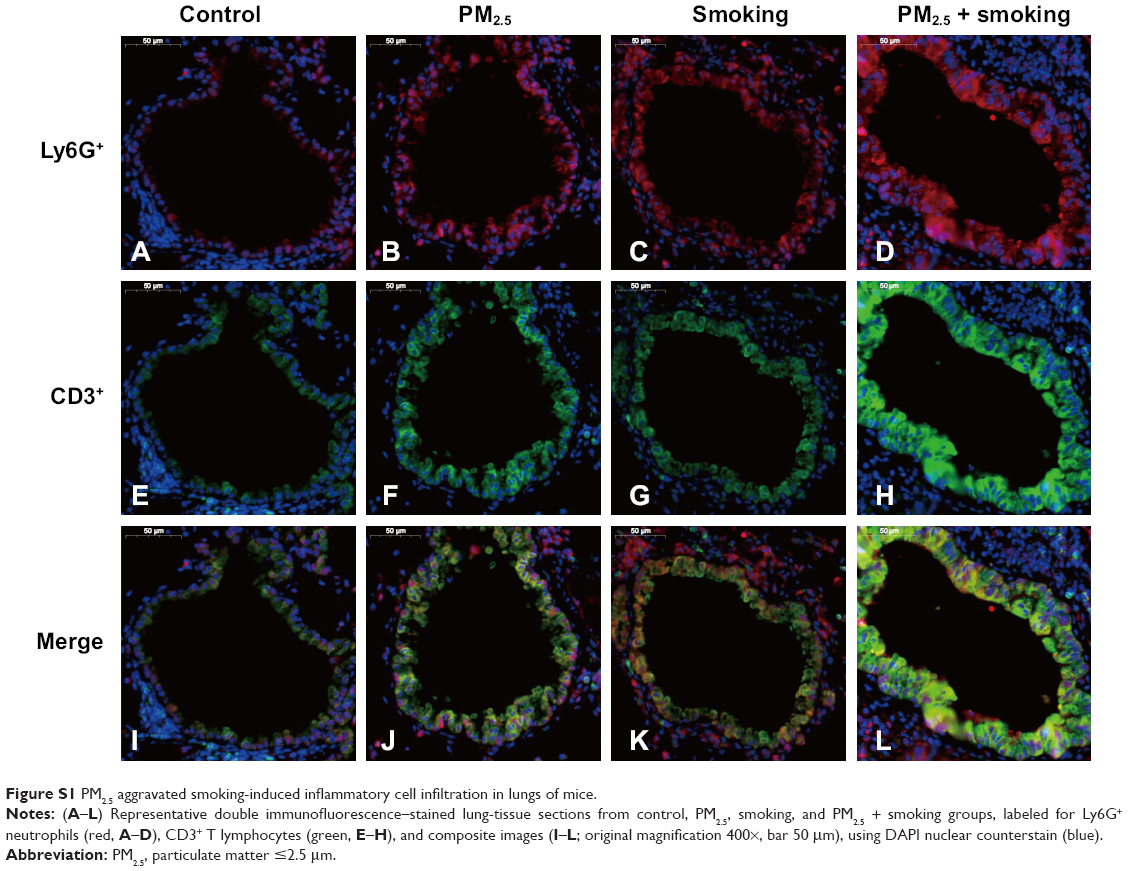 | Figure S1 PM2.5 aggravated smoking-induced inflammatory cell infiltration in lungs of mice. |
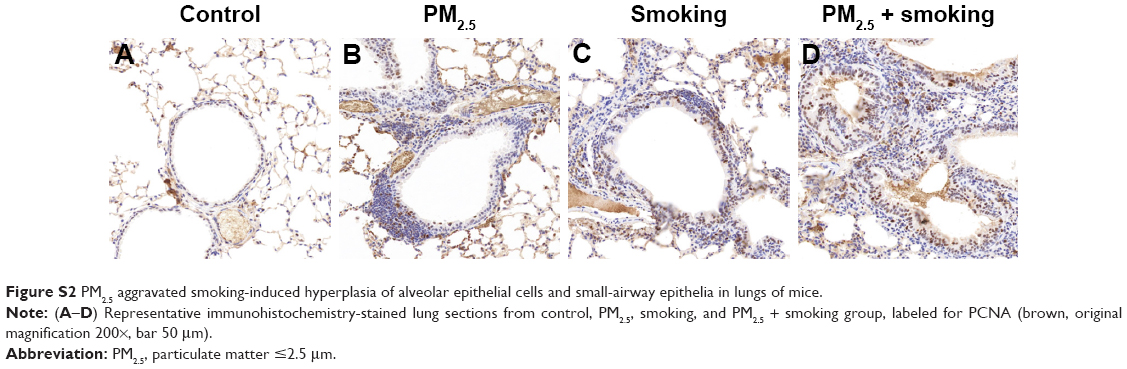 | Figure S2 PM2.5 aggravated smoking-induced hyperplasia of alveolar epithelial cells and small-airway epithelia in lungs of mice. |
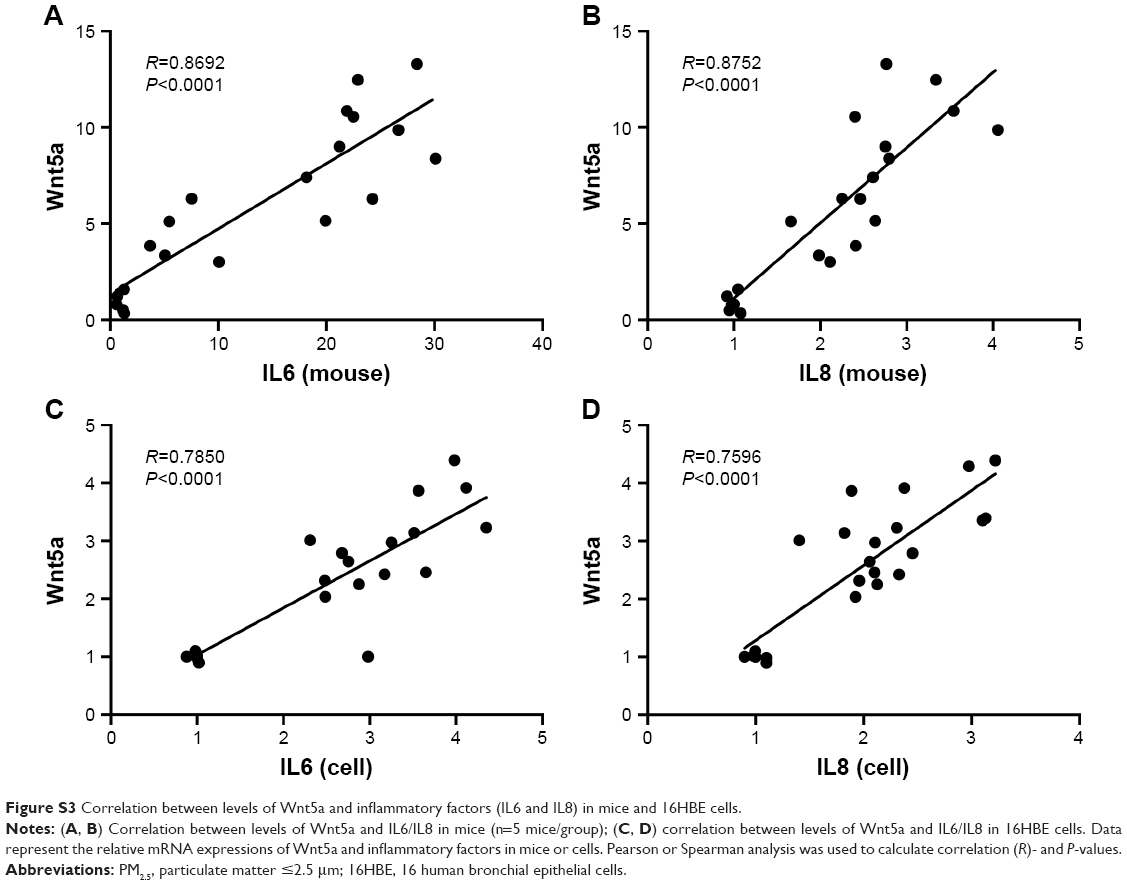 | Figure S3 Correlation between levels of Wnt5a and inflammatory factors (IL6 and IL8) in mice and 16HBE cells. |
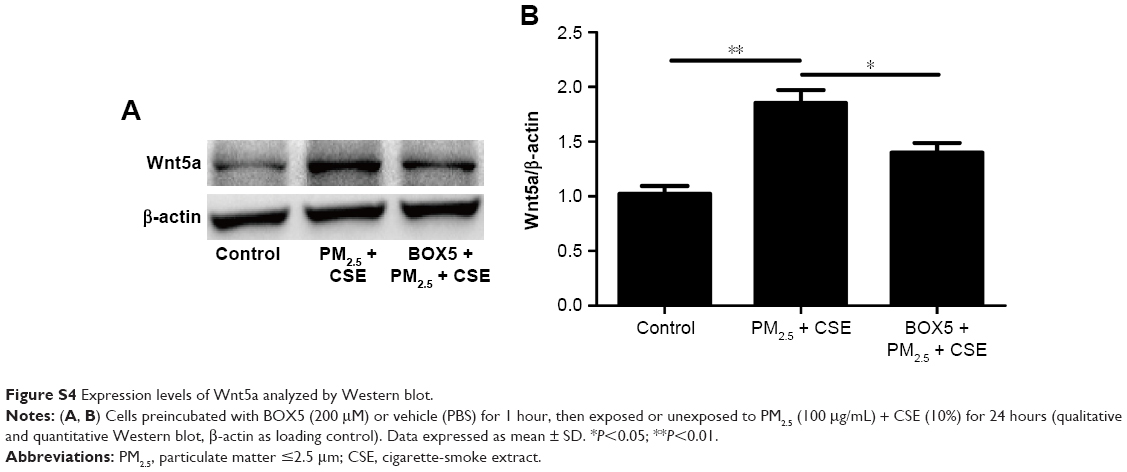 | Figure S4 Expression levels of Wnt5a analyzed by Western blot. |
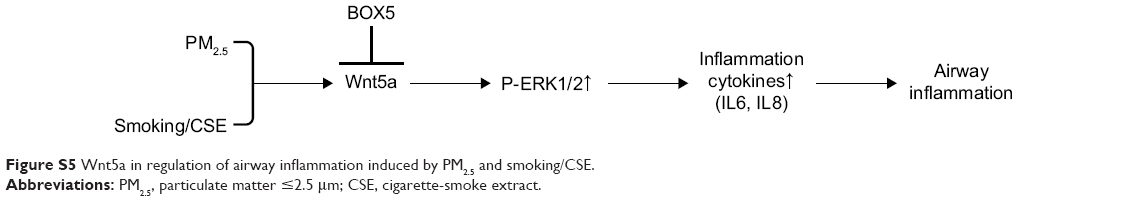 | Figure S5 Wnt5a in regulation of airway inflammation induced by PM2.5 and smoking/CSE. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.