Back to Journals » Journal of Inflammation Research » Volume 16
Fibroblast Insights into the Pathogenesis of Ankylosing Spondylitis
Authors Liu Z, Cai M, Ke H, Deng H ![]() , Ye W, Wang T, Chen Q, Cen S
, Ye W, Wang T, Chen Q, Cen S
Received 7 October 2023
Accepted for publication 3 December 2023
Published 22 December 2023 Volume 2023:16 Pages 6301—6317
DOI https://doi.org/10.2147/JIR.S439604
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tara Strutt
Zhenhua Liu,1,* Mingxi Cai,2,* Haoteng Ke,2,* Huazong Deng,2 Weijia Ye,2 Tao Wang,1 Qifan Chen,1 Shuizhong Cen1
1Department of Spinal Surgery, Orthopedic Medical Center, Zhujiang Hospital, Southern Medical University, Guangzhou, 510280, People’s Republic of China; 2The Second Clinical School, Zhujiang Hospital, Southern Medical University, Guangzhou, 510280, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shuizhong Cen, Department of Spinal Surgery, Orthopedic Medical Center, Zhujiang Hospital, Southern Medical University, Guangzhou, People’s Republic of China, Tel +86 13480297160, Email [email protected]
Purpose of the Review: Emerging evidence has shown that ankylosing spondylitis fibroblasts (ASFs) act as crucial participants in inflammation and abnormal ossification in ankylosing spondylitis (AS). This review examines the investigations into ASFs and their pathological behavior, which contributes to inflammatory microenvironments and abnormal bone formation. The review spans the period from 2000 to 2023, with a primary focus on the most recent decade. Additionally, the review provides an in-depth discussion on studies on ASF ossification at the cellular level.
Recent Findings: ASFs organize immune functions by recruiting immune cells and influencing their differentiation and activation, thus mediate the inflammatory response in the early phase of disease. ASFs promote joint destruction at sites of cartilage and actively promote abnormal ossification by recruiting osteoblasts, differentiation into myofibroblasts or ossification directly. Many signaling pathways and cytokines such as Wnt signaling and BMP/TGF-β signaling are involved in ASF ossification.
Summary: ASFs play a key role in AS inflammation and osteogenesis. Further studies are required to elucidate molecular mechanisms behind that and provide new targets and directions for AS diagnosis and treatment from a new perspective of fibroblasts.
Keywords: ankylosing spondylitis, fibroblast, inflammation, ossification
Introduction
Ankylosing spondylitis (AS), a chronic inflammatory arthritis that mainly affects the spine and sacroiliac joints, leads to structural and functional impairments and progressive ankylosis of the axial skeleton.1 As a special inflammatory disease, inflammation in AS has a widespread influence. Axial inflammation, including sacroiliac arthritis, leads to irreversible structural damage and restrict spinal mobility. Peripheral manifestations such as arthritis in lower extremities and enthesitis at the insertion of the Achilles tendon and plantar fascia are also commonly observed.2 Moreover, patients may concurrently exhibit extra-musculoskeletal manifestations, specifically uveitis, Inflammatory Bowel Disease (IBD), and psoriasis.3 And unlike other systemic autoimmune diseases, AS is characterized by a dominant role of the innate immune system. This is marked by aberrant activity of innate and innate-like immune cells, which leads to their unique inflammatory conditions.4 Then, as MRI results showed, abnormal bone formation is more likely to occur in sites with previous inflammation.5 When osteophytes span the entire joint cavity, this results in the immobilization of the affected joint, a condition known as bony ankylosis. This can potentially lead to restricted spinal motion and even result in permanent disability.6 The clinical criteria for AS include Sacroiliitis on imaging plus one or more spondyloarthritis (SpA) features. Or, being HLA-B27 positive plus two or more other SpA features. The histologic SpA features of AS always include enthesis, sacroiliitis, syndesmophyte formation, ectopic ossification, and so on, all of which contribute to the progression of the disease.7 Intriguingly, fibroblasts were frequently observed among the above lesions, participating in early sacroiliitis,8,9 mediating the invasion of the subchondral bone,8,10,11 and even contributing to adipocyte accumulation.12 This suggests that ankylosing spondylitis fibroblasts (ASFs) may play a crucial role in the etiology of AS. In vitro, the feature displayed in ASF culture also supports this and relates ASF to structural damage and bony ankylosis in AS.13–16
Fibroblasts exist in all organizations of the body and maintain the structural integrity and tissue health of connective tissue by secreting collagen and extracellular matrix. In recent years, an increasing number of researchers have realized that fibroblasts are involved in inflammation, tissue damage, bone erosion, and the destruction of articular cartilage in autoimmune diseases. For example, fibroblasts are considered to be a reservoir that can provide specialized activated fibroblasts that lead to a pathological process in RA,17 which would lead to inflammatory and joint erosion by forming pannus and other factors.18 Similarly, fibroblasts, as possible target cells in the pathology of AS, have attracted increasing attention from many researchers in recent years, while there have been no systematic reviews on ASFs to lead to further research.
Thus, to obtain a further understanding of the special inflammation conditions and bony ankylosis in AS, we focus on the recent investigation of ASFs and their pathological behaviour that contribute to inflammatory microenvironments and abnormal bone formation. Additionally, the review provides an in-depth discussion on studies on ASF ossification at the cellular level.
Fibroblasts in AS
Previously, fibroblasts were considered to be a homogeneous cell population, while emerging evidence indicates that fibroblasts can serve as a reservoir that can provide tissue-specialized fibroblasts and pathological fibroblasts in disease.17 In AS, with THY1 (also known as CD90),19 vimentin,20 alpha smooth muscle actin, prolyl 4-hydroxylase β,21 S100A4, etc., used as markers in recent studies, ASFs were reported to mainly regulate the inflammatory environment and mediate abnormal ossification.22
First, similar to rheumatoid arthritis synovial fibroblasts (RASFs) in RA, ASFs can cause certain pathological responses in diseases by regulating factor secretion, coordinating inflammatory responses, regulating tissue homeostasis, and mediating joint remodeling. Currently, fibroblast research in RA is well-established,23 with key findings in fibroblast subpopulations, fibroblast interactions,18 and fibroblast-targeted therapies,24,25 among others. AS and RA are autoimmune diseases with significant differences but certain similarities, especially in inflammation. Leveraging the successful practices from RA research may help us elucidate the specific mechanisms of ASFs, particularly in the context of AS inflammation.
As both the major cells in joint remodeling, osteoclasts are terminally differentiated myeloid with distinct morphological and phenotypic characteristics,26 while osteoblasts differentiated from mesenchymal stromal cells (MSCs).27 Though with significant difference in differentiation, cellular communication between them is essential for bone remodeling, which maintains bone homeostasis.28 Meanwhile, fibroblasts derived from the same source of bone marrow MSCs as osteoblasts,29 with considerable evidence showing significant expression of osteogenic markers, were observed in ASFs, suggesting that ASFs may also play a key role in the abnormal bone proliferation of AS.
According to the series of studies that are discussed in this review, it is clear that fibroblasts are inextricably linked to the different stages of structural damage progress in AS: inflammation → erosive destruction → syndesmophyte formation→ ankylosis.11
Histological Evidence of ASFs
Emerging histological evidence has revealed that ASFs from subchondral bone marrow may play a role in the aetiology of AS by recruiting immune cells to regulate the inflammatory environment and bone metabolism in joint remodeling (Figure 1).
 |
Figure 1 ASFs in AS joint remodeling. ASFs promote joint destruction at sites of cartilage and actively promote abnormal ossification by recruiting osteoblasts, differentiation into myofibroblasts or ossification directly. (a) ASFs affect osteoclasts to promote bone erosion. ASFs recruit a cohort of osteoclasts towards the joint surface and secret RANKL and inflammatory cytokines to induce the function of osteoclasts with the effect of Th17. The inflammatory environment will be the key to lead to this phenomenon. (b) ASFs affect osteoblasts to promote bone formation. Evidence suggested that ASFs recruit osteoblasts into proximity of the remaining cartilage islands and lead to bone formation. The overexpression of BMP in ASFs will facilitate the cellular differentiation of osteoblasts. (c) ASFs contribute to fat enrichment. Evidence shows that the lipid accumulation in ASF-rich tissue occasionally in the form of spindle-like–shaped lipid accumulation directly within the ASFs. ASFs also activate as myofibroblasts and differentiate into adipocyte induced by BMPs. (d) ASFs activate as myofibroblasts to form collagen-rich extracellular matrix independent of inflammation, which contribute to abnormal bone formation. (e) Direct transformation of ASFs into new bone. Considerable cell-level studies suggest that ASFs have the potential for osteogenic differentiation themselves with some signaling pathways and cytokines involved. Created with Biorender.com. |
ASFs Mediate the Inflammatory Response
In earlier disease stages of AS, morning stiffness and inflammatory back pain are major symptoms. Radiographs of the sacroiliac joints could appear normal in the early phase of disease many years before structural changes become apparent.1 In this disease state, fibroblasts have been reported to act as dynamic participants in immune processes.30
Emerging histological evidence has shown that subchondral ASF-rich tissue is infiltrated with inflammatory cells in early sacroiliitis without definite structural damage.8,9 The inflammatory cells reported include CD45+ lymphocytes that seem to invade a degenerate cartilaginous area31 and cell populations of CD4 and CD8 T cells that infiltrate the cartilage from the subchondral bone marrow along with osteoclast-mediated resorption of the bone end plate. Remarkably, it seems that the infiltration of subchondral T-cell substantially declined simultaneously with the cartilage destruction during disease progression, which suggests its correlation with the existence of cartilage at the joint surface.
This result indicated that ASFs’ potential to enhance the bone resorption of osteoclasts by participating in pro-inflammatory milieu formation through secreting inflammatory factors such as Tumour Necrosis Factor (TNF),32 a mechanism that might resemble animal models of arthritis in which mice overexpressing TNF develop destructive arthritis caused by activated osteoclasts.33 Based on clinical trials, anti-TNF therapy has been proven to improve clinical symptoms in AS.34 So far adalimumab, etanercept, golimumab, and infliximab have been used for the treatment of AS,35 and among them Infliximab showed a relatively better effect.36 However, there remains a subset of patients who do not respond to this treatment.37 Additionally, a significant proportion of patients experience a reduction in efficacy after an initial period of response, intolerance, or adverse reactions, and might need a switch to an alternative treatment regimen.38 Therefore, in recent years, researchers have been making continuous efforts to improve and try other targets such as IL-1739 and Janus kinase (JAK).40 By and large, a series of further researches in ASFs involved TNF secretion may provide a fibroblast new insight into AS anti-TNF treatment.
Appel et al observed a significantly higher number of Th17 cells present in the bone marrow of subchondral ASF-rich tissue-affected facet joints.41 In the past few years, the cytokine Interleukin-17A (IL-17A) has been shown to play an important role in the pathogenesis of human chronic inflammatory diseases. Based on clinical trials performed with IL-17 blockade in AS, IL-17 inhibition has been supported to be an effective treatment for AS.42 Inflammatory factors such as TNFα, IL-1 and IL-6 synergize with IL-17 to induce further proinflammatory cytokine and chemokine secretion by fibroblasts, amplifying inflammatory reactions and increasing the aggregation of inflammatory cells.18,43 These effects have been proven in RA,18 and we suggest determining the exact relationship among ASFs, IL-17 and inflammation. In addition, IL-17 is thought to enhance the function of osteoclasts via ASFs, which will be discussed below.
Dense CD163+ macrophages were also reported to infiltrate subchondral ASF-rich tissue.9,32,44 However, in AS patients of long-standing, bone marrow macrophages appear to have a minor effect on inflammation or repair due to the fact that the number of bone marrow macrophages shows no significant differences with non-AS controls.45
In addition, there are CD20+ B-cell infiltrates in subchondral ASF-rich tissue, especially in AS patients with persistent inflammation in the spine.45 B-cells have been overlooked in the discussion of AS pathogenesis for so long, mainly due to the fact that autoantibodies seem to have a minor role in AS. However, the resistance of B cells after anti–tumour necrosis factor α (anti-TNFα) therapy46 might explain why relapses occur soon after TNF-α therapy has been discontinued. In addition, a few small clinical trials used rituximab in AS and achieved successful treatment response,47 again pointing to a possible role of B cells in the immunopathology of the disease.45 An inhibition of B cell antigen-presenting function might be a possible explanation. It is reported that B cells are able to mediate osteoclastogenesis through regulating the level of osteoprotegerin (OPG) and receptor activator of nuclear factor-κB ligand (RANKL),48 which could be a possible mechanism to relate B cells and ASFs.
Fibroblasts are now thought to organize immune functions by recruiting immune cells and influencing their differentiation and activation. Specific mechanisms may include cytokine and chemokine secretion and behavioural modulation through extracellular matrix remodeling. In addition, the transfer of extracellular matrix microenvironments was recently described.30 The research of Cambré et al described another interesting mechanism: it showed that biomechanical forces in joints would cause fibroblasts to secrete C–C motif ligand 2 (CCL2) among other chemokines responsible for the attraction of inflammatory monocytes towards biomechanically exposed sites and lead to the differentiation of these cells into osteoclasts.49
In rheumatoid arthritis (RA), the relationship between RASFs and inflammation has been reviewed in detail. In RA, the destructive properties of RASFs can be activated by IL-6, TNF-α and IL-1b, which are elevated in AS inflammation as well. The activated RASFs then further amplify the inflammatory cycle by producing inflammatory factors themselves.23,50 Furthermore, RASFs can enhance their activation and cytokine release through direct contact with inflammatory cells, such as T cells and macrophages. The factors above, in the meantime, recruit, activate, and promote the differentiation of other, engaged in the inflammatory cycle in RA, cell types.18 Given the similar relationship between inflammation and fibroblasts in AS and RA, studies on RA may support us in determining the specific mechanism by which ASFs relate to inflammation.
With the subchondral ASF-rich tissue forming up with infiltrated immune cells, a high density of CD34+ microvessels was also observed in AS subchondral bone. It was reported to increase only when there was still cartilage at the surface of the femoral heads.32 Growth factors such as vascular endothelial growth factor (VEGF) released by ASFs may support angiogenesis, which is supposed to allow inflammatory factors and immune cells to access and participate in the AS inflammatory environment and even lead to bone erosion.8,51,52 Another report suggested that microvascular aggregation was observed not only near lymphocytic infiltration but also at sites of new bone formation, where osteoblasts lined the trabecular bone.45 It implied that the formation of microvessels had a different role in AS, beyond just contributing to inflammation. This role could be pivotal for bony ankylosis in AS, potentially due to increased nutritional needs. However, the exact mechanism behind this process still necessitates further exploration (Figure 2).
 |
Figure 2 ASFs mediate the inflammatory response. Subchondral ASF-rich tissue is infiltrated with inflammatory cells in the early phase of disease. Vascular endothelial growth factor (VEGF) `released by ASFs may support angiogenesis, which is supposed to allow inflammatory factors and immune cells to access and participate in the inflammatory environment. Biomechanical forces in joints would cause fibroblasts to secrete CCL2 among other chemokines responsible for the attraction of inflammatory monocytes towards biomechanically exposed sites and lead to the differentiation of these cells into osteoclasts. Inflammatory factors may enhance the destructive properties of ASFs, including tumour necrosis factor (TNF)-α, IL-6 and IL-1b. In turn, activated ASFs further enhance the inflammatory cycle by producing inflammatory factors themselves. Moreover, cell–cell contact between ASFs and inflammatory cells enhances the activation of ASFs, increasing the release of inflammatory cytokines. These factors, in turn, recruit, activate and promote the differentiation of multiple cell types that contribute to the inflammatory cycle, which may promote the inflammatory response. Created with Biorender.com. |
ASFs Promote Joint Remodeling
According to recent studies discussed in this review, it is clear that fibroblasts are inextricably linked to the different stages of structural damage progress in AS:
1. Inflammation: The disease begins with inflammation, typically in the sacroiliac joints and spreads to the spine in most patients. 2. Erosive destruction: These refer to the wearing away of the bone, which are considered minor signs of progression. 3. Syndesmophyte formation: syndesmophytes are bony growths inside the ligaments of the spine. They are a characteristic feature of AS and can lead to reduced physical function and quality of life.53 4. Ankylosis: This is the final stage where the vertebrae fuse together due to the growth of syndesmophytes, leading to a rigid and inflexible spine.11 Unlike Ossification of the Posterior Longitudinal Ligament (OPLL), whose most common location happens in the cervical spine region and can compress the spinal cord and cause neurological deficits,54,55 symptoms like pain and stiffness caused by AS always start from the lower back. Radiographs of sacroiliac arthritis, one of the most common causes of low back pain, are the hallmarks of AS structural damage progress, which is presented as ASF-related subchondral sclerosis, uniform joint space narrowing and erosions in early and ankylosis in later progress.56–58
ASFs Affect Osteoclasts to Promote Bone Erosion
In AS-mediated joint destruction, especially erosion of the subchondral bone plate of the joint, ASFs may promote osteoclast function or initiate their recruitment to promote bone erosion (Figure 1a). Some pathological studies have shown that ASFs can mediate the invasion of the subchondral bone plate by forming a cohort of osteoclasts towards the joint surface.8,10,11 In monocyte lineage, osteoclasts express receptor activator of nuclear factor (NF)-κB (RANK), whereas cells of the mesenchymal lineage, such as osteoblasts, fibroblasts, and synoviocytes, express RANKL.59 As a key mediator of osteoclast formation, RANKL directly induces osteoclast development and bone resorption when binding to RANK.60,61 The mechanism by which ASFs mediate osteoclast enhancement may be the secretion of RANKL as mesenchymal cells, which enhances osteoclast function.42
Based on histological evidence, the presence of osteoclasts seems to be related to inflammation since they always increase and then decrease simultaneously.11,32,62 Moreover, studies have shown that inflammatory cytokines such as TNF, interleukin-1 (IL-1), and IL-17 stimulate RANKL expression on mesenchymal cells while also increasing RANK’s action on osteoclasts by CD40 ligand, as well as by Toll-like receptor 2 (TLR-2) and TLR-4 ligands in vitro, thus inducing bone destruction and causing a downregulation of osteoblast function.63–65 Both of the above results led us to the conclusion that inflammation preceding bone formation would enhance osteoclast function by upregulating RANKL secretion. However, the AS expression of RANKL, osteoprotegerin (OPG), and RANK in another study appeared to be largely independent of the levels of systemic and local inflammation.65 Results that seemed contradictory require further study.
ASFs Affect Osteoblasts to Promote Bone Formation
Depending on some evidence, ASFs with osteoclasts towards the joint surface might bring osteoblasts closer to the cartilage by creating a channel through the subchondral bone plate. It is noteworthy that this hypothesis corresponds to the irregular ankylosis observed on radiographs of AS joints (Figure 1b). This suggests that the remaining cartilage islands in joints with broken cartilage are transformed into bone by the invading groups of ASFs and other functional cells mediated by them.9,12 However, the specific mechanism is still unclear, although the possibility is that TNF and IL-17 can cause bony proliferation when there is no contact between osteoblasts and osteoclasts by upregulating receptor activator of nuclear factor-κ B Ligand (RANKL) secretion of mesenchymal lineages, such as osteoblasts, fibroblasts, and synoviocytes.42,63 In addition, osteogenic differentiation characteristics that have been proven to be overexpressed by ASFs, such as bone morphogenetic protein (BMP), may facilitate cellular differentiation of osteoblasts,66 and the function of Cx43 discussed in a later section may provide another potential mechanism.
Therefore, based on histological evidence and the close relationship between fibroblasts and osteoblasts,29 it would be important to understand AS aetiology to demonstrate how ASFs affect osteoblasts through specific molecular mechanisms.
ASFs Contribute to Fat Enrichment
Several MRI studies have demonstrated that “inflammation → fat deposition → new bone formation” can be observed in MRI and X-ray findings during follow-up of AS patients over time.67 An obvious correlation was found between new fat deposition and new bone formation. Once AS patients have developed fat deposits, new bone formation continues to occur more frequently in both the clinical trial and the observational cohort.68,69 Taken together, the above results suggest that fat deposition plays an important role in new bone formation in AS.
Previous studies have suggested that fat deposition is mainly composed of adipocytes. However, adipocytes alone could not explain the “increased local fat deposition but lower body fat level in AS”; this opposite phenomenon suggests that other cells may be involved in fat accumulation.70 Recent studies have shown that ASFs also play a role in fat deposition in two ways (Figure 1c).
In the first place, Bleil et al found evidence of adipocyte accumulation in ASF-rich tissue, manifested either as cells containing a large fat vacuole resembling adipocyte or as spindle-shaped direct lipid accumulation within the ASFs occasionally.12
In addition, some researchers observed the conversion of myofibroblasts to adipocytes, which could be a theoretical basis for the hypothesis that lipid accumulation occurs within the extracellular matrix or within ASFs. Interestingly, BMP signaling is necessary for this process.71 Overexpression of BMP signaling in ASFs strengthens the belief that ASFs may be involved in fat deposition.
ASFs Activate as Myofibroblasts to Form Extracellular Matrix Independent of Inflammation
Currently, the results from multiple randomized clinical trials showed that new bone formation or radiographic progression is not inhibited by the usage of various TNF-α blockers for 2–4 years,72–75 suggesting that bone formation in AS is at least partly dissociated from inflammation. The study of Yeremenko et al suggested that ASFs’ participation in bone remodeling is mostly independent from the inflammatory environment and instead seems to be caused by an intrinsic transcriptional signature.21 Their analysis of AS biopsy samples showed that a large majority of the genes that were overexpressed in AS synovium were related to muscle–myocyte–myofibroblast biology, and α-smooth muscle actin (α-SMA) showed substantial colocalization with CD146 and CD90, which are markers of myofibroblasts, pericytes, and mesenchymal stem cells, accompanied by the expression of the fibroblast markers vimentin and prolyl 4-hydroxylase β.21 In addition, the levels of Transforming Growth Factor Beta-1 (TGF-β1), which is perhaps the most impactful morphogen in mediating pathological fibroblasts in myofibroblast differentiation,76 were increased in ligamentum flava and paraspinal muscle tissues of AS patients.77 Therefore, it is natural to hypothesize that ASFs may play a role by activating myofibroblasts and hypothetically forming collagen-rich extracellular matrix adjacent to the articular bone (Figure 1d). Calcification of these extracellular matrices, when Ca2+ and Pi present at physiologic concentrations,78 might result in syndesmophyte formation and progressive ankylosis. Beyer and JHW Distler thought the findings above provide a new idea and a basic dataset to build upon in new research projects in AS. In their hypothesis, the pathogenetic feature of fibrotic diseases may relate to AS, and thus, the disease mechanisms from fibrotic disease may be translated to the pathogenesis of AS. If this hypothesis could be proven by future studies, blocking extracellular matrix formation and calcification, and thus blocking syndesmophyte formation and ankylosis in AS,78–80 may be more readily achieved by using the therapeutic toolbox of fibrotic disease to target fibroblasts.79,80 Surprisingly, Stougaard et al demonstrated that the antifibrotic drug pirfenidone could inhibit fibroblast-like synoviocyte-related osteoblast mineralization in spondyloarthritis.81
Direct Transformation of ASFs into New Bone
Progressive ankylosis, the most unique phenomenon in AS, is due to abnormal bone formation. Recent studies have shown that ASFs have the potential for osteogenic differentiation themselves, either by observation at the tissue level9 or by isolated cultures of fibroblasts from patients13–15 (Figure 1e). Osteogenic markers are markedly upregulated in ASF cultures.16 There is no doubt that there is a theoretical basis since fibroblasts are of the same origin as osteoblasts.29 In this review, we will report the results of cell-level studies in detail.
Cell-Level Studies of ASFs
Signaling Pathways Mainly Involved in ASF Ossification
Wnt/β-Catenin
Having been historically subdivided into three main branches, Wnt signaling contains the canonical Wnt pathway, the Wnt/PCP pathway, and the Wnt/Ca2+ pathway. The fact that canonical pathways are a major component of Wnt signaling in bone cells has led to a relatively clear understanding of the mechanisms by which Wnt affects bone.82 We focus on how Wnt signaling relates to the ossification of ASFs.
Some target molecules have been reported to affect ASFs via Wnt signaling (Figure 3).
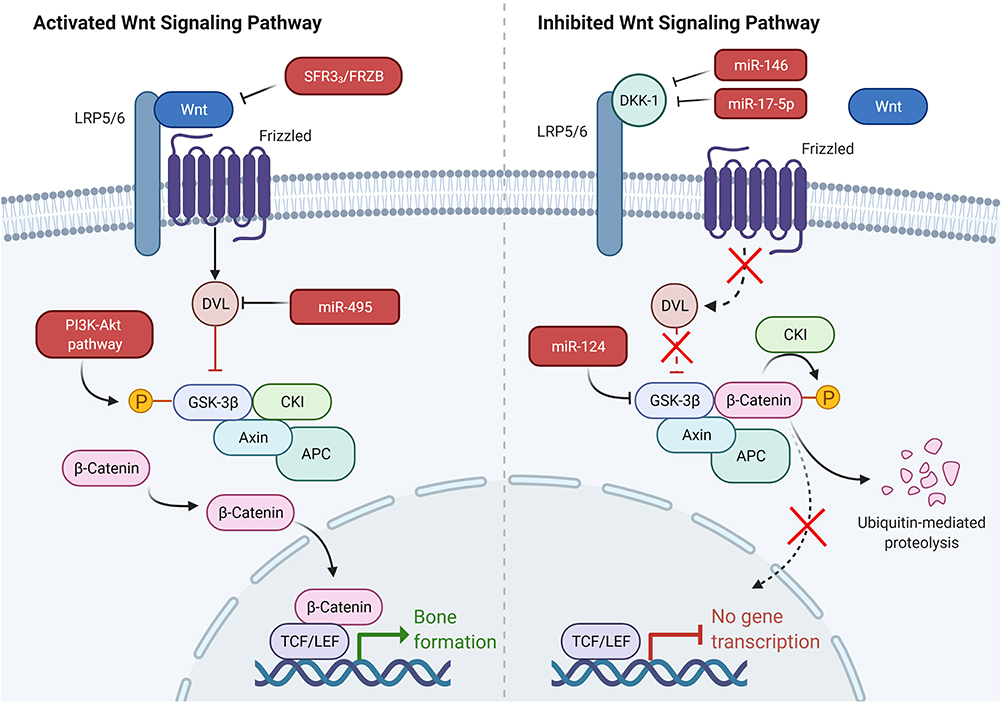 |
Figure 3 Some target molecules that affect ASFs via Wnt signaling. Downregulation of miR-124 expression enhanced GSK-3β expression, weakened Wnt/β-catenin pathway activity. PGE-2 activated the trimeric G-protein by binding to its EP2 receptor and then activated PI3-kinase, which in turn activated Akt. Then, Akt phosphorylated GSK-3β, leading to the inhibition of its phosphorylation of β-catenin. DVL-2 prevented β-catenin by titrating GSK-3β from Axin complex degradation and was identified as the target of miR-495 and highly expressed in AS. MiR-495 and si-DVL-2 upregulated the expression of β-catenin and downregulated the p-β-catenin level in synovial ASFs. Dickkopf (Dkk) family bind with high affinity to lipoprotein receptor related protein 5/6 (LRP5/6) and thereby directly prevent Wnt binding. MiR-17-5p and miR-146a affect the proliferation and osteogenic potential of ASFs by regulating DKK-1 expression. sFRP3/FRZB (members of the sFRP family) directly bind extracellular Wnt. IL-22 has the capacity to increase the expression of sFRP3/FRZB and thereby inhibit Wnt signaling. Created with Biorender.com. |
First, a recent study showed that a low expression of miR-124 enhanced GSK-3β expression and in turn weakened Wnt/β-catenin pathway activity, leading to the inhibition of osteogenic differentiation of ligament ASFs.83 In the absence of Wnt, the destruction complex containing Gsk-3β phosphorylated β-catenin, leading to the proteasomal degradation of β-catenin. In addition, PGE-2 activated the trimetric G-protein by binding to its EP2 receptor and then activated PI3-kinase, which in turn activated Akt. Then, Akt phosphorylated GSK-3β, leading to the inhibition of its phosphorylation of β-catenin. Based on this, celastrol (an active compound isolated from Tripterygium wilfordii) was found to inhibit PGE-2-induced osteogenic differentiation of ASFs in vitro.84 Second, DVL-2 prevented β-catenin by titrating GSK-3β from Axin complex degradation and was identified as the target of miR-495 and highly expressed in AS. Du et al found that miR-495 and si-DVL-2 upregulated the expression of β-catenin and downregulated the p-β-catenin level in synovial ASFs.85 Third, the canonical Wnt signaling pathway can be antagonized by secreted proteins from the Dickkopf (Dkk) family, which bind with high affinity to lipoprotein receptor-related protein 5/6 (LRP5/6) and thereby directly prevent Wnt binding.86 Several recent studies have also found that the level of DKK-1 bound to LRP-6 is lower in AS patients than in healthy controls. Downregulation of DKK-1 enhances the proliferation and osteogenic potential of ASFs via the Wnt/β-catenin signaling pathway in vitro.87 MiR-17-5p and miR-146a have been proven to affect the proliferation and osteogenic potential of ASFs by regulating DKK-1 expression.20,88 In addition, some other factors involved in Wnt signaling directly bind extracellular Wnt, such as sFRP3/FRZB (members of the sFRP family). It has been reported that IL-22 has the capacity to increase the expression of sFRP3/FRZB and thereby inhibit Wnt signaling in fibroblasts in both in vitro and ex vivo models.89
In addition, some factors have been proven to affect ASFs’ proliferation and ossification by Wnt signaling through an exact mechanism. C‑X‑C chemokine receptor type 4 (CXCR4) was upregulated in AS and led to increasing ossification and growth rates of ASFs.90,91 In contrast, inorganic pyrophosphate transport regulator Gene (ANKH) overexpression inhibited viability, mineralization, and ossification in ASFs.92 Both of the above findings may indicate a new way to study ASFs via Wnt signaling.
BMP/TGFβ-Smads
The TGF‐β superfamily comprises a group of polypeptide factors with similar structures, including TGF‐β, BMPs, and over 30 other members, that play indispensable roles in many cellular functions, such as bone reconstruction. Both TGF‐β1 and BMP2 are thought to be involved in bone formation as important osteogenic mediators.93 Recent studies have suggested that BMP/TGF-β signaling contributes to the progression of AS and emerging evidence has shown high expression of the BMP/TGF-β signaling pathway in ASFs.94
BMP receptors contain type I and type II receptors. Activated through binding with a ligand to induce autophosphorylation, the type II receptor activates the type I receptor, which in turn forms a complex receptor and finally leads to the BMP activation.95 Core-binding factor a1 (Cbfa1)/runt-related transcription factor 2 (Runx2), which is an essential transcription factor for osteoblastic differentiation and osteogenesis, is reported to be dependent on Smad1/Smad5 activation caused by BMP-2 stimulation.96 mRNA expression of Cbfa1 was induced by continuous activation of Smad1 or Smad5. In contrast, transfecting the cells with dominant-negative Smad1, Smad4, Smad5, or Smad6 significantly reduced the BMP-2-induced expression of Cbfa1.
Cbfa1/Runx2, bone morphogenetic protein receptors (BMPR-I/II), and Smad family protein receptor-regulated Smads (Smad1/5/8), common-partner Smad (Smad4), and phosphorylated Smads (pSmad1/5/8) were found to be overexpressed in ASFs in multiple studies. TGF-β1 and p-Smad2/3 were also found to be overexpressed by Zhang et al.97 These data, together with the findings summarized in Table 1,16,77,97–104 confirm that ASFs are target cells of BMP/TGF-β signaling and indicate that a highly activated BMP/TGF-β signal transduction pathway exists in ASFs. Concurrently, based on the evidence that the Smad6 expression is lower in ASFs than normal, it reveals the lack of self-regulation and inhibition in the BMP/TGF-β signaling of ASFs.
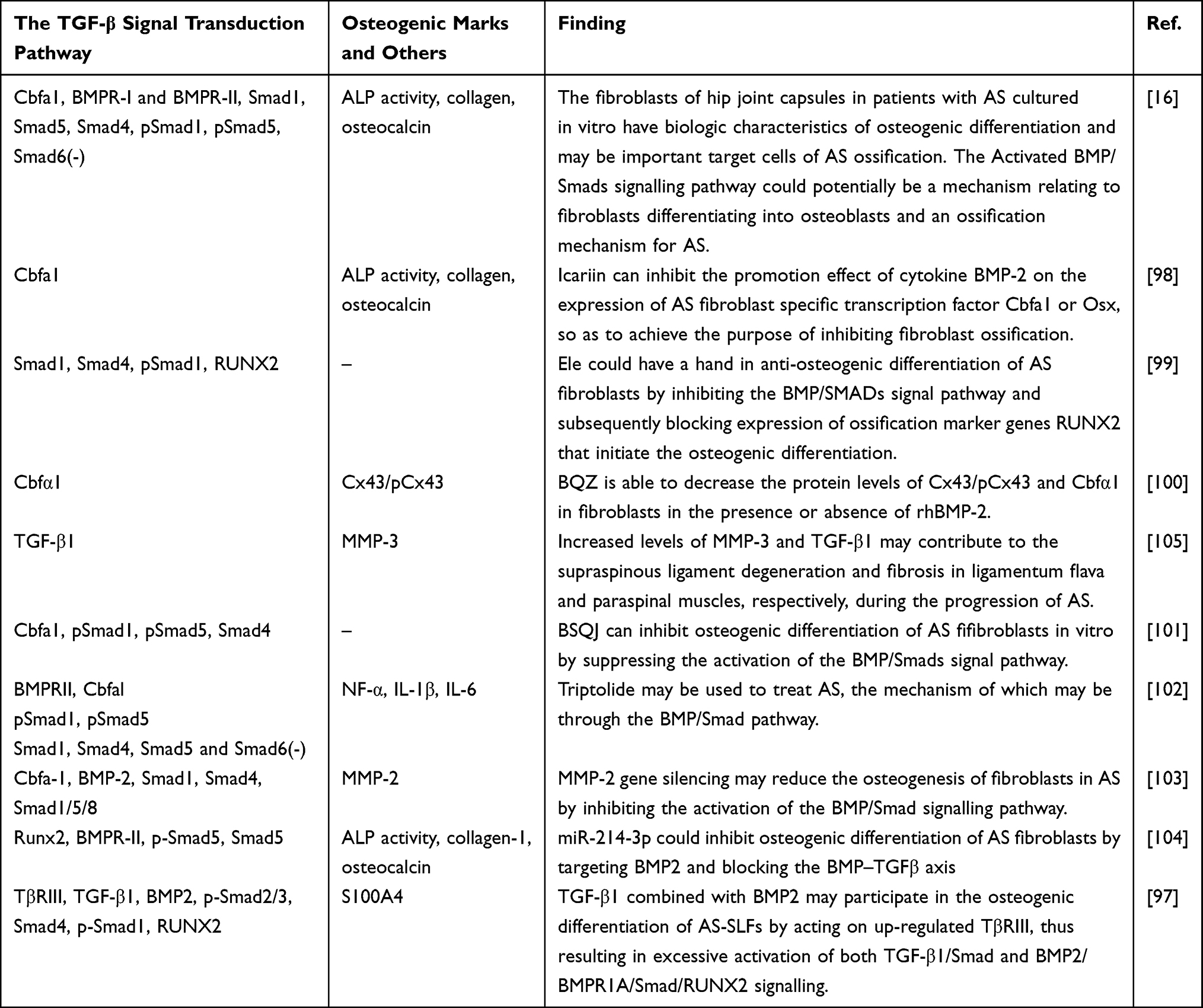 |
Table 1 BMP/TGF-β Signaling in ASFs |
β-proteoglycan (TβRIII), originally defined as an auxiliary receptor of the TGF-β superfamily, was an abundant membrane-anchoring protein. A recent study has reported that TβRIII plays a critical role in TGF-β/Smad signaling. Another report suggested that using a biosensor method, BMP2, BMP4, and BMP7 can bind to TβRIII. BMP2 activity was significantly downregulated due to the loss of TβRIII.106 TβRIII binds all three TGF-β ligands and BMP2, and Zhang et al further elaborated that the expression of TGF‐β1, BMP2, and TβRIII was markedly increased in ASFs. Moreover, TGF‐β1 combined with BMP2 significantly upregulated the expression of TβRIII but not TβRI or TβRII, further suggesting that TβRIII upregulation may participate in the osteogenic differentiation of ASFs.97
Matrix metalloproteinase (MMP)-2 has recently been demonstrated to be associated with AS, the mRNA expression level of which in the ASFs group was approximately four times higher than that in the control group. Moreover, the expression of Cbfa-1 was significantly downregulated by MMP-2 gene silencing and in turn inhibited the activation of the BMP/Smad signaling pathway. The results revealed that MMP-2 gene silencing may reduce the osteogenesis of ASFs by inhibiting the activation of the BMP/Smad signaling pathway.103 Connexin43 (Cx43) is a major gap junction protein in bone and has been shown to play a critical role in osteoblast differentiation in recent studies. A study suggests that cell communication mediated by gap junctions is indispensable for osteoblast differentiation induced by BMP-2.107 Fibroblasts from AS and Ossification of the Posterior Longitudinal Ligament (OPLL) patients were reported to have osteogenic characteristics that were upregulated by Cx43.100,108 These studies indicated that Cx43 may be a potential target to explain how ASFs affect osteoblasts.
Pi3k/Akt and AMPK
Regarding the inflammatory factors that are overexpressed in ASFs, Qin et al reported that as a kind of specific agonist for adenosine 5’-monophosphate activated protein kinase (AMPK), which was blocked in AS, metformin could decrease these inflammatory factors and inhibit ASF ossification.109 This finding indicates the potential of the Pi3k/Akt and AMPK pathways to mediate inflammation and bone formation in AS. In addition, rapamycin blockade of mammalian target of rapamycin (mTOR) in AS also ameliorated ASF-mediated pathological progression independent of IL-17A and TNF-α cytokines,110 which also confirmed the role of the Pi3k/Akt and AMPK pathways in ASF ossification.
MAPK-ERK
In ASFs, the high expression of Annexin A2 upregulated by IL-6 drew the attention of Li et al. They performed further research and observed that silencing of Annexin A2 ameliorated the ASF ossification induced by IL-6. They also proved that Annexin A2 might activate extracellular signal-regulated kinase (ERK) signaling and induce ASF ossification by mitogen-activated protein kinase (MEK) inhibitor experiments.111 In addition, mitogen-activated protein kinase 1 (MAPK1) was also observed to increase in synovial ASFs.91
Other Cytokines in ASFs
Apart from investigating signaling pathways, many other researchers have attempted to explore and identify the correlations between ASFs and inflammatory and bone ankylosing processes in AS by studying microRNAs112,113 (summarized in Table 2 20,83,85,88,103,104,114–118) and other cytokines (summarized in Table 3). The results are obviously based on the fact that with cytokine secretion and the function of mediating the inflammatory microenvironment, ASFs have been proven to have apparent osteogenic potential. Thus, it is natural to hypothesize that ASFs would be a bridge between inflammatory and abnormal bone formation in AS by secreting cytokines or being affected by other cytokines.
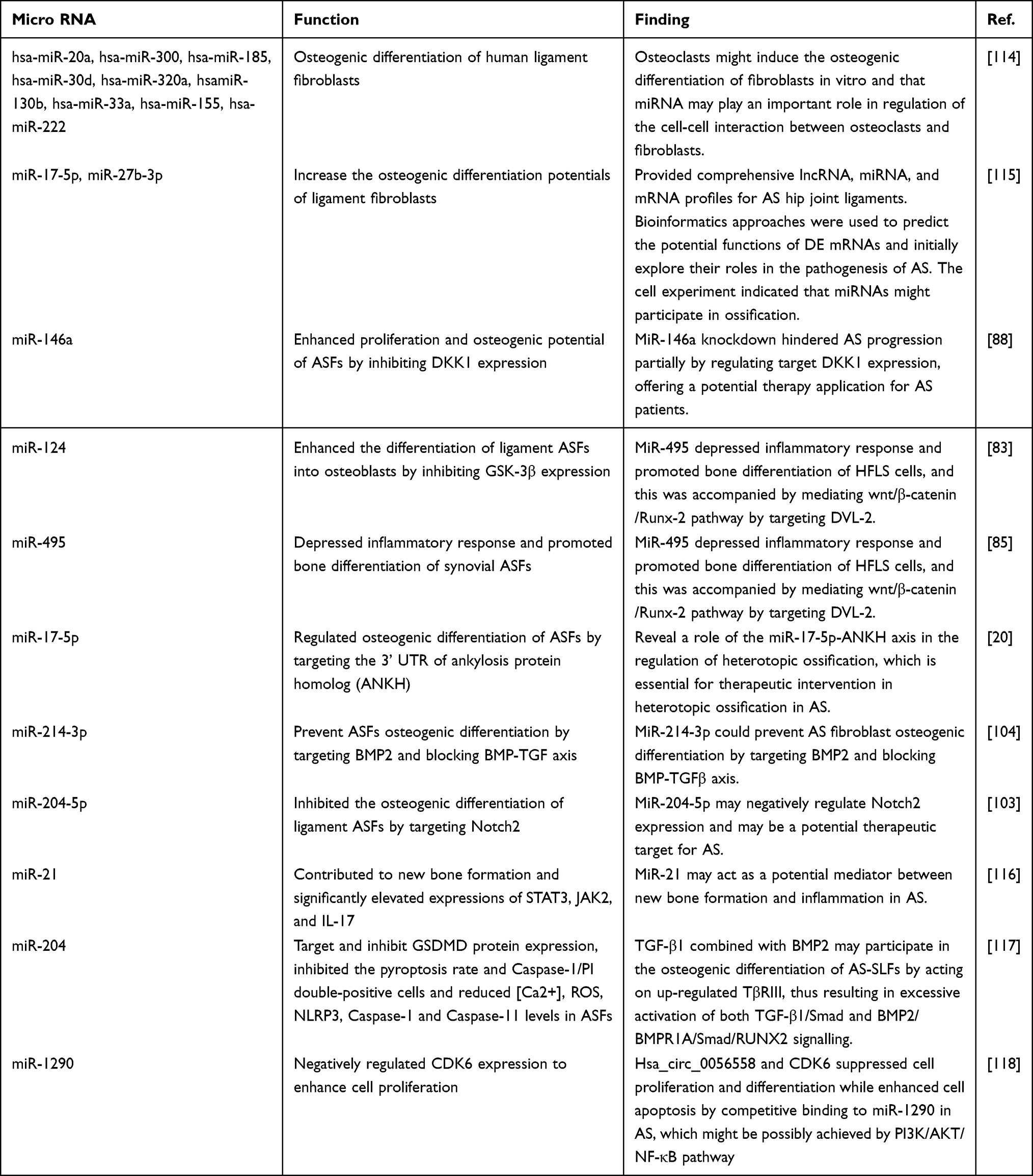 |
Table 2 Micro RNA in ASFs |
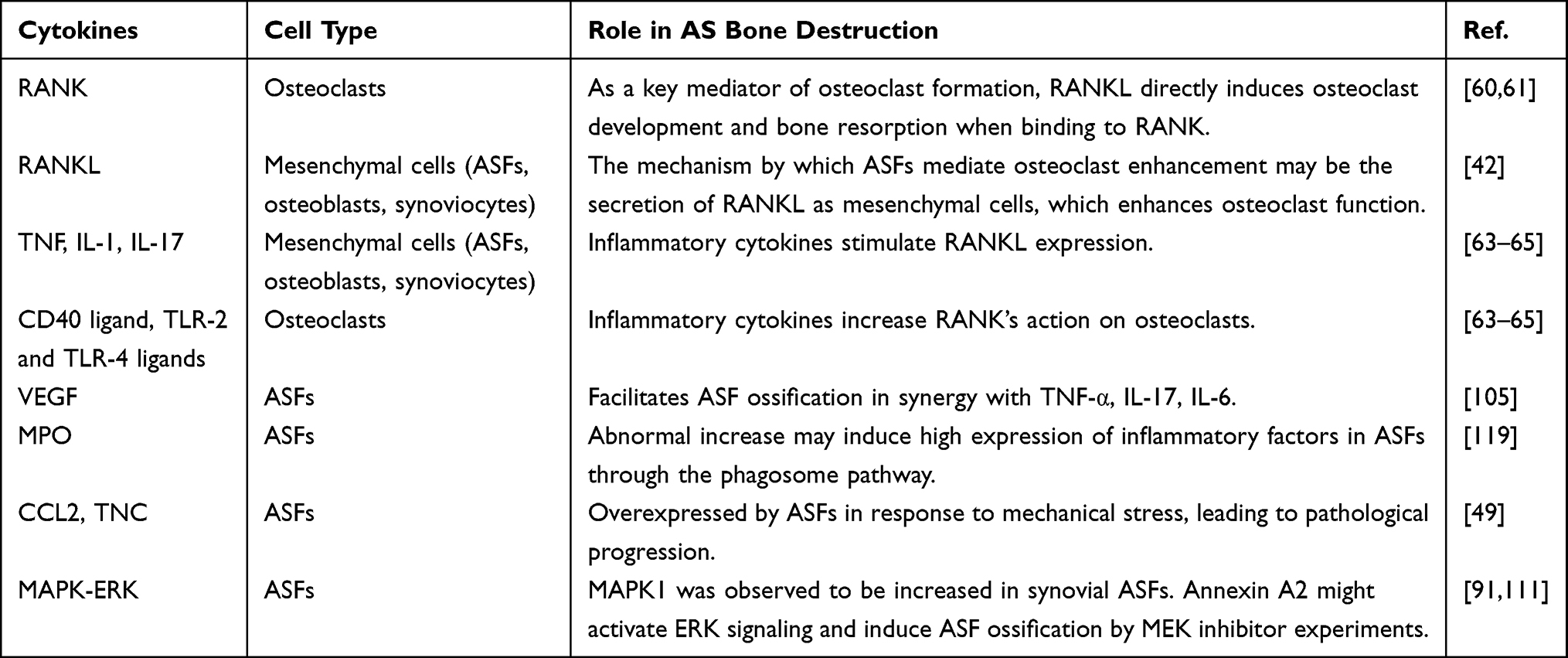 |
Table 3 Several Cytokine and Cell Types Involved in as Bone Homeostasis |
The overexpression of TNF-α, IL-17, IL-6 and other cytokines is believed to play an important role in the abnormal pathological changes in ASFs.110,120 These cytokines synergized with VEGF to facilitate ASF ossification, and the mRNA levels were markedly positively correlated with the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI).105 Concurrently, Yu et al supposed that the abnormal increase in myeloperoxidase (MPO) may induce the high expression of inflammatory factors in ASFs through the phagosome pathway.119
Recently, based on anatomical characteristics and mechanobiology in AS-affected areas, such as sacroiliac joints and entheses, researchers have started to search for a correlation between mechanical force and AS pathogenesis.121 Emerging evidence has shown that in response to mechanical stress, ASFs overexpress some cytokines, such as CCL249 and Tenascin-C (TNC),122,123 leading to pathological progression. The activation of the Hippo/YAP signaling pathway would be a novel mechanism, although more research is required to confirm this hypothesis.
Concluding Remarks
As the pathological mechanism of AS has been further investigated, there is a growing awareness of the important role that ASFs may play in the regulation of the AS-specific inflammatory environment and in mediating the subsequent progression of ankylosing bone proliferation. This review looks at previous studies and for the first time summarizes the role of fibroblasts in AS pathology, suggesting that ASFs play a crucial role in AS. We also attempt to theoretically elucidate the unspecified issues of “bone metabolism”, “inflammation and osteogenesis” in AS at the fibroblast level by reviewing the literature.
In conclusion, ASFs, as a group of frequently observed cells among the histologic AS features,8,9 organize immune functions by recruiting immune cells and influencing their differentiation and activation, thus mediate the inflammatory response in the early phase of disease. ASFs promote joint destruction at sites of cartilage and actively promote abnormal ossification by recruiting osteoblasts, differentiation into myofibroblasts or ossification directly. Besides, evidence of adipocyte accumulation in ASF-rich tissue also relates ASFs to fat deposition in AS.12 A growing body of evidence has pointed towards the involvement of many signaling pathways and cytokines. The mediation of bone erosion is illustrated by ASFs’ secretion of RANKL as mesenchymal cells,42 which directly induces osteoclast development and bone resorption when binding to RANK.60,61 When it comes to in vitro ASF ossification research, Wnt signaling and90,91 BMP/TGF-β signaling16,98 are proved to be the major factors. In addition to being involved in ossification, TGF-β is also an impactful factor in ASFs’ myofibroblast differentiation.21,76 Among the molecular mechanism study of ASFs in AS inflammation and osteogenesis, TNF and IL-17 are active factors that draw our attention,42,63 which may even be the link that connects them.
However, most of the current studies on ASFs are limited to the cellular level or histology, and the candidate targets and gene expression modifications discussed above are primarily obtained from in vitro cell studies. The vast discrepancy between the in vitro cell culture and the in vivo complex pathological microenvironment may lead to large differences in the relevant studies and thus limits further understanding. Besides, a number of the in vitro studies above lack good data and stringent experimental conditions including proper controls, which makes them only offer some reference value that still requires further study to confirm. Another major limitation may be that the distinction between pathogenic subtypes of ASFs is unclear. It is still unknown which ASF subtypes exerting pathological effects may present optimal targets. In particular, when fibroblasts are used as therapeutic targets, identifying such pathological subtypes will also contribute to avoiding the impact on normal fibroblasts. Encouragingly, rapid advances in RNA sequencing, tissue single-cell profiling, and spatial transcriptomic techniques would enable the exact identification of ASFs.43
With increasing attention given to ASF investigations, elucidating the key roles and molecular mechanisms of ASFs in AS inflammation and osteogenesis would provide new targets and directions for AS diagnosis and treatment from a new perspective of fibroblasts.
Abbreviations
ASFs, ankylosing spondylitis fibroblasts; AS, ankylosing spondylitis; SpA, spondyloarthritis; RASFs, rheumatoid arthritis synovial fibroblasts; MSCs, mesenchymal stromal cells; TNF, Tumour Necrosis Factor; IL-17A, Interleukin-17A; CCL2, C–C motif ligand 2; VEGF, vascular endothelial growth factor; BMP, Bone morphogenetic protein; RANKL, receptor activator of nuclear factor-κ B Ligand; IL-1, interleukin-1; TLR-2, Toll-like receptor 2; OPG, Osteoprotegerin; LRP5/6, lipoprotein receptor-related protein 5/6; CXCR4, C‑X‑C chemokine receptor type 4; ANKH, inorganic pyrophosphate transport regulator Gene; ERK, extracellular signal-regulated kinase; MEK, mitogen-activated protein kinase; MMP, Matrix metalloproteinase; OPLL, Ossification of the Posterior Longitudinal Ligament; TGF-β1, Transforming Growth Factor Beta-1; AMPK, adenosine 5’-monophosphate activated protein kinase; mTOR, mammalian target of rapamycin; TNC, Tenascin-C.
Data Sharing Statement
The data that support the findings of this study are available. The authors will supply the relevant data in response to reasonable requests.
Acknowledgments
The authors thank American Journal Experts for providing English language editing of the manuscript and BioRender for providing icons in our figures.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval for the version to be published, and agree to be accountable for all aspects of the work.
Funding
This study was supported by the Guangdong Basic and Applied Basic Research Foundation (2022A1515012118 and 2020A1515110930), the Dean’s Fund youth Development Program of Zhujiang Hospital, Southern Medical University (yzjj2020qn06), and the National Science Fund for Distinguished Young Scholars (82202657).
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Braun J, Sieper J. Ankylosing spondylitis. Lancet. 2007;369(9570):1379–1390. doi:10.1016/S0140-6736(07)60635-7
2. de Winter JJ, van Mens LJ, van der Heijde D, Landewé R, Baeten DL. Prevalence of peripheral and extra-articular disease in ankylosing spondylitis versus non-radiographic axial spondyloarthritis: a meta-analysis. Arthritis Res Ther. 2016;18(1):196. doi:10.1186/s13075-016-1093-z
3. Navarro-Compán V, Sepriano A, El-Zorkany B, et al. Axial spondyloarthritis. Ann Rheum Dis. 2021;80(12):1511–1521. doi:10.1136/annrheumdis-2021-221035
4. Mauro D, Thomas R, Guggino G, Lories R, Brown MA, Ciccia F. Ankylosing spondylitis: an autoimmune or autoinflammatory disease?. Nat Rev Rheumatol. 2021;17(7):387–404. doi:10.1038/s41584-021-00625-y
5. Baraliakos X, Listing J, Rudwaleit M, Sieper J, Braun J. The relationship between inflammation and new bone formation in patients with ankylosing spondylitis. Arthritis Res Ther. 2008;10(5):R104. doi:10.1186/ar2496
6. Shao F, Liu Q, Zhu Y, et al. Targeting chondrocytes for arresting bony fusion in ankylosing spondylitis. Nat Commun. 2021;12(1):6540. doi:10.1038/s41467-021-26750-6
7. Sieper J, Rudwaleit M, Baraliakos X, et al. The Assessment of SpondyloArthritis international Society (ASAS) handbook: a guide to assess spondyloarthritis. Ann Rheum Dis. 2009;68(Suppl 2):ii1–i44. doi:10.1136/ard.2008.104018
8. Min Wang D, Lin L, Hua Peng J, et al. Pannus inflammation in sacroiliitis following immune pathological injury and radiological structural damage: a study of 193 patients with spondyloarthritis. Arthritis Res Ther. 2018;20(1):120. doi:10.1186/s13075-018-1594-z
9. François RJ, Gardner DL, Degrave EJ, Bywaters EGL. Histopathologic evidence that sacroiliitis in ankylosing spondylitis is not merely enthesitis: systematic study of specimens from patients and control subjects. Arthritis Rheum. 2000;43(9):2011–2024. doi:10.1002/1529-0131(200009)43:9<2011::AID-ANR12>3.0.CO;2-Y
10. Bleil J, Maier R, Hempfing A, et al. Histomorphologic and histomorphometric characteristics of zygapophyseal joint remodeling in ankylosing spondylitis. Arthritis Rheumatol. 2014;66(7):1745–1754. doi:10.1002/art.38404
11. Sieper J, Appel H, Braun J, Rudwaleit M. Critical appraisal of assessment of structural damage in ankylosing spondylitis: implications for treatment outcomes. Arthritis Rheum. 2008;58(3):649–656. doi:10.1002/art.23260
12. Bleil J, Maier R, Hempfing A, Sieper J, Appel H, Syrbe U. Granulation tissue eroding the subchondral bone also promotes new bone formation in ankylosing spondylitis. Arthritis Rheumatol. 2016;68(10):2456–2465. doi:10.1002/art.39715
13. Lories RJ, Luyten FP. Activated fibrocytes: circulating cells that populate the arthritic synovium? Rheumatology. 2010;49(4):617–618. doi:10.1093/rheumatology/kep365
14. Lories RJU, Matthys P, de Vlam K, Derese I, Luyten FP. Ankylosing enthesitis, dactylitis, and onychoperiostitis in male DBA/1 mice: a model of psoriatic arthritis. Ann Rheum Dis. 2004;63(5):595–598. doi:10.1136/ard.2003.013599
15. Yang M, Yuan H, Miao M, Xu W. The osteogenic potential of ligament fibroblasts is greater in ankylosing spondylitis patients than in patients with osteoarthritis. Zeitschrift fur Rheumatol. 2015;74(4):340–345. doi:10.1007/s00393-014-1394-z
16. Jiang N, Liu H-X, Liang H-Y, Feng X-H, Liu B-Y, Zhou Y-Y. Osteogenic differentiation characteristics of Hip joint capsule fibroblasts obtained from patients with ankylosing spondylitis. Ann Transl Med. 2021;9(4):331. doi:10.21037/atm-20-7817
17. Buechler MB, Pradhan RN, Krishnamurty AT, et al. Cross-tissue organization of the fibroblast lineage. Nature. 2021;593(7860):575–579. doi:10.1038/s41586-021-03549-5
18. Neumann E, Lefèvre S, Zimmermann B, et al. Rheumatoid arthritis progression mediated by activated synovial fibroblasts. Trends Mol Med. 2010;16(10):458–468. doi:10.1016/j.molmed.2010.07.004
19. Zhang X-P, Jian-Da M, Ying-Qian M, et al. Addition of fibroblast-stromal cell markers to immune synovium pathotypes better predicts radiographic progression at 1 year in active rheumatoid arthritis. Front Immunol. 2021;12:778480. doi:10.3389/fimmu.2021.778480
20. Qin X, Zhu B, Jiang T, et al. miR-17-5p regulates heterotopic ossification by targeting ANKH in ankylosing spondylitis. Mol Ther Nucleic Acids. 2019;18:696–707. doi:10.1016/j.omtn.2019.10.003
21. Yeremenko N, Noordenbos T, Cantaert T, et al. Disease-specific and inflammation-independent stromal alterations in spondylarthritis synovitis. Arthritis Rheum. 2013;65(1):174–185. doi:10.1002/art.37704
22. Mauro D, Simone D, Bucci L, Ciccia F. Novel immune cell phenotypes in spondyloarthritis pathogenesis. Semi Immunopathol. 2021;43(2):265–277. doi:10.1007/s00281-021-00837-0
23. Komatsu N, Takayanagi H. Mechanisms of joint destruction in rheumatoid arthritis - immune cell-fibroblast-bone interactions. Nat Rev Rheumatol. 2022;18(7):415–429. doi:10.1038/s41584-022-00793-5
24. Nygaard G, Firestein GS. Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes. Nat Rev Rheumatol. 2020;16(6):316–333. doi:10.1038/s41584-020-0413-5
25. Chu C-Q. Highlights of strategies targeting fibroblasts for novel therapies for rheumatoid arthritis. Front Med. 2022;9:846300. doi:10.3389/fmed.2022.846300
26. Teitelbaum SL, Patrick Ross F. Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003;4(8):638–649. doi:10.1038/nrg1122
27. Canalis E. Notch signaling in osteoblasts. Sci Signal. 2008;1(17):e17. doi:10.1126/stke.117pe17
28. Nakahama K-I. Cellular communications in bone homeostasis and repair. Cell Mol Life Sci. 2010;67(23):4001–4009. doi:10.1007/s00018-010-0479-3
29. Ducy P, Schinke T, Karsenty G. The osteoblast: a sophisticated fibroblast under central surveillance. Science. 2000;289(5484):1501–1504. doi:10.1126/science.289.5484.1501
30. Correa‐Gallegos D, Jiang D, Rinkevich Y. Fibroblasts as confederates of the immune system. Immunol Rev. 2021;302(1):147–162. doi:10.1111/imr.12972
31. Bollow M, Fischer T, Reisshauer H, et al. Quantitative analyses of sacroiliac biopsies in spondyloarthropathies: t cells and macrophages predominate in early and active sacroiliitis- cellularity correlates with the degree of enhancement detected by magnetic resonance imaging. Ann Rheum Dis. 2000;59(2):135–140. doi:10.1136/ard.59.2.135
32. Appel H, Kuhne M, Spiekermann S, et al. Immunohistochemical analysis of Hip arthritis in ankylosing spondylitis: evaluation of the bone-cartilage interface and subchondral bone marrow. Arthritis Rheum. 2006;54(6):1805–1813. doi:10.1002/art.21907
33. Keffer J, Probert L, Cazlaris H, et al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991;10(13):4025–4031. doi:10.1002/j.1460-2075.1991.tb04978.x
34. Ward MM, Bruckel J, Colbert R, et al. Summary of the 2005 annual research and education meeting of the Spondyloarthritis Research and Therapy Network (SPARTAN). J Rheumatol. 2006;33(5):978–982.
35. Péntek M, Poór G, Wiland P, et al. Biological therapy in inflammatory rheumatic diseases: issues in Central and Eastern European countries. Eur J Health Econom. 2014;15(Suppl 1):S35–S43. doi:10.1007/s10198-014-0592-6
36. Baji P, Péntek M, Szántó S, et al. Comparative efficacy and safety of biosimilar infliximab and other biological treatments in ankylosing spondylitis: systematic literature review and meta-analysis. Eur J Health Econom. 2014;15(Suppl 1):S45–S52. doi:10.1007/s10198-014-0593-5
37. Danve A, Deodhar A. Treatment of axial spondyloarthritis: an update. Nat Rev Rheumatol. 2022;18(4):205–216. doi:10.1038/s41584-022-00761-z
38. Deodhar A, David Y. Switching tumor necrosis factor inhibitors in the treatment of axial spondyloarthritis. Semin Arthritis Rheumatism. 2017;47(3):343–350. doi:10.1016/j.semarthrit.2017.04.005
39. Baeten D, Baraliakos X, Braun J, et al. Anti-interleukin-17A monoclonal antibody secukinumab in treatment of ankylosing spondylitis: a randomised, double-blind, placebo-controlled trial. Lancet. 2013;382(9906):1705–1713. doi:10.1016/S0140-6736(13)61134-4
40. van der Heijde D, Song I-H, Pangan AL, et al. Efficacy and safety of upadacitinib in patients with active ankylosing spondylitis (SELECT-AXIS 1): a multicentre, randomised, double-blind, placebo-controlled, Phase 2/3 trial. Lancet. 2019;394(10214):2108–2117. doi:10.1016/S0140-6736(19)32534-6
41. Appel H, Maier R, Peihua W, et al. Analysis of IL-17(+) cells in facet joints of patients with spondyloarthritis suggests that the innate immune pathway might be of greater relevance than the Th17-mediated adaptive immune response. Arthritis Res Ther. 2011;13(3):R95. doi:10.1186/ar3370
42. Sieper J, Poddubnyy D, Miossec P. The IL-23-IL-17 pathway as a therapeutic target in axial spondyloarthritis. Nat Rev Rheumatol. 2019;15(12):747–757. doi:10.1038/s41584-019-0294-7
43. Wei K, Nguyen HN, Brenner MB. Fibroblast pathology in inflammatory diseases. J Clin Invest. 2021;131(20). doi:10.1172/JCI149538
44. Peng J, Gong Y, Zhang Y, Wang D, Xiao Z. Immunohistological analysis of active sacroiliitis in patients with axial spondyloarthritis. Medicine. 2017;96(16):e6605. doi:10.1097/MD.0000000000006605
45. Appel H, Kuhne M, Spiekermann S, et al. Immunohistologic analysis of zygapophyseal joints in patients with ankylosing spondylitis. Arthritis Rheum. 2006;54(9):2845–2851. doi:10.1002/art.22060
46. Kruithof E, Baeten D, Van den Bosch F, Mielants H, Veys EM, De Keyser F. Histological evidence that infliximab treatment leads to downregulation of inflammation and tissue remodelling of the synovial membrane in spondyloarthropathy. Ann Rheum Dis. 2005;64(4):529–536. doi:10.1136/ard.2003.018549
47. Song IH, Heldmann F, Rudwaleit M, et al. Different response to rituximab in tumor necrosis factor blocker-naive patients with active ankylosing spondylitis and in patients in whom tumor necrosis factor blockers have failed: a twenty-four-week clinical trial. Arthritis Rheum. 2010;62(5):1290–1297. doi:10.1002/art.27383
48. Liu L, Yuan Y, Zhang S, Jiake X, Zou J. Osteoimmunological insights into the pathogenesis of ankylosing spondylitis. J Cell Physiol. 2021;236(9):6090–6100. doi:10.1002/jcp.30313
49. Cambré I, Gaublomme D, Burssens A, et al. Mechanical strain determines the site-specific localization of inflammation and tissue damage in arthritis. Nat Commun. 2018;9(1):4613. doi:10.1038/s41467-018-06933-4
50. Gibbons LJ, Hyrich KL. Biologic therapy for rheumatoid arthritis: clinical efficacy and predictors of response. BioDrugs. 2009;23(2):111–124. doi:10.2165/00063030-200923020-00004
51. Batko B, Maga P, Urbanski K, et al. Microvascular dysfunction in ankylosing spondylitis is associated with disease activity and is improved by anti-TNF treatment. Sci Rep. 2018;8(1):13205. doi:10.1038/s41598-018-31550-y
52. Miotla J, Maciewicz R, Kendrew J, Feldmann M, Paleolog E. Treatment with soluble VEGF receptor reduces disease severity in murine collagen-induced arthritis. Lab Invest. 2000;80(8):1195–1205. doi:10.1038/labinvest.3780127
53. Liu C-H, Raj S, Chen C-H, et al. HLA-B27-mediated activation of TNAP phosphatase promotes pathogenic syndesmophyte formation in ankylosing spondylitis. J Clin Invest. 2019;129(12):5357–5373. doi:10.1172/JCI125212
54. Saetia K, Cho D, Lee S, Kim DH, Kim SD. Ossification of the posterior longitudinal ligament: a review. Neurosurgical Focus. 2011;30(3):E1. doi:10.3171/2010.11.FOCUS10276
55. Le HV, Wick JB, Van BW, Klineberg EO. Ossification of the posterior longitudinal ligament: pathophysiology, diagnosis, and management. J Am Aca Orthopa Surg. 2022;30(17):820–830. doi:10.5435/JAAOS-D-22-00049
56. Braun J, Sieper J, Bollow M. Imaging of sacroiliitis. Clin Rheumatol. 2000;19(1):51–57. doi:10.1007/s100670050011
57. Laloo F, Herregods N, Jaremko JL, et al. MRI of the axial skeleton in spondyloarthritis: the many faces of new bone formation. Insights Imaging. 2019;10(1):67. doi:10.1186/s13244-019-0752-4
58. Tam L-S, Jieruo G, David Y. Pathogenesis of ankylosing spondylitis. Nat Rev Rheumatol. 2010;6(7):399–405. doi:10.1038/nrrheum.2010.79
59. Gravallese EM, Schett G. Effects of the IL-23-IL-17 pathway on bone in spondyloarthritis. Nat Rev Rheumatol. 2018;14(11):631–640. doi:10.1038/s41584-018-0091-8
60. Lacey DL, Timms E, Tan HL, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93(2):165–176. doi:10.1016/S0092-8674(00)81569-X
61. Danks L, Komatsu N, Guerrini MM, et al. RANKL expressed on synovial fibroblasts is primarily responsible for bone erosions during joint inflammation. Ann Rheum Dis. 2016;75(6):1187–1195. doi:10.1136/annrheumdis-2014-207137
62. Baraliakos X, Boehm H, Bahrami R, et al. What constitutes the fat signal detected by MRI in the spine of patients with ankylosing spondylitis? A prospective study based on biopsies obtained during planned spinal osteotomy to correct hyperkyphosis or spinal stenosis. Ann Rheum Dis. 2019;78(9):1220–1225. doi:10.1136/annrheumdis-2018-214983
63. Osta B, Lavocat F, Eljaafari A, Miossec P. Effects of Interleukin-17A on osteogenic differentiation of isolated human mesenchymal stem cells. Front Immunol. 2014;5:425. doi:10.3389/fimmu.2014.00425
64. Shin HR, Kim BS, Kim HJ, et al. Excessive osteoclast activation by osteoblast paracrine factor RANKL is a major cause of the abnormal long bone phenotype in Apert syndrome model mice. J Cell Physiol. 2022;237(4):2155–2168.
65. Vandooren B, Cantaert T, Noordenbos T, Tak PP, Baeten D. The abundant synovial expression of the RANK/RANKL/Osteoprotegerin system in peripheral spondylarthritis is partially disconnected from inflammation. Arthritis Rheum. 2008;58(3):718–729. doi:10.1002/art.23290
66. Bharadwaz A, Jayasuriya AC. Osteogenic differentiation cues of the bone morphogenetic protein-9 (BMP-9) and its recent advances in bone tissue regeneration. Mater Sci Eng C. 2021;120:111748. doi:10.1016/j.msec.2020.111748
67. Maksymowych WP, Wichuk S, Chiowchanwisawakit P, Lambert RG, Pedersen SJ. Fat metaplasia and backfill are key intermediaries in the development of sacroiliac joint ankylosis in patients with ankylosing spondylitis. Arthritis Rheumatol. 2014;66(11):2958–2967. doi:10.1002/art.38792
68. Chiowchanwisawakit P, Lambert RGW, Conner-Spady B, Maksymowych WP. Focal fat lesions at vertebral corners on magnetic resonance imaging predict the development of new syndesmophytes in ankylosing spondylitis. Arthritis Rheum. 2011;63(8):2215–2225. doi:10.1002/art.30393
69. Baraliakos X, Heldmann F, Callhoff J, et al. Which spinal lesions are associated with new bone formation in patients with ankylosing spondylitis treated with anti-TNF agents? A long-term observational study using MRI and conventional radiography. Ann Rheum Dis. 2014;73(10):1819–1825. doi:10.1136/annrheumdis-2013-203425
70. Wei-Dong X, Yang X-Y, Da-He L, et al. Up-regulation of fatty acid oxidation in the ligament as a contributing factor of ankylosing spondylitis: a comparative proteomic study. J proteo. 2015;113:57–72. doi:10.1016/j.jprot.2014.09.014
71. Plikus MV, Guerrero-Juarez CF, Ito M, et al. Regeneration of fat cells from myofibroblasts during wound healing. Science. 2017;355(6326):748–752. doi:10.1126/science.aai8792
72. van der Heijde D, Landewé R, Baraliakos X, et al. Radiographic findings following two years of infliximab therapy in patients with ankylosing spondylitis. Arthritis Rheum. 2008;58(10):3063–3070. doi:10.1002/art.23901
73. van der Heijde D, Landewé R, Einstein S, et al. Radiographic progression of ankylosing spondylitis after up to two years of treatment with etanercept. Arthritis Rheum. 2008;58(5):1324–1331. doi:10.1002/art.23471
74. van der Heijde D, Salonen D, Weissman BN, et al. Assessment of radiographic progression in the spines of patients with ankylosing spondylitis treated with Adalimumab for up to 2 years. Arthritis Res Ther. 2009;11(4):R127. doi:10.1186/ar2794
75. Braun J, Baraliakos X, Hermann K-GA, et al. The effect of two golimumab doses on radiographic progression in ankylosing spondylitis: results through 4 years of the GO-RAISE trial. Ann Rheum Dis. 2014;73(6):1107–1113. doi:10.1136/annrheumdis-2012-203075
76. Pakshir P, Noskovicova N, Lodyga M, et al. The myofibroblast at a glance. J Cell Sci. 2020;133(13). doi:10.1242/jcs.227900
77. Zhang Y, Hongfei X, Hu X, Zhang C, Chu T, Zhou Y. Histopathological changes in supraspinous ligaments, ligamentum flava and paraspinal muscle tissues of patients with ankylosing spondylitis. Int J Rheum Dis. 2016;19(4):420–429. doi:10.1111/1756-185X.12305
78. Murshed M. Mechanism of Bone Mineralization. Cold Spring Harb Perspect Med. 2018;8(12):a031229. doi:10.1101/cshperspect.a031229
79. Beyer C, Distler JHW. Changing paradigms in spondylarthritis: the myofibroblast signature. Arthritis Rheum. 2013;65(1):24–27. doi:10.1002/art.37703
80. Nanthakumar CB, Hatley RJD, Lemma S, Gauldie J, Marshall RP, Macdonald SJF. Dissecting fibrosis: therapeutic insights from the small-molecule toolbox. Nat Rev Drug Discov. 2015;14(10):693–720. doi:10.1038/nrd4592
81. Stougaard J, Lomholt S, Ommen P, Kelsen J, Kragstrup TW. The antifibrotic drug pirfenidone inhibits spondyloarthritis fibroblast-like synoviocytes and osteoblasts in vitro. BMC Rheumatol. 2018;2:33.
82. Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19(2):179–192. doi:10.1038/nm.3074
83. Tang SL, Huang QH, Wu LG, Liu C, Cai AL. MiR-124 regulates osteoblast differentiation through GSK-3β in ankylosing spondylitis. Eur Rev Med Pharmacol Sci. 2018;22(20):6616–6624. doi:10.26355/eurrev_201810_16136
84. Zou Y-C, Yang X-W, Yuan S-G, Zhang P, Yi-Kai L. Celastrol inhibits prostaglandin E2-induced proliferation and osteogenic differentiation of fibroblasts isolated from ankylosing spondylitis Hip tissues in vitro. Drug Design Dev Ther. 2016;10:933–948. doi:10.2147/DDDT.S97463
85. Du W, Yin L, Tong P. MiR-495 targeting dvl-2 represses the inflammatory response of ankylosing spondylitis. Am J Transl Res. 2019;11(5):2742–2753.
86. Patel MS, Karsenty G. Regulation of bone formation and vision by LRP5. New Eng J Med. 2002;346(20):1572–1574. doi:10.1056/NEJM200205163462011
87. Zou Y-C, Yang X-W, Yuan S-G, Zhang P, Yong-Liang Y, Yi-Kai L. Downregulation of dickkopf-1 enhances the proliferation and osteogenic potential of fibroblasts isolated from ankylosing spondylitis patients via the Wnt/β-catenin signaling pathway in vitro. Connect Tissue Res. 2016;57(3):200–211. doi:10.3109/03008207.2015.1127916
88. Di G, Kong L, Zhao Q, Ding T. MicroRNA-146a knockdown suppresses the progression of ankylosing spondylitis by targeting dickkopf 1. Biomed Pharmacother. 2018;97:1243–1249. doi:10.1016/j.biopha.2017.11.067
89. Resende GG, Machado CRL, Rocha MA, et al. IL-22 increases the production of sFRP3 by FLS in inflammatory joint diseases. Braz J Med Biol. 2020;53(9):e9880. doi:10.1590/1414-431x20209880
90. Xindong H, Dong Y. Ankylosis progressive homolog upregulation inhibits cell viability and mineralization during fibroblast ossification by regulating the Wnt/β‑catenin signaling pathway. Mol Med Rep. 2020;22(6):4551–4560. doi:10.3892/mmr.2020.11576
91. Feng X, Zhu S, Yan Z, Wang C, Tong W, Weidong X. PDGFRB as a potential therapeutic target of ankylosing spondylitis: validation following bioinformatics analysis. Cell Mol Biol. 2020;66(6):127–134. doi:10.14715/cmb/2020.66.6.23
92. Chongru H, Dahe L, Gao J, Jia L, Liu Z, Weidong X. Inhibition of CXCR4 inhibits the proliferation and osteogenic potential of fibroblasts from ankylosing spondylitis via the Wnt/β‑catenin pathway. Mol Med Rep. 2019;19(4):3237–3246. doi:10.3892/mmr.2019.9980
93. Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science. 2002;296(5573):1646–1647. doi:10.1126/science.1071809
94. Lories RJU, Derese I, Luyten FP. Modulation of bone morphogenetic protein signaling inhibits the onset and progression of ankylosing enthesitis. J Clin Invest. 2005;115(6):1571–1579. doi:10.1172/JCI23738
95. Thatcher JD. The TGF-beta signal transduction pathway. Sci Signal. 2010;3(119):tr4. doi:10.1126/scisignal.3119tr4
96. Nishimura R, Hata K, Harris SE, Ikeda F, Yoneda T. Core-binding factor alpha 1 (Cbfa1) induces osteoblastic differentiation of C2C12 cells without interactions with Smad1 and Smad5. Bone. 2002;31(2):303–312. doi:10.1016/S8756-3282(02)00826-8
97. Zhang Y, Chen W-G, Yang S-Z, et al. Up-regulation of TβRIII facilitates the osteogenesis of supraspinous ligament-derived fibroblasts from patients with ankylosing spondylitis. J Cell Mol Med. 2021;25(3):1613–1623. doi:10.1111/jcmm.16262
98. Jia C, Liu H, Min L, Zhikui W, Feng X. Effects of icariin on cytokine-induced ankylosing spondylitis with fibroblastic osteogenesis and its molecular mechanism. Int J Clin Exp Pathol. 2014;7(12):9104–9109.
99. Zhou Y-Y, Liu H-X, Jiang N, et al. Elemene, the essential oil of Curcuma wenyujin, inhibits osteogenic differentiation in ankylosing spondylitis. Joint Bone Spine. 2015;82(2):100–103. doi:10.1016/j.jbspin.2014.05.004
100. Zhou Y-Y, Huang R-Y, Lin J-H, Xu -Y-Y, He X-H, He Y-T. Bushen-Qiangdu-Zhilv decoction inhibits osteogenic differentiation of rat fibroblasts by regulating connexin 43. Exp Ther Med. 2016;12(1):347–353. doi:10.3892/etm.2016.3292
101. Liu H-X, Jiang N, Liang H-Y, et al. Bushen Qiangji Granule medicated serum inhibits osteogenic differentiation of fibroblasts in ankylosing spondylitis by inhibiting the BMP/Smads signal pathway in vitro. Chin J Integr Med. 2016;22(11):817–822. doi:10.1007/s11655-016-2268-9
102. Wang G, Cai J, Zhang J, Cuiyun L. Mechanism of triptolide in treating ankylosing spondylitis through the anti‑ossification effect of the BMP/Smad signaling pathway. Mol Med Rep. 2018;17(2):2731–2737. doi:10.3892/mmr.2017.8117
103. Yuan B, Zhiming W. MMP-2 silencing reduces the osteogenic transformation of fibroblasts by inhibiting the activation of the BMP/Smad pathway in ankylosing spondylitis. Oncol Letters. 2018;15(3):3281–3286. doi:10.3892/ol.2017.7714
104. Ding L, Yin Y, Hou Y, et al. microRNA-214-3p suppresses ankylosing spondylitis fibroblast osteogenesis BMP-TGF axis and BMP2. Front Endocrinol. 2020;11:609753. doi:10.3389/fendo.2020.609753
105. Liu KG, He QH, Tan JW, Liao GJ. Expression of TNF-α, VEGF, and MMP-3 mRNAs in synovial tissues and their roles in fibroblast-mediated osteogenesis in ankylosing spondylitis. Gene Mole Res. 2015;14(2):6852–6858. doi:10.4238/2015.June.18.28
106. Kirkbride KC, Townsend TA, Bruinsma MW, Barnett JV, Blobe GC. Bone morphogenetic proteins signal through the transforming growth factor-beta type III receptor. J Biol Chem. 2008;283(12):7628–7637. doi:10.1074/jbc.M704883200
107. Hashida Y, Nakahama K-I, Shimizu K, Akiyama M, Harada K, Morita I. Communication-dependent mineralization of osteoblasts via gap junctions. Bone. 2014;61:19–26. doi:10.1016/j.bone.2013.12.031
108. Yang H-S, Xu-hua L, Chen D-Y, et al. Upregulated expression of connexin43 in spinal ligament fibroblasts derived from patients presenting ossification of the posterior longitudinal ligament. Spine. 2011;36(26):2267–2274. doi:10.1097/BRS.0b013e31820ccfc6
109. Qin X, Jiang T, Liu S, et al. Effect of metformin on ossification and inflammation of fibroblasts in ankylosing spondylitis: an in vitro study. J Cell Biochem. 2018;119(1):1074–1082. doi:10.1002/jcb.26275
110. Chen S, van Tok MN, Knaup VL, et al. mTOR blockade by rapamycin in spondyloarthritis: impact on inflammation and new bone formation and. Front Immunol. 2019;10:2344. doi:10.3389/fimmu.2019.02344
111. Li DH, He CR, Liu F-P, et al. Annexin A2, up-regulated by IL-6, promotes the ossification of ligament fibroblasts from ankylosing spondylitis patients. Biomed Pharmacother. 2016;84:674–679. doi:10.1016/j.biopha.2016.09.091
112. Assadiasl S, Rajabinejad M, Soleimanifar N, et al. MicroRNAs-mediated regulation pathways in rheumatic diseases. Inflammopharmacology. 2023;31(1):129–144. doi:10.1007/s10787-022-01097-6
113. Najm A, Blanchard F, Le Goff B. Micro-RNAs in inflammatory arthritis: from physiopathology to diagnosis, prognosis and therapeutic opportunities. Biochem Pharmacol. 2019;165:134–144. doi:10.1016/j.bcp.2019.02.031
114. Fangcang Y, Cui Y, Zhou X, Zhang X, Han J. Osteogenic differentiation of human ligament fibroblasts induced by conditioned medium of osteoclast-like cells. Biosci Trends. 2011;5(2):46–51. doi:10.5582/bst.2011.v5.2.46
115. Zhang C, Wang C, Jia Z. Differentially expressed mRNAs, lncRNAs, and miRNAs with associated co-expression and ceRNA networks in ankylosing spondylitis. Oncotarget. 2017;8(69):113543–113557. doi:10.18632/oncotarget.22708
116. Zou Y-C, Yan L-M, Gao Y-P, et al. miR-21 may act as a potential mediator between inflammation and abnormal bone formation in ankylosing spondylitis based on TNF-α concentration-dependent manner through the JAK2/STAT3 pathway. Dose-Response. 2020;18(1):1559325819901239. doi:10.1177/1559325819901239
117. Guangxue G, Huo Y, Gang X, et al. MicroRNA-204-GSDMD interaction regulates pyroptosis of fibroblast-like synoviocytes in ankylosing spondylitis. Int Immunopharmacol. 2021;91:107227. doi:10.1016/j.intimp.2020.107227
118. Xia L, Zhou W, Zhen L, Guan F. Hsa_circ_0056558 regulates cyclin-dependent kinase 6 by sponging microRNA-1290 to suppress the proliferation and differentiation in ankylosing spondylitis. Autoimmunity. 2021;54(2):114–128. doi:10.1080/08916934.2021.1894417
119. Yu C, Zhan X, Liang T, et al. Mechanism of hip arthropathy in ankylosing spondylitis: abnormal myeloperoxidase and phagosome. Front Immunol. 2021;12:572592. doi:10.3389/fimmu.2021.572592
120. van Tok MN, van Duivenvoorde LM, Kramer I, et al. Interleukin-17A inhibition diminishes inflammation and new bone formation in experimental spondyloarthritis. Arthritis Rheumatol. 2019;71(4):612–625. doi:10.1002/art.40770
121. Gracey E, Burssens A, Cambré I, et al. Tendon and ligament mechanical loading in the pathogenesis of inflammatory arthritis. Nat Rev Rheumatol. 2020;16(4):193–207. doi:10.1038/s41584-019-0364-x
122. Zihao L, Chen S, Cui H, et al. Tenascin-C-mediated suppression of extracellular matrix adhesion force promotes entheseal new bone formation through activation of Hippo signalling in ankylosing spondylitis. Ann Rheum Dis. 2021;80(7):891–902. doi:10.1136/annrheumdis-2021-220002
123. Van Mechelen M, Lories R. Tenascin-C, a novel target to inhibit new bone formation in axial spondyloarthritis, linked with inflammation, mechanical strain and tissue damage. Ann Rheum Dis. 2021;80(7):823–824. doi:10.1136/annrheumdis-2021-220443
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Advances in cGAS-STING Signaling in Fibrosis Diseases: Therapeutic Target in Pathological Scars
Zhao W, Angspatt A, Kitkumthorn N, Meevassana J
Journal of Inflammation Research 2025, 18:10777-10793
Published Date: 9 August 2025