Back to Journals » Journal of Hepatocellular Carcinoma » Volume 1
Fibroblast growth factor family as a potential target in the treatment of hepatocellular carcinoma
Authors Coleman S, Grose R, Kocher H
Received 25 February 2014
Accepted for publication 9 April 2014
Published 29 May 2014 Volume 2014:1 Pages 43—54
DOI https://doi.org/10.2147/JHC.S48958
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Stacey J Coleman,1 Richard P Grose,1 Hemant M Kocher1,2
1Centre for Tumour Biology, Barts Cancer Institute – a CRUK Centre of Excellence, Queen Mary University of London, 2Barts and the London HPB Centre, The Royal London Hospital, Barts Health NHS Trust, London, UK
Abstract: Hepatocellular cancer (HCC) is currently the third leading cause of cancer death worldwide. The prognosis of patients diagnosed with late-stage disease is dismal due to high resistance to conventional systemic therapies. The introduction of sorafenib, despite its limited efficacy, as the standard systemic therapy for advanced HCC has paved a way for targeted molecular therapies for HCC. Fibroblast growth factor (FGF) signaling plays an important role in the developing embryo and the adult. The FGF signaling pathway is often hijacked by cancer cells, including HCC. Several alterations in FGF signaling correlate with poor outcome in HCC patients, suggesting that this family of signaling molecules plays an important role in the development of HCC. Multikinase inhibitors targeting FGF signaling are currently under investigation in clinical trials. This review discusses the current understanding of the biological and clinical implications of aberrant FGF signaling in the prognosis, diagnosis, and treatment of HCC.
Keywords: signaling, stroma, stellate, tumor, cross-talk
Introduction
The mortality rate for hepatocellular carcinoma (HCC), despite a plethora of treatment options, remains high. HCC is the fifth most common cancer worldwide and is currently the third leading cause of cancer mortality.1 Eighty percent of new cases occur in developing countries, but the incidence is increasing in economically developed regions such as Japan, Western Europe, and the USA, which may be attributable to the greater prevalence of hepatitis C virus (HCV) infection as well as an increase in obesity and diabetes levels in these areas.2–4 The 5-year survival rate for patients with HCC is only 7%, and very few patients survive more than 12 months.5 Most HCC patients present with late-stage disease, only 30% are surgically resectable (although resection can improve 5-year survival rates of up to 70%, even in these resectable patients, there is a high risk of tumor recurrence, as high as 50% after 5 years)6 given that more than 80% have underlying cirrhosis,7 a known predisposing factor for development of new HCC,8 and only 30% of patients are eligible for repeat resection.7 The treatment options available for patients with late-stage disease include ablation, selective chemotherapy and/or embolization, selective radiotherapy, and systemic chemotherapy, but the prognosis is dismal (5-year survival rate approximately 10%).9
Patients with advanced HCC have very limited treatment options, and sorafenib is the only known targeted therapy approved for those with advanced, unresectable, or metastatic HCC, after demonstrating significant improvement in overall survival of patients in two pivotal randomized Phase III clinical trials (the Sorafenib HCC Assessment Randomized Protocol [SHARP] and Asia-Pacific trials).10,11 Sorafenib was developed as an inhibitor of vascular endothelial growth factor (VEGF) receptors 2 and 3, platelet-derived growth factor (PDGF) receptor, and intracellular Raf kinases; however, its mechanism of action remains unknown, and sorafenib demonstrates only modest efficacy even in patients with preserved liver function (10.7 months with sorafenib versus 7.9 months with placebo in the SHARP trial). Further, sorafenib is associated with significant toxicity, which may impact on its efficacy.10 Thus, it is imperative that new alternative therapeutics are investigated in order to optimize the outcomes for patients with advanced HCC (this may include synergistic combinations to block several pathways or agents, which will offer hope to patients who are refractory to treatment with sorafenib).12 As such, other small molecules such as brivanib (a VEGF receptor 2 inhibitor) and erlotinib (an epidermal growth factor inhibitor), and monoclonal antibodies such as bevacizumab (a VEGF A inhibitor) and cetuximab (an epidermal growth factor inhibitor), are currently being studied in patients with HCC.13
Increasing knowledge of the oncogenic processes and signaling pathways that regulate tumor development in HCC, as well as the advent of genome wide studies, have identified potential therapeutic targets. Further, several molecular subtypes of HCC have been identified.14 Recent data highlight the interplay between surrounding tissue (stroma) and the tumor, suggesting that HCC is a complex process leading to alterations in several signaling pathways.15,16 Among these pathways, it appears the fibroblast growth factor (FGF) signaling axis may play an important role in the development of HCC.17–21
FGF signaling
The first FGF was discovered as a mitogen for cultured fibroblasts over three decades ago.22 FGFs are crucial modulators of cellular proliferation, differentiation, embryonic development, and organogenesis.23 The mammalian FGF family comprises 18 secreted FGF ligands that signal through four high affinity transmembrane FGF receptors. A fifth receptor, FGF receptor 5, has no tyrosine kinase activity and is thought to negatively regulate signaling by dimerizing with FGF receptors 1–4 and blocking transphosphorylation.24 FGFs also bind to heparan sulfate proteoglycans, low affinity receptors that do not transmit a biological signal but function as accessory molecules.25,26 Unlike the canonical FGFs that act as autocrine and paracrine factors, FGF15/19, FGF21, and FGF23 have a low affinity for heparan sulfate proteoglycans, function as endocrine hormones, and exert actions distant to the tissues from which they are secreted. These molecules require single pass transmembrane glycoproteins, known as klotho or βklotho proteins, to act as coreceptors to bind and activate FGF receptors.27
FGF receptors are comprised of an extracellular ligand-binding domain linked to an intracellular catalytic protein kinase core via a single pass transmembrane domain. Normally, the extracellular ligand binding domain of the receptor consists of three immunoglobulin domains (designated D1–3), a stretch of 7–8 acidic residues in the region connecting D1 to D2 (designated the “acid box”), and a conserved positively charged region in D2 that binds heparin.28 A feature unique to the FGF receptor family of receptor tyrosine kinases is the variety of isoforms that are generated by alternative splicing of FGF receptor mRNAs.29 Many splice isoforms have been described, the principal ones being alternative splicing of the D3 domain of FGF receptors 1–3, which determines the sequence of the carboxy-terminal half of the third membrane-proximal immunoglobulin domain and strongly dictates ligand-receptor binding (Table 1). In general, FGF receptor b isoforms are expressed on epithelial cells, while FGF receptor c isoforms are restricted to mesenchymal cell types, this being particularly true for FGF receptors 2 and 3.30 This lineage-specific expression of the FGF receptor b and c isoforms allows establishment of paracrine signaling loops between epithelial and mesenchymal tissues during development.
 | Table 1 Specificity of ligands for FGF receptor isoforms |
Ligand-induced dimerization of FGF receptors leads to a conformational shift in receptor structure and release of kinase autoinhibition. This results in a 50–100-fold increase in kinase activity of the receptor, resulting in activation through transphosphorylation of several tyrosine residues within the intracellular domain.31,32 This phosphorylation increases the receptor kinase activity, generating docking sites for downstream signaling molecules and consequent activation of multiple signal transduction pathways. Activation of the FGF receptor leads to phosphorylation of a number of intracellular proteins, such as FGF receptor substrate 2 and phospholipase Cγ.32,33 Further downstream signaling is believed to be due to a combination of mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt signaling, along with other pathways, being dominant in specific cell types or context.32,34,35
Role of FGF signaling in physiology and pathology
FGF signaling plays an important role in both embryogenesis and the adult organism, and during development FGF receptor signaling orchestrates a plethora of processes.35 FGF signaling is key to mesenchymal–epithelial interaction, and FGF receptors are well known inducers of mesoderm. In particular, endodermal to mesenchymal FGF signaling plays an important role in the development of the liver.36 Using an endoderm tissue explants system from mouse embryos, Jung et al showed that FGF signaling from the cardiac mesoderm is necessary for the induction of hepatic fate.37 Prior to hepatic induction, the cardiac mesoderm secretes FGF8, and at the time of hepatic development, secretes FGF1 and FGF2.38,39 Detailed studies of embryonic tissue explants of the ventral foregut endoderm have shown that, in the absence of FGF signaling from the cardiac mesoderm, the domain of the ventral foregut that is fated to become the liver rapidly defaults to the pancreas fate, by inhibiting pancreatic gene expression and inducing liver genes.37 Thus, the ventral foregut endoderm contains a bipotential precursor cell population for liver and pancreas, and FGF signaling diverts the endoderm from a default pancreas fate to that for the liver. In addition to the importance of FGF signaling for development of the liver, FGF receptor signaling is important for formation of the nervous system, the limbs, the midbrain, and the lungs.40 FGF signaling also plays an important role in the development of the embryonic mammary gland.41 In the adult, FGF receptor signaling regulates tissue repair, angiogenesis, and inflammation.40
Given the role of FGF signaling in the developing embryo and the adult, it is not surprising that this pathway is often hijacked by cancer cells.29 Indeed, the importance of FGF signaling in tumor pathogenesis was highlighted by a screen of more than 1,000 somatic mutations found in the coding exons of 518 protein kinase genes from over 200 different cancers. Of the non-synonymous mutations, FGF signaling was one of the most commonly mutated pathways.42 Several FGF receptor tyrosine kinase inhibitors and FGF receptor blocking antibodies are under various stages of preclinical and clinical development43 because of the oncogenic role of FGF in driving proliferation, survival, migration, invasion, and angiogenesis.44
Aberrant FGF signaling in HCC
HCC often emerges on a background of persistent liver injury, inflammation, and hepatocellular proliferation induced by cirrhosis or chronic hepatitis.45,46 This persistent injury induces aberrations in signaling pathways that give rise to early precursor lesions of HCC.32 These lesions respond to growth stimulatory cytokines and show increased proliferation and reduced cell death, providing a perfect environment for growth and expansion of cancer cells.47 Hepatocarcinogenesis is dependent on the development of a tumor-specific microenvironment in the cirrhotic liver composed of inflammatory cells, small vessels, myofibroblasts, and extracellular matrix components. These epithelial–mesenchymal interactions in the early and advanced stages of HCC are driven by various growth factors and their receptors.48 In addition to the signaling pathways induced by hepatocyte growth factor and insulin-like growth factors, aberrant FGF signaling is emerging as an important player in the development of liver tumors and their stroma.20,49 The main FGF receptors expressed in liver tissues are FGF receptors 3 and 4, and under normal conditions, FGF receptors 1 and 2 are expressed at low levels.50 Mounting evidence suggests that aberrant signaling of these receptors and their ligands may be involved in the mechanisms underlying the tumorigenesis of HCC, and as such will be discussed in detail in the following section.
FGF8
The FGF8 subfamily ligands (FGF8, 17, 18) are thought to bind with high affinity to the IIIC isoforms of FGF receptors 2 and 3 as well as FGF receptor 4.51 In normal adult homeostasis, FGF8 expression is restricted to steroid hormone target tissues. However, studies have shown that FGF8 is overexpressed in hormone-responsive tumors such as prostate and breast cancer.52 Further, FGF17 is also deregulated in prostate cancer and FGF18 is frequently overexpressed in ovarian and colon cancers.53–55 In the liver, FGF18 expression can induce hepatocyte proliferation, leading to increased liver weight, and is upregulated in rat HCC, suggesting a gain of autocrine function of FGF18 in HCC.56 Importantly, analysis of human HCC cases has shown that the FGF8 subfamily, as well as FGF receptors 2, 3, and 4, are upregulated in epithelial HCC cells.20 In vitro, members of the FGF8 subfamily are overexpressed in human HCC cells and contribute to the aggressive behavior of malignant hepatocytes under hypoxic conditions.20 Moreover, FGF8, FGF17, and FGF18 were able to induce neo-angiogenesis of hepatic endothelium and promoted growth of hepatic stellate cell-like cells found in the stroma of HCC patients,57 suggesting that aberrant autocrine and paracrine FGF signaling may drive development of HCC.20 Hepatic stellate cells were first described by Karl van Kuppfer in the 19th century as “sternzellen”, which were able to store vitamin A (retinol) as droplets in their cytoplasm.58 Hepatic stellate cells are the most abundant collagen-producing myofibroblast cells in the stroma of liver fibrosis, and are a target for therapy across different types of liver disease where fibrosis is prominent, including HCC.59 Involvement of stellate cells in the fibrotic response to liver injury has been recognized for several years.60,61 In response to repeated liver injury, hepatic stellate cells are activated and transdifferentiate into myofibroblast-like cells. Activated stellate cells are responsible for the production of cytokines, growth factors such as FGF2 and epidermal growth factor, two potent epithelial growth factors that play an important role in the proliferation of hepatocytes as well as extensive production of extracellular matrix.57 Studies have shown activation and consequent proliferation of stellate cells in regions of greatest injury.62,63
Indeed, in HCC and biliary malignancy, activated stellate cells contribute to accumulation of the tumor stroma and play a role in driving hepatic metastasis.64,65 Further, in vitro studies have also shown that stellate cells cross-talk with tumoral cells, fuelling a vicious cycle of paracrine activation and proliferation.66 Indeed, we have recently identified a potential novel mechanism by which FGF signaling regulates pancreatic stromal cell behavior and cross-talk with pancreatic cancer cells.67 Other studies have shown that lipid-storing pancreatic stellate cells are abundant in the areas of fibrosis and are capable of secreting extracellular matrix proteins in patients with chronic alcoholic pancreatitis.68,69 These studies suggest that pancreatic stellate cells are a possible source of pancreatic fibrosis, similar to that observed in alcohol-related liver fibrosis. Further, pancreatic stellate cells are thought to be the key cell type driving the desmoplasia characteristic of pancreatic ductal adenocarcinoma and conferring resistance to conventional therapies.68,70 We have shown that both FGF2 and FGF receptor 1 colocalize to the nucleus exclusively in pancreatic stellate cells at the invasive front of human pancreatic ductal adenocarcinoma.67 Using a three-dimensional organotypic model of pancreatic ductal adenocarcinoma developed in our laboratory to explore the cross-talk between pancreatic stellate cells and cancer cells, we showed that pancreatic stellate cells with nuclear FGF receptor 1 and FGF2 led cancer cells to invade the underlying extracellular matrix.71 Thus, nuclear FGF receptor 1 and FGF2 in activated stromal pancreatic stellate cells may facilitate invasion of pancreatic stellate cells at the invasive front of pancreatic ductal adenocarcinoma. FGF receptor inhibition profoundly blocked invasion of both pancreatic stellate cells and cancer cells, and resulted in cytoplasmic localization of FGF receptor 1 and FGF2. These findings suggest a novel therapeutic approach, where preventing nuclear FGF/FGF receptor-mediated proliferation and invasion in pancreatic stellate cells leads to disruption of the tumor microenvironment, preventing invasion of pancreatic cancer cells. Pancreatic stellate cells are nearly identical to hepatic stellate cells, and both are thought to share a common origin,72 thus targeting FGF signaling in hepatic stellate cells may offer a therapeutic target in HCC.
FGF receptor 1
FGF receptor 1 expression is low in normal liver epithelium. Recently, Wang et al have identified high expression of FGF receptor 1 in the tumor epithelium of patients with HCC.73 In the prostate epithelium, FGF receptor 1 regulates matrix turnover and cell invasion by affecting expression of matrix-degrading enzymes, and overexpression of FGF receptor 1 is associated with an aggressive phenotype in prostate and breast cancer.74,75 Thus, FGF receptor 1 may function in invasion of cancer cells. How overexpression of FGF receptor 1 is regulated in HCC is not well understood. Recent studies have suggested that FGF receptor 1 may be under the control of the microRNA, miR-214.73 Deregulation of microRNAs is involved in the development of cancer and can be related to clinical outcome in cancer patients, including those with HCC.76,77 While microRNA overexpression has been seen in some cancers, including HCC, it appears that expression of miR-214 may inhibit development of HCC by target regulation of FGF receptor 1 gene expression.73 Indeed, it has been previously reported that reduced expression of miR-214 contributes to intrahepatic metastasis of cholangiocarcinoma by targeting Twist.78 In HCC cells, miR-214 has been reported to directly interact with FGF receptor 1, and its downregulation in HCC correlates with high FGF receptor 1 expression promoting invasion of HCC cells.79 Thus, during tumor progression, decreased miR-214 levels and subsequent overexpression of FGF receptor 1 may play a role in promoting the aggressiveness of HCC.
FGF19 and FGF receptor 4
The FGF receptor 4–FGF19 signaling axis is an attractive target for progression of HCC for several reasons.17,18,80,81 Firstly, the main FGF receptors expressed in the liver are FGF receptors 3 and 4, and the hepatocyte is the only human cell type in which FGF receptor 4 is the predominant isoform of FGF receptors.82 Secondly, it is reported that the liver has the highest transcript expression of FGF receptor 4 compared with other organs, as well as high expression of βklotho, which is required for the liver-specific activities of FGF19.82 Thirdly, in the normal liver, hepatocyte FGF19 and FGF receptor 4 regulate biosynthesis in the bile duct by repression of the bile acid enzyme, cholesterol 7 α-hydroxylase.83,84 However, ectopic expression of FGF19 (in skeletal muscle) but not FGF21 in mice, is sufficient to induce hepatocyte proliferation, dysplasia, and neoplasia, suggesting that specific aberration in FGF19-FGF receptor 4 signaling may have a strong pathophysiological impact in the liver.85,86 Indeed, recent reports have shown that a neutralizing antibody that selectively blocks the interaction between FGF receptor 4 and FGF19 inhibits the growth of xenograft colon and liver tumors in vivo.81
Performing a comprehensive mutation analysis of 57 human HCC and normal tissue samples, Ho et al found that FGF receptor 4 harbored eight tumor-associated genetic alterations, including two very common single nucleotide polymorphisms, ie, V101 and G338R. Approximately one third of the HCC samples overexpressed FGF receptor 4 when compared with normal tissues. Further, patients who harbored the G338R single nucleotide polymorphism secreted greater amounts of alpha-fetoprotein, a widely used biomarker for HCC. In vitro studies have shown that this polymorphism is important for production of alpha-fetoprotein as well as proliferation and survival of HCC cells.87 Moreover, the G388R mutation is more common in Asian populations than in other ethnicities. This single nucleotide polymorphism association, if validated in a larger study, could have important consequences for development of HCC and could possibly act as a predictive marker, since these single nucleotide polymorphisms may modify FGF receptor function, particularly in Asian populations with a high incidence of HCC.88 Using a genome wide approach, Sawey et al identified 18 tumor-promoting genes that are amplified in human HCC, of which FGF19 was one of the most important.18 Gene amplification does not always correspond to increased expression of FGF19, and this may be tissue-specific. For example, FGF19 was found not to be overexpressed despite gene amplification in oral cancer, lung cancer, and melanoma.18,89 However, in HCC, amplification of FGF19 correlates with strong overexpression. Orthotopic transplantation of hepatocytes with amplified FGF19 resulted in highly proliferative tumors. Furthermore, following inhibition of FGF19 with RNA interference or a FGF19 monoclonal antibody, the clonal growth and tumorigenicity of human HCC cells harboring the FGF19 amplicon was blocked.18 This suggests that FGF19 is an oncogene which is amplified and overexpressed in HCC and is a promising target for therapy. Indeed, overexpression of FGF19 in human HCC was found to be an independent prognostic factor for a poor response.17
FGF receptor 2IIIb
In the normal liver, FGF receptor 2IIIb is expressed on hepatocytes and plays a role in liver regeneration and homeostasis.90 However, expression of FGF receptor 2IIIb is downregulated or lost in many HCC cell lines and tissues, and as a consequence can induce growth of HCC cells in vitro and in tumor xenografts,91 suggesting a tumor-suppressive role. This seemingly paradoxical role of FGF receptors in tumor development is not well understood, particularly as FGF receptors are reported to be potent oncogenes in tumorigenesis.29 However, HCC is not unique in this respect. FGF receptor 2IIIb is downregulated in several cancers, including those of the bladder and prostate, and loss of function mutations in the FGF receptor 2 gene has been detected in malignant melanoma.92 One possible explanation for this phenomenon is that the non-phosphotyrosine-containing region within the C-terminal part of FGF receptor 2IIIb may play a key role in FGF receptor 2IIIb-induced inhibitory signals in some tumors, such as HCC.93
Cross-talk of FGF and other signaling pathways in HCC
An additional manner by which signaling molecules can elicit distinct responses in different cell types is through activation or repression of other signaling pathways. This cross-talk between signaling pathways results from specific interactions between signal transducing molecules, and convergence or divergence of the programs for gene expression activated by each pathway. Importantly, FGFs have been shown to interact with a number of signaling pathways in a variety of developmental systems and, in some cases, simultaneous activation of these signaling pathways leads to effects that are distinct from the individual effects of each factor.94
One such example is that of the interaction between the WNT and FGF signaling pathways. WNT family members are secreted glycoproteins that bind to Frizzled transmembrane receptors and the LRP5/LRP6 coreceptor on the cell surface.95 A key event in the canonical WNT pathway is the activation of β-catenin, which subsequently regulates transcription of specific target genes that modulate cell proliferation and apoptosis.96 β-catenin is a dual function protein that plays a key role in maintaining cell–cell adhesion via association of E-cadherin and linking cadherins to the cytoskeleton as well as the canonical and noncanonical WNT signaling cascade.97 Activation of WNT signaling in carcinogenesis leads to induction of FGF signaling activation and induces epithelial–mesenchymal transition.98 Thus, coactivation of the WNT and FGF signaling pathways leads to a more malignant phenotype in carcinogenesis. For example, treatment with FGF2 promotes translocation of β-catenin to the nucleus and maintains the proliferation of multipotent neural stem cells.99 Indeed, recent results from our group suggest that pancreatic cancer cells show active WNT signaling. Given that FGF2 is readily secreted by pancreatic stellate cells in pancreatic ductal adenocarcinoma, this may be one way in which FGF signaling may modulate the effect of WNT signaling in cancer cells and fuel an increase in tumor cell growth.100
In certain contexts, for example, in colorectal carcinogenesis, coactivation of WNT and FGF signaling pathways in tumors, such as FGF19, directly modulating β-catenin signaling by loss of β-catenin/E-cadherin binding, leads to a more malignant phenotype.101 Further, inhibition of FGF19 signaling reduces β-catenin signaling. This cross-talk is apparent in HCC, whereby overexpression of FGF19 can induce β-catenin activity and lead to elevation of the β-catenin downstream target cyclin D1 protein.18 Along with FGF19, cyclin D1 is an important oncogene in HCC and can drive tumorigenesis.18 Importantly, blocking FGF19 signaling abolishes β-catenin activation, and when HCC cells are treated with small interfering RNA to β-catenin, FGF19 can no longer induce cyclin D1 activity. Thus, in addition to the well established pathway involving RAS/RAF/mitogen-activated protein kinase signaling, FGF19 is able to induce expression of cyclin D1 through β-catenin signaling in HCC cells.18 Further, deregulated Wnt/β-catenin signaling is able to transform stem/progenitor cells in the liver and as such may play an important role in the maintenance of liver cancer stem cells.102 WNT/β-catenin can directly target the hepatic progenitor cell marker EpCAM and induce its expression in HCC.103 A number of EpCAM-regulated target genes have been identified, including c-myc and cyclins, which regulate cell proliferation, growth, and survival.104 Studies by Yang et al have shown that poorly differentiated HCC cells characterized by the hepatic progenitor marker OV6 are enriched in response to overexpression of WNT/β-catenin. Importantly, in these studies, silencing of β-catenin prevents the development of chemoresistant HCC populations, which demonstrate progenitor-like characteristics.105 Further, Mavila et al have recently shown that treatment of well characterized tumor-initiating liver stem cells (CD133-expressing tumor-initiating cells isolated from the livers of methionine adenosyltransferase 1A null mice106) with recombinant FGF7 or FGF10 is sufficient to induce nuclear translocation of β-catenin and subsequent proliferation of progenitor cells.107 Thus, further understanding of the cross-talk between FGF and WNT signaling could aid in the development of therapeutics that target this signaling pathway in progenitor/cancer stem cells, reducing chemoresistance in HCC.
Abnormal vascularity of HCC is harnessed in the key diagnostic test by analyzing the differential perfusion of liver lesions on computed tomography and magnetic resonance scans. HCC vessels are excessively leaky and have arteriovenous shunts. Although HCC is a highly angiogenic cancer, it is also characterized by hypoxia which may promote growth and resistance of HCC to therapies.108 Studies have shown that inducing vessel normalization and preventing hypoxia can reduce growth of HCC.109 Amongst some of the best characterized angiogenic factors that are often secreted by cancer cells are FGF2 and VEGF.110 FGFs are key factors, particularly in tumor angiogenesis.111,112 FGF2 is a known mitogen for a number of cells, including myofibroblasts and vascular endothelial cells, and is often overexpressed in a number of cancers, including HCC.19,110 Indeed, in a clinical study of patients undergoing resection of HCC, a high preoperative serum FGF2 level was predictive of tumor invasion and recurrence.19 In addition to FGF2, levels of VEGF in serum are elevated in patients with HCC and correlate with a poor response to chemoembolization.113
Studies have shown that VEGF and FGF2 have synergistic effects as inducers of angiogenesis.114 FGF2 is able to induce overexpression of VEGF and its receptor (VEGF receptor 1) in endothelial cells and several other cell types. Addition of exogenous VEGF and FGF2 to three-dimensional microvascular endothelial cell cultures can induce cellular invasion and capillary-like tubule formation to a greater extent than addition of each growth factor alone. In vivo, xenografts simultaneously treated with FGF2 and VEGF show fast growing tumors with high blood vessel density, patency, and permeability. Further, angiogenesis enhanced by FGF2 can be significantly inhibited using a VEGF neutralizing antibody and, under certain circumstances, FGF receptor signaling may mediate resistance to VEGF receptor targeting.114,115 In HCC, the combined effect of FGF2 and VEGF increases tumor growth and angiogenesis as tested by inducible expression of FGF2 or VEGF in vivo.116 Further, overexpression of FGF2 can induce expression of VEGF, which leads to an increase in tumor development, and FGF2-induced augmentation is suppressed using the monoclonal antibody to VEGF receptor 1.117 These results suggest that VEGF is located downstream of FGF2 and together they can synergistically increase VEGF-mediated development of HCC.
Finally, studies by Ueba et al have shown that FGF2 may be under the control of mutated p53 in HCC.118 p53 is a nuclear phosphoprotein that regulates expression of various genes in the manner of sequence-specific DNA binding and or/protein-protein interactions, and is often mutated in cancers, including HCC.119,120 Using HCC cell lines transfected with a dominant negative mutant p53, they showed that mutant p53 was able to induce FGF2 promoter activity, while wild-type p53 repressed it.118 These findings demonstrate one possible mechanism for FGF2 activity in HCC tumor progression. Loss of normal function of p53 may trigger activation of FGF2 transcription, resulting in tumor progression.
Therapeutic options
Several chemotherapeutic agents have been evaluated for the treatment of HCC; however, no single or combination therapy regimen is particularly effective.121 Prior to the advent of sorafenib, doxorubicin was routinely used as a single drug for advanced HCC. However, despite initial encouraging reports for single-agent doxorubicin, this has proven to lack efficacy, with a response rate of about 15%–20%.122,123 Other chemotherapy agents, such as epirubicin, cisplatin, 5-fluorouracil, etoposide, and their combinations demonstrate even lower efficacy.123 Sorafenib, a multitargeted tyrosine kinase inhibitor, is the first targeted therapy to improve the survival of patients with advanced stage HCC in Phase III trials.124 Sorafenib was designed to target VEGF receptors 1–3; however, the exact mechanism of action of this drug in HCC patients is still unknown.108,125 No effective second-line treatment options currently exist for patients who are resistant or refractory to and/or intolerant of sorafenib.125 Given the pathogenetic role of FGF/FGF receptor signaling in HCC, treatment targeting FGF signaling may benefit patients. Several novel FGF receptor-targeted agents (multikinase inhibitors and monoclonal antibodies) are currently being developed and explored for HCC (Table 2).
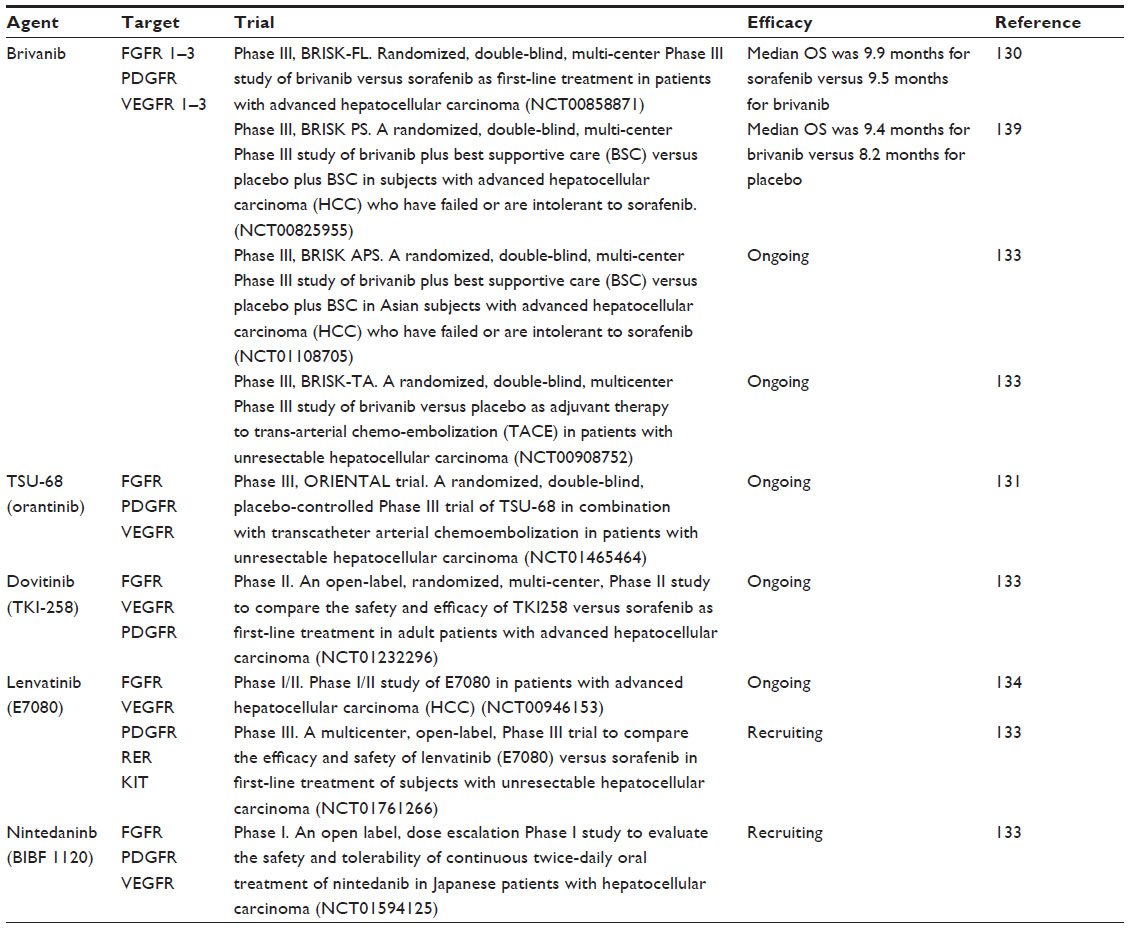 | Table 2 Novel targeted multikinase inhibitors currently being investigated in clinical trials for hepatocellular carcinoma |
Dual inhibition of VEGF and FGF receptors
Adding to the challenges of single-target inhibition of VEGF, recent data suggest that although VEGF inhibitors reduce primary tumor growth, they also promote tumor invasiveness and metastasis.126 Given that FGF signaling may contribute to acquired resistance or compensatory signaling during anti-VEGF receptor therapy, simultaneous inhibition of these two pathways may provide a mechanism to overcome resistance to VEGF-targeted agents in HCC.127 TSU-68 (SU6668; Taiho Pharmaceuticals Co, Ltd, Tokyo, Japan) is a dual inhibitor of VEGF receptor 2 and FGF receptors 1–3.79 Preclinical reports show that treatment with brivanib can inhibit growth of HCC and that TSU-68 can normalize tumor vasculature in xenograft mouse models.128 Brivanib has demonstrated activity against several types of solid tumors in clinical trials and is currently under investigation in many cancer types.129 Data from a single-arm Phase II study in advanced HCC have demonstrated the antitumor activity of brivanib in first-line and second-line therapy.128 Brivanib is being evaluated in the first-line setting versus sorafenib in Phase III studies and in the second-line setting in patients with refractory advanced stage HCC by the Brivanib Study in Patients at Risk trials (BRISK-FL, BRISK-PS, and BRISK-APS).125 Despite brivanib showing promise in early Phase II trials, recent results show that the BRISK-PS study did not meet its primary endpoint of improving overall survival. However, treatment with brivanib did result in overall improvement in cancer response rates.130 Similarly, the BRISK-FL trial, directly comparing clinical outcomes of brivanib versus sorafenib in patients with advanced HCC who had received no prior systemic therapy, failed to demonstrate a statistically significant difference in overall survival (9.5 months versus 9.9 months).125 TSU-68 has demonstrated some clinical efficacy in a Phase I/II trial of heavily pretreated patients with advanced HCC who had a mean survival of 13.1 months, suggesting that TSU-68 may have a clinical benefit in patients with advanced HCC. A larger randomized Phase III study is now recruiting patients with unresectable HCC to evaluate transcatheter arterial chemoembolization in combination with either TSU-68 or placebo.131
Multitarget tyrosine receptor kinase inhibitors
Dovitinib (TKI 258; Novartis, Basel, Switzerland) is an inhibitor of FGF, PDGF, and VEGF receptors and has shown promising activity in early-phase trials in solid tumors.132 Dovitinib is currently being tested in a Phase II study to compare its safety and efficacy versus that of sorafenib as first-line treatment in adult patients with advanced HCC, with overall survival as the primary endpoint. Patient recruitment has finished and the study is planned to be completed later in the year.133
Lenvatinib (E7080; Eisai Co, Ltd, Tokyo, Japan) is an orally administered tyrosine kinase inhibitor that targets VEGF receptors 1–3 and FGF receptors 1–4. After showing preliminary antitumor activity and safety profile in a Phase I/II study, a randomized, double-blind Phase III study is currently comparing the efficacy and safety of lenvatinib versus sorafenib in the first-line treatment of patients with unresectable HCC (ClinicalTrials.gov identifier NCT01761266).133,134 Finally, early-phase studies are ongoing in HCC patients to test the safety and tolerability of the multikinase inhibitor nintedanib (BIBF 1220; Boehringer Ingelheim, Rhineland-Palatinate, Germany) which targets VEGF receptors 1–3, FGF receptors 1–3, and PDGF receptors (ClinicalTrials.gov identifier NCT01594125).
Conclusion
To date, many of these targeted therapies have shown little liver toxicity, which is surprising given the role of FGF receptors in normal liver physiology. This may be explained by the failure of many multikinase inhibitors to target FGF receptor 4, which has a major role in normal hepatocytes. FGF receptor 4 shows the lowest degree of homology with other FGF receptor family members, and as such none of the small-molecule tyrosine kinase inhibitors developed so far show specificity for FGF receptor 4.135 This may also explain why some of these targeted therapies, such as sravanib, have not achieved overall survival endpoints in larger Phase III trials, given that FGF receptor 4 has been shown to play a major role in the development and aggressiveness of HCC. Thus, patients may need to be selected on the basis of their FGF status to achieve more efficacious results in the trial setting. Nevertheless, given the important metabolic functions of FGF receptor 4 in the liver, the side effects of an FGF receptor 4-specific inhibitor should be carefully considered.136 FGF receptor 4 null mice show elevated cholesterol metabolism and bile acid synthesis and were vulnerable to liver damage.83 However, studies have shown that blocking FGF receptor 4 activity (either biochemically or genetically, as discussed above) has an anticancer effect, particularly in cancers overexpressing FGF receptor 4. Therefore, it is important to develop an FGF receptor 4-specific inhibitor in order to clarify the importance of this therapeutic strategy in patients harboring FGF receptor 4 mutations. Recently, in silico design has identified FGF receptor 4 inhibitors as potential anticancer agents that have shown promise in early in vitro studies.137 Further, blocking FGF receptor 4 in some instances has led to selection of subpopulations of treatment-refractory cells, and should be considered when targeting FGF receptor 4-mediated signaling in HCC in order to prevent development of resistance to therapy.138
However, given the importance of FGF receptor signaling in the development of HCC, and the clinical trial testing of promising new kinase inhibitors that target FGF receptors, there is promise for improving the prognosis of HCC patients who have limited therapeutic options.
Disclosure
The authors report no conflicts of interest in this work.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | |
Dyer Z, Peltekian K, van Zanten SV. Review article: the changing epidemiology of hepatocellular carcinoma in Canada. Aliment Pharmacol Ther. 2005;22(1):17–22. | |
El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–2576. | |
Altekruse SF, McGlynn KA, Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol. 2009;27(9):1485–1491. | |
Bosch FX, Ribes J, Diaz M, Cleries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127(5 Suppl 1):S5–S16. | |
Poon RT. Optimal initial treatment for early hepatocellular carcinoma in patients with preserved liver function: transplantation or resection? Ann Surg Oncol. 2007;14(2):541–547. | |
Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362(9399):1907–1917. | |
Thomas MB, Zhu AX. Hepatocellular carcinoma: the need for progress. J Clin Oncol. 2005;23(13):2892–2899. | |
Altekruse SF, McGlynn KA, Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol. 2009;27(9):1485–1491. | |
Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. | |
Cheng AL, Kang YK, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a Phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10(1):25–34. | |
Worns MA, Koch S, Niederle IM, et al. The impact of patient and tumour baseline characteristics on the overall survival of patients with advanced hepatocellular carcinoma treated with Sorafenib. Dig Liver Dis. 2013;45(5):408–413. | |
El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118–1127. | |
Lee JS, Heo J, Libbrecht L, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med. 2006;12(4):410–416. | |
Hoshida Y, Villanueva A, Llovet JM. Molecular profiling to predict hepatocellular carcinoma outcome. Expert Rev Gastroenterol Hepatol. 2009;3(2):101–103. | |
Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6(9):674–687. | |
Miura S, Mitsuhashi N, Shimizu H, et al. Fibroblast growth factor 19 expression correlates with tumor progression and poorer prognosis of hepatocellular carcinoma. BMC Cancer. 2012;12:56. | |
Sawey ET, Chanrion M, Cai C, et al. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by Oncogenomic screening. Cancer Cell. 2011;19(3):347–358. | |
Poon RT, Ng IO, Lau C, Yu WC, Fan ST, Wong J. Correlation of serum basic fibroblast growth factor levels with clinicopathologic features and postoperative recurrence in hepatocellular carcinoma. Am J Surg. 2001;182(3):298–304. | |
Gauglhofer C, Sagmeister S, Schrottmaier W, et al. Up-regulation of the fibroblast growth factor 8 subfamily in human hepatocellular carcinoma for cell survival and neoangiogenesis. Hepatology. 2011;53(3):854–864. | |
Ho HK, Pok S, Streit S, et al. Fibroblast growth factor receptor 4 regulates proliferation, anti-apoptosis and alpha-fetoprotein secretion during hepatocellular carcinoma progression and represents a potential target for therapeutic intervention. J Hepatol. 2009;50(1):118–127. | |
Gospodarowicz D. Localisation of a fibroblast growth factor and its effect alone and with hydrocortisone on 3T3 cell growth. Nature. 1974;249(453):123–127. | |
Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8(3):235–253. | |
Wiedemann M, Trueb B. Characterization of a novel protein (FGF receptorL1) from human cartilage related to FGF receptors. Genomics. 2000;69(2):275–279. | |
Harmer NJ, Ilag LL, Mulloy B, Pellegrini L, Robinson CV, Blundell TL. Towards a resolution of the stoichiometry of the fibroblast growth factor (FGF)-FGF receptor-heparin complex. J Mol Biol. 2004;339(4):821–834. | |
Mohammadi M, Olsen SK, Ibrahimi OA. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005;16(2):107–137. | |
Kuro-o M. Klotho and betaKlotho. Adv Exp Med Biol. 2012;728: 25–40. | |
Schlessinger J, Plotnikov AN, Ibrahimi OA, et al. Crystal structure of a ternary FGF-FGF receptor-heparin complex reveals a dual role for heparin in FGF receptor binding and dimerization. Mol Cell. 2000;6(3):743–750. | |
Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10(2):116–129. | |
Kornmann M, Beger HG, Korc M. Role of fibroblast growth factors and their receptors in pancreatic cancer and chronic pancreatitis. Pancreas. 1998;17(2):169–175. | |
Dailey L, Ambrosetti D, Mansukhani A, Basilico C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 2005;16(2):233–247. | |
Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16(2):139–149. | |
Peters KG, Marie J, Wilson E, et al. Point mutation of an Fgf receptor abolishes phosphatidylinositol turnover and Ca2+ flux but not mitogenesis. Nature. 1992;358(6388):678–681. | |
Klint P, Claesson-Welsh L. Signal transduction by fibroblast growth factor receptors. Front Biosci. 1999;4:D165–D177. | |
Haugsten EM, Wiedlocha A, Olsnes S, Wesche J. Roles of fibroblast growth factor receptors in carcinogenesis. Mol Cancer Res. 2010;8(11):1439–1452. | |
Fukuda-Taira S. Hepatic induction in the avian embryo: specificity of reactive endoderm and inductive mesoderm. J Embryol Exp Morphol. 1981;63:111–125. | |
Jung J, Zheng M, Goldfarb M, Zaret KS. Initiation of mammalian liver development from endoderm by fibroblast growth factors. Science. 1999;284(5422):1998–2003. | |
Crossley PH, Martin GR. The mouse Fgf8 gene encodes a family of polypeptides and is expressed in regions that direct outgrowth and patterning in the developing embryo. Development. 1995;121(2):439–451. | |
Zhu X, Sasse J, McAllister D, Lough J. Evidence that fibroblast growth factors 1 and 4 participate in regulation of cardiogenesis. Dev Dyn. 1996;207(4):429–438. | |
Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer. 2000;7(3):165–197. | |
Parsa S, Ramasamy SK, De Langhe S, et al. Terminal end bud maintenance in mammary gland is dependent upon FGF receptor2b signaling. Dev Biol. 2008;317(1):121–131. | |
Greenman C, Stephens P, Smith R, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446(7132):153–158. | |
Knights V, Cook SJ. De-regulated FGF receptors as therapeutic targets in cancer. Pharmacol Ther. 2010;125(1):105–117. | |
Jeffers M, LaRochelle WJ, Lichenstein HS. Fibroblast growth factors in cancer: therapeutic possibilities. Expert Opin Ther Targets. 2002;6(4):469–482. | |
Berasain C, Castillo J, Perugorria MJ, Latasa MU, Prieto J, Avila MA. Inflammation and liver cancer: new molecular links. Ann NY Acad Sci. 2009;1155:206–221. | |
Grasl-Kraupp B, Luebeck G, Wagner A, et al. Quantitative analysis of tumor initiation in rat liver: role of cell replication and cell death (apoptosis). Carcinogenesis. 2000;21(7):1411–1421. | |
Drucker C, Parzefall W, Teufelhofer O, et al. Non-parenchymal liver cells support the growth advantage in the first stages of hepatocarcinogenesis. Carcinogenesis. 2006;27(1):152–161. | |
Imbeaud S, Ladeiro Y, Zucman-Rossi J. Identification of novel oncogenes and tumor suppressors in hepatocellular carcinoma. Semin Liver Dis. 2010;30(1):75–86. | |
Berasain C, Perugorria MJ, Latasa MU, et al. The epidermal growth factor receptor: a link between inflammation and liver cancer. Exp Biol Med (Maywood). 2009;234(7):713–725. | |
Hughes SE. Differential expression of the fibroblast growth factor receptor (FGF receptor) multigene family in normal human adult tissues. J Histochem Cytochem. 1997;45(7):1005–1019. | |
Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem. 2006;281(23):15694–15700. | |
Mattila MM, Harkonen PL. Role of fibroblast growth factor 8 in growth and progression of hormonal cancer. Cytokine Growth Factor Rev. 2007;18(3–4):257–266. | |
Sonvilla G, Allerstorfer S, Stattner S, et al. FGF18 in colorectal tumour cells: autocrine and paracrine effects. Carcinogenesis. 2008;29(1):15–24. | |
Tchagang AB, Tewfik AH, DeRycke MS, Skubitz KM, Skubitz AP. Early detection of ovarian cancer using group biomarkers. Mol Cancer Ther. 2008;7(1):27–37. | |
Heer R, Douglas D, Mathers ME, Robson CN, Leung HY. Fibroblast growth factor 17 is over-expressed in human prostate cancer. J Pathol. 2004;204(5):578–586. | |
Hu MC, Qiu WR, Wang YP, et al. FGF-18, a novel member of the fibroblast growth factor family, stimulates hepatic and intestinal proliferation. Mol Cell Biol. 1998;18(10):6063–6074. | |
Leonardi GC, Candido S, Cervello M, et al. The tumor microenvironment in hepatocellular carcinoma (review). Int J Oncol. 2012;40(6):1733–1747. | |
Wake K. “Sternzellen” in the liver: perisinusoidal cells with special reference to storage of vitamin A. Am J Anat. 1971;132(4):429–462. | |
Mederacke I, Hsu CC, Troeger JS, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823. | |
Okanoue T, Burbige EJ, French SW. The role of the Ito cell in perivenular and intralobular fibrosis in alcoholic hepatitis. Arch Pathol Lab Med. 1983;107(9):459–463. | |
Bronfenmajer S, Schaffner F, Popper H. Fat-storing cells (lipocytes) in human liver. Arch Pathol. 1966;82(5):447–453. | |
Johnson SJ, Hines JE, Burt AD. Macrophage and perisinusoidal cell kinetics in acute liver injury. J Pathol. 1992;166(4):351–358. | |
Tanaka Y, Nouchi T, Yamane M, et al. Phenotypic modulation in lipocytes in experimental liver fibrosis. J Pathol. 1991;164(3):273–278. | |
Torimura T, Ueno T, Inuzuka S, et al. The extracellular matrix in hepatocellular carcinoma shows different localization patterns depending on the differentiation and the histological pattern of tumors: immunohistochemical analysis. J Hepatol. 1994;21(1):37–46. | |
Schmitt-Graff A, Chakroun G, Gabbiani G. Modulation of perisinusoidal cell cytoskeletal features during experimental hepatic fibrosis. Virchows Arch A Pathol Anat Histopathol. 1993;422(2):99–107. | |
Faouzi S, Lepreux S, Bedin C, et al. Activation of cultured rat hepatic stellate cells by tumoral hepatocytes. Lab Invest. 1999;79(4):485–493. | |
Coleman SJ, Chioni AM, Ghallab M, et al. Nuclear translocation of FGF receptor1 and FGF2 in pancreatic stellate cells facilitates pancreatic cancer cell invasion. EMBO Mol Med. 2014;6(4):467–481. | |
Apte MV, Park S, Phillips PA, et al. Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas. 2004;29(3):179–187. | |
Bachem MG, Schunemann M, Ramadani M, et al. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology. 2005;128(4):907–921. | |
Mantoni TS, Lunardi S, Al-Assar O, Masamune A, Brunner TB. Pancreatic stellate cells radioprotect pancreatic cancer cells through beta1-integrin signaling. Cancer Res. 2011;71(10):3453–3458. | |
Froeling FE, Mirza TA, Feakins RM, et al. Organotypic culture model of pancreatic cancer demonstrates that stromal cells modulate E-cadherin, beta-catenin, and Ezrin expression in tumor cells. Am J Pathol. 2009;175(2):636–648. | |
Buchholz M, Kestler HA, Holzmann K, et al. Transcriptome analysis of human hepatic and pancreatic stellate cells: organ-specific variations of a common transcriptional phenotype. J Mol Med (Berl). 2005;83(10):795–805. | |
Wang J, Li J, Wang X, Zheng C, Ma W. Downregulation of microRNA-214 and overexpression of FGF receptor-1 contribute to hepatocellular carcinoma metastasis. Biochem Biophys Res Commun. 2013;439(1):47–53. | |
Tiburcio M, Costa SM, De Fatima Duarte M, Schmitt FC, Longatto Filho A. Characterization of PAR1 and FGF receptor1 expression in invasive breast carcinomas: prognostic significance. Oncol Lett. 2012;4(4):647–657. | |
Yang F, Zhang Y, Ressler SJ, et al. FGF receptor1 is essential for prostate cancer progression and metastasis. Cancer Res. 2013;73(12):3716–3724. | |
Nicoloso MS, Spizzo R, Shimizu M, Rossi S, Calin GA. MicroRNAs – the micro steering wheel of tumour metastases. Nat Rev Cancer. 2009;9(4):293–302. | |
Law PT, Wong N. Emerging roles of microRNA in the intracellular signaling networks of hepatocellular carcinoma. J Gastroenterol Hepatol. 2011;26(3):437–449. | |
Li B, Han Q, Zhu Y, Yu Y, Wang J, Jiang X. Down-regulation of miR-214 contributes to intrahepatic cholangiocarcinoma metastasis by targeting Twist. FEBS J. 2012;279(13):2393–2398. | |
Cheng AL, Shen YC, Zhu AX. Targeting fibroblast growth factor receptor signaling in hepatocellular carcinoma. Oncology. 2011;81(5–6):372–380. | |
French DM, Lin BC, Wang M, et al. Targeting FGF receptor4 inhibits hepatocellular carcinoma in preclinical mouse models. PLoS One. 2012;7(5):e36713. | |
Desnoyers LR, Pai R, Ferrando RE, et al. Targeting FGF19 inhibits tumor growth in colon cancer xenograft and FGF19 transgenic hepatocellular carcinoma models. Oncogene. 2008;27(1):85–97. | |
Kan M, Wu X, Wang F, McKeehan WL. Specificity for fibroblast growth factors determined by heparan sulfate in a binary complex with the receptor kinase. J Biol Chem. 1999;274(22):15947–15952. | |
Yu C, Wang F, Kan M, et al. Elevated cholesterol metabolism and bile acid synthesis in mice lacking membrane tyrosine kinase receptor FGF receptor4. J Biol Chem. 2000;275(20):15482–15489. | |
Song KH, Li T, Owsley E, Strom S, Chiang JY. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology. 2009;49(1):297–305. | |
Nicholes K, Guillet S, Tomlinson E, et al. A mouse model of hepatocellular carcinoma: ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am J Pathol. 2002;160(6):2295–2307. | |
Wu X, Ge H, Lemon B, et al. FGF19-induced hepatocyte proliferation is mediated through FGF receptor4 activation. J Biol Chem. 2010;285(8):5165–5170. | |
Ho HK, Pok S, Streit S, et al. Fibroblast growth factor receptor 4 regulates proliferation, anti-apoptosis and alpha-fetoprotein secretion during hepatocellular carcinoma progression and represents a potential target for therapeutic intervention. J Hepatol. 2009;50(1):118–127. | |
Deuffic S, Poynard T, Buffat L, Valleron AJ. Trends in primary liver cancer. Lancet. 1998;351(9097):214–215. | |
Huang X, Godfrey TE, Gooding WE, McCarty KS Jr, Gollin SM. Comprehensive genome and transcriptome analysis of the 11q13 amplicon in human oral cancer and synteny to the 7F5 amplicon in murine oral carcinoma. Genes Chromosomes Cancer. 2006;45(11):1058–1069. | |
Steiling H, Wustefeld T, Bugnon P, et al. Fibroblast growth factor receptor signalling is crucial for liver homeostasis and regeneration. Oncogene. 2003;22(28):4380–4388. | |
Amann T, Bataille F, Spruss T, et al. Reduced expression of fibroblast growth factor receptor 2IIIb in hepatocellular carcinoma induces a more aggressive growth. Am J Pathol. 2010;176(3):1433–1442. | |
Gartside MG, Chen H, Ibrahimi OA, et al. Loss-of-function fibroblast growth factor receptor-2 mutations in melanoma. Mol Cancer Res. 2009;7(1):41–54. | |
Bernard-Pierrot I, Ricol D, Cassidy A, et al. Inhibition of human bladder tumour cell growth by fibroblast growth factor receptor 2b is independent of its kinase activity. Involvement of the carboxy-terminal region of the receptor. Oncogene. 2004;23(57):9201–9211. | |
Moon RT, Brown JD, Yang-Snyder JA, Miller JR. Structurally related receptors and antagonists compete for secreted Wnt ligands. Cell. 1997;88(6):725–728. | |
Katoh M. Networking of WNT, FGF, Notch, BMP, and Hedgehog signaling pathways during carcinogenesis. Stem Cell Rev. 2007;3(1):30–38. | |
Moon RT, Bowerman B, Boutros M, Perrimon N. The promise and perils of Wnt signaling through beta-catenin. Science. 2002;296(5573):1644–1646. | |
Kemler R. From cadherins to catenins: cytoplasmic protein interactions and regulation of cell adhesion. Trends Genet. 1993;9(9):317–321. | |
Katoh M. Cross-talk of WNT and FGF signaling pathways at GSK3beta to regulate beta-catenin and SNAIL signaling cascades. Cancer Biol Ther. 2006;5(9):1059–1064. | |
Israsena N, Hu M, Fu W, Kan L, Kessler JA. The presence of FGF2 signaling determines whether beta-catenin exerts effects on proliferation or neuronal differentiation of neural stem cells. Dev Biol. 2004;268(1):220–231. | |
Froeling FE, Feig C, Chelala C, et al. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-beta-catenin signaling to slow tumor progression. Gastroenterology. 2011;141(4):1486–1497, 1497. e1–e14. | |
Pai R, Dunlap D, Qing J, Mohtashemi I, Hotzel K, French DM. Inhibition of fibroblast growth factor 19 reduces tumor growth by modulating beta-catenin signaling. Cancer Res. 2008;68(13):5086–5095. | |
Cairo S, Armengol C, De Reynies A, et al. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell. 2008;14(6):471–484. | |
Yamashita T, Budhu A, Forgues M, Wang XW. Activation of hepatic stem cell marker EpCAM by Wnt-beta-catenin signaling in hepatocellular carcinoma. Cancer Res. 2007;67(22):10831–10839. | |
Maetzel D, Denzel S, Mack B, et al. Nuclear signalling by tumour-associated antigen EpCAM. Nat Cell Biol. 2009;11(2):162–171. | |
Yang W, Yan HX, Chen L, et al. Wnt/beta-catenin signaling contributes to activation of normal and tumorigenic liver progenitor cells. Cancer Res. 2008;68(11):4287–4295. | |
Rountree CB, Senadheera S, Mato JM, Crooks GM, Lu SC. Expansion of liver cancer stem cells during aging in methionine adenosyltransferase 1A-deficient mice. Hepatology. 2008;47(4):1288–1297. | |
Mavila N, James D, Utley S, et al. Fibroblast growth factor receptor-mediated activation of AKT-beta-catenin-CBP pathway regulates survival and proliferation of murine hepatoblasts and hepatic tumor initiating stem cells. PLoS One. 2012;7(11):e50401. | |
Zhu AX, Duda DG, Sahani DV, Jain RK. HCC and angiogenesis: possible targets and future directions. Nat Rev Clin Oncol. 2011;8(5):292–301. | |
Van de Veire S, Stalmans I, Heindryckx F, et al. Further pharmacological and genetic evidence for the efficacy of PlGF inhibition in cancer and eye disease. Cell. 2010;141(1):178–190. | |
Nugent MA, Iozzo RV. Fibroblast growth factor-2. Int J Biochem Cell Biol. 2000;32(2):115–120. | |
Presta M, Dell’Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16(2):159–178. | |
Folkman J, Shing Y. Angiogenesis. J Biol Chem. 1992;267(16):10931–10934. | |
Poon RT, Lau C, Yu WC, Fan ST, Wong J. High serum levels of vascular endothelial growth factor predict poor response to transarterial chemoembolization in hepatocellular carcinoma: a prospective study. Oncol Rep. 2004;11(5):1077–1084. | |
Pepper MS, Ferrara N, Orci L, Montesano R. Potent synergism between vascular endothelial growth factor and basic fibroblast growth factor in the induction of angiogenesis in vitro. Biochem Biophys Res Commun. 1992;189(2):824–831. | |
Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8(4):299–309. | |
Yoshiji H, Kuriyama S, Yoshii J, et al. Synergistic effect of basic fibroblast growth factor and vascular endothelial growth factor in murine hepatocellular carcinoma. Hepatology. 2002;35(4):834–842. | |
Seghezzi G, Patel S, Ren CJ, et al. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: an autocrine mechanism contributing to angiogenesis. J Cell Biol. 1998;141(7):1659–1673. | |
Ueba T, Nosaka T, Takahashi JA, et al. Transcriptional regulation of basic fibroblast growth factor gene by p53 in human glioblastoma and hepatocellular carcinoma cells. Proc Natl Acad Sci U S A. 1994;91(19):9009–9013. | |
Ginsberg D, Mechta F, Yaniv M, Oren M. Wild-type p53 can down-modulate the activity of various promoters. Proc Natl Acad Sci U S A. 1991;88(22):9979–9983. | |
Oda T, Tsuda H, Scarpa A, Sakamoto M, Hirohashi S. p53 gene mutation spectrum in hepatocellular carcinoma. Cancer Res. 1992;52(22):6358–6564. | |
Zhu AX. Systemic therapy of advanced hepatocellular carcinoma: how hopeful should we be? Oncologist. 2006;11(7):790–800. | |
Sciarrino E, Simonetti RG, Le Moli S, Pagliaro L. Adriamycin treatment for hepatocellular carcinoma. Experience with 109 patients. Cancer. 1985;56(12):2751–2755. | |
Cao H, Phan H, Yang LX. Improved chemotherapy for hepatocellular carcinoma. Anticancer Res. 2012;32(4):1379–1386. | |
Cheng AL, Kang YK, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a Phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10(1):25–34. | |
Shin JW, Chung YH. Molecular targeted therapy for hepatocellular carcinoma: current and future. World J Gastroenterol. 2013;19(37):6144–6155. | |
Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15(3):232–239. | |
Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8(8):592–603. | |
Huynh H, Ngo VC, Fargnoli J, et al. Brivanib alaninate, a dual inhibitor of vascular endothelial growth factor receptor and fibroblast growth factor receptor tyrosine kinases, induces growth inhibition in mouse models of human hepatocellular carcinoma. Clin Cancer Res. 2008;14(19):6146–6153. | |
Jonker DJ, Rosen LS, Sawyer MB, et al. A phase I study to determine the safety, pharmacokinetics and pharmacodynamics of a dual VEGFR and FGF receptor inhibitor, brivanib, in patients with advanced or metastatic solid tumors. Ann Oncol. 2011;22(6):1413–1419. | |
Llovet JM, Decaens T, Raoul JL, et al. Brivanib in patients with advanced hepatocellular carcinoma who were intolerant to sorafenib or for whom sorafenib failed: results from the randomized phase III BRISK-PS study. J Clin Oncol. 2013;31(28):3509–3516. | |
Kanai F, Yoshida H, Tateishi R, et al. A Phase I/II trial of the oral antiangiogenic agent TSU-68 in patients with advanced hepatocellular carcinoma. Cancer Chemother Pharmacol. 2011;67(2):315–324. | |
Sarker D, Molife R, Evans TR, et al. A Phase I pharmacokinetic and pharmacodynamic study of TKI258, an oral, multitargeted receptor tyrosine kinase inhibitor in patients with advanced solid tumors. Clin Cancer Res. 2008;14(7):2075–2081. | |
Shen YC, Lin ZZ, Hsu CH, Hsu C, Shao YY, Cheng AL. Clinical trials in hepatocellular carcinoma: an update. Liver Cancer. 2013;2(3–4):345–364. | |
Mitsunaga S, Ikeda M, Ueno H, et al. Phase I/II study of lenvatinib (E7080), a multitargeted tyrosine kinase inhibitor, in patients with advanced hepatocelluar carcinoma: Phase I results. J Clin Oncol. 2012;30 Suppl 4:Abstr 231. | |
Heinzle C, Erdem Z, Paur J, et al. Is fibroblast growth factor receptor 4 a suitable target of cancer therapy? Curr Pharm Des. August 12, 2013. [Epub ahead of print.] | |
Yu C, Wang F, Jin C, Wu X, Chan WK, McKeehan WL. Increased carbon tetrachloride-induced liver injury and fibrosis in FGF receptor4-deficient mice. Am J Pathol. 2002;161(6):2003–2010. | |
Ho HK, Nemeth G, Ng YR, et al. Developing FGF receptor4 inhibitors as potential anti-cancer agents via in silico design, supported by in vitro and cell-based testing. Curr Med Chem. 2013;20(10):1203–1217. | |
Poh W, Wong W, Ong H, et al. Klotho-beta overexpression as a novel target for suppressing proliferation and fibroblast growth factor receptor-4 signaling in hepatocellular carcinoma. Mol Cancer. 2012;11:14. | |
Johnson PJ, Qin S, Park JW, et al. Brivanib versus sorafenib as first-line therapy in patients with unresectable, advanced hepatocellular carcinoma: results from the randomized phase III BRISK-FL study. J Clin Oncol. 2013;31(28):3517–3524. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.