Back to Journals » OncoTargets and Therapy » Volume 13
Fibroblast Activation Protein (FAP) Overexpression Induces Epithelial–Mesenchymal Transition (EMT) in Oral Squamous Cell Carcinoma by Down-Regulating Dipeptidyl Peptidase 9 (DPP9)
Authors Wu Q ![]() , Zhao M, Huang G, Zheng Z, Chen Y, Zeng W, Lv X
, Zhao M, Huang G, Zheng Z, Chen Y, Zeng W, Lv X
Received 23 December 2019
Accepted for publication 2 March 2020
Published 27 March 2020 Volume 2020:13 Pages 2599—2611
DOI https://doi.org/10.2147/OTT.S243417
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Nicola Silvestris
Qing-qing Wu, 1 Meng Zhao, 1 Guang-zhao Huang, 1 Ze-nan Zheng, 1 Yuechuan Chen, 1 Wei-sen Zeng, 2 Xiao-zhi Lv 1
1Department of Oral & Maxillofacial Surgery, NanFang Hospital, Southern Medical University, Guangzhou, People’s Republic of China; 2Department of Cell Biology, School of Basic Medical Science, Southern Medical University, Guangzhou, People’s Republic of China
Correspondence: Xiao-zhi Lv
Tel +86 20 6164 2025
Email [email protected]
Wei-sen Zeng
Tel +86 20 6164 7073
Email [email protected]
Purpose: Fibroblast activation protein (FAP) acts as a tumor promoter via epithelial–mesenchymal transition (EMT) in human oral squamous cell carcinoma (OSCC). The present study was designed to investigate the FAP targeting proteins and explore the precise mechanism by which FAP promotes EMT in OSCC.
Patients and Methods: Proteins interacting with FAP were found and filtered by immunoprecipitation-mass spectrometry (IP-MS). Both DPP9 protein and mRNA were examined in 90 paired OSCC samples and matched normal tissue. DPP9 knockdown was conducted to determine its function in OSCC in vitro and in vivo.
Results: Dipeptidyl peptidase 9 (DPP9) was identified as interacting with FAP intracellularly by IP-MS. The levels of both DPP9 protein and mRNA were down-regulated in OSCC tissue. Lower DPP9 expression was correlated with unfavorable survival rates of OSCC patients. DPP9 knockdown accelerates the proliferation of OSCC cells in vitro and in vivo. Overexpression of FAP leads to a reduction in DPP9 expression. Likewise, DPP9 overexpression reverses the proliferation, migration, invasion and EMT induced by FAP during OSCC.
Conclusion: Our study finds that FAP promotes EMT of OSCC by down-regulating DPP9 in a non-enzymatic manner. FAP-DPP9 pathway could be a potential therapeutic target of OSCC.
Keywords: FAP, DPP9, EMT, OSCC, oral cancer
Introduction
OSCC is one of the most common malignant cancers of the oral cavity, as well as an important cause of morbidity and death.1 OSCC can be divided into three major subtypes: buccal mucosal squamous cell carcinoma (BMSCC), tongue squamous cell carcinoma (TSCC), and gingival squamous cell carcinoma (GSCC).2 OSCC accounts for more than 90% of all oral cancers with the main risk factors being the consumption of tobacco and/or alcohol and chewing areca. At a histopathological level, OSCC is characterized by squamous differentiation, nuclear pleomorphisms, invasive growth, and metastasis.3 Despite major advances in diagnosis and treatment, the prognosis of OSCC is poor due to its invasion, metastasis, and recurrence. Although it is easily detected, up to 60% of OSCC cases are undiagnosed in early clinical stages. The biomarkers4 for early diagnosis of OSCC are therefore crucial to improving patient prognosis and survival rates.
FAP is a member of the dipeptidyl peptidase (DPP) family.5 FAP is highly expressed in cancer-associated fibroblasts (CAFs). It is also highly expressed in cancer cells and has been demonstrated to have pro-tumorigenic activity.6,7 Some studies8,10,9 indicated that FAP can induce EMT in various human cancers. However, the exact mechanism of FAP in EMT and OSCC carcinogenesis is still unknown. Structurally, FAP consists of a cytoplasmic tail, a transmembrane domain, and an extracellular domain.5 FAP has post-proline exopeptidase activity and gelatinase activity.11 Its dual enzymatic activity gives it a range of putative substrates.12 Although many studies12 have suggested that FAP can enhance various carcinogenesis processes, it is still not clear whether the observed carcinogenesis is simply based on enhanced enzymatic activity. Emerging evidence15,13,14 has suggested that FAP plays a non-enzymatic role in cancer. We reason that FAP may play its role in cancer promotion not only by enzymatic effects but also by non-enzymatic effects.
After immunoprecipitation-mass spectrometry (IP-MS), we indicated DPP9 is an intracellular target of FAP. DPP9, the FAP homologous protein, shares the same subcellular localization, protein domain and Gene Ontology (GO) function. DPP9 belongs to the DPP gene family,16 localizes in cell cytosol, expresses ubiquitously in human tissues, and is mainly enriched in lymphocytes and epithelial cells.29,17 Emerging evidence also suggests that abnormal expression of DPP9 may play a key role in the development and progression of cancer. The functional role of DPP9 in OSCC remains to be elucidated. Thus, this study was designed to explore the possible molecular mechanism of FAP through DPP9 in OSCC.
Materials and Methods
Cell Culture, Tissue Collection, and Ethics Statement
OSCC cell lines SCC9, SCC25, SCC15 were purchased from ATCC and maintained in DMEM/F12 supplemented with 10% fetal bovine serum (FBS) (Gibco Company, USA). A total of 118 untreated OSCC tumor specimens (TUM) and matched normal tissues (MNT) were obtained from Nanfang Hospital of Southern Medical University, Guangzhou, from 2015 to 2018. Of the 118 cases, there were 86 males and 32 females. All patients were informed with written consents and the Ethics Committees of Nanfang Hospital approved the collection and use of all clinical specimens (NO: NFEC-2018-027). All specimens were staged according to the 2009 UICC-TNM Classification of Malignant Tumors.
Transient Transfection with siRNAs for FAP and DPP9
Small interfering RNAs (siRNA) for FAP and DPP9 were designed and synthesized (GenePharma Inc., Suzhou, PR China). siRNAs were transfected into cells by Lipofectamine3000 Transfection Reagent (Thermos Fishers Co, Ltd., USA) according to the manufacturer’s protocol. Cells were collected after 48–72 h for further experiments. siRNA sequences are listed in Table A1.
RNA Isolation, Reverse Transcription, and qRT-PCR
Total RNA was extracted from the cells using Trizol (RNA Isolator (Vazyme Biotech Co., Ltd, Nanjing)). Reverse transcription (RT) and qPCR were performed in accordance with the manufacturer’s instructions (Vazyme Biotech Co., Ltd, Nanjing). RT-qPCR for each gene was repeated three times. Quantification amounts were normalized to GAPDH levels. Primers are listed in Table A2.
Plasmid Construction
PFU enzyme (Thermo Fisher, Inc; USA) was used for the PCR program. Using cDNA from OSCC samples as a template, fragmented wildtype FAP (wFAP) with HIS-tag was cloned with primer A and primer B (See Table 1 for the list of primers). Using wFAP as a template for intracellular site deletion (tFAP), we used primers B and C to excise fragment tFAP. To delete 624–704 (mFAP), we overlapped primers D and E. After using primers A and D, and primers B and C, respectively, to excise fragments F1 and F2, both fragments were added together as templates and used to clone out mFAP with primer A and primer E. These three fragments (wFAP, tFAP, mFAP) were cloned into plasmid pcDNA3.1(+) using restriction enzymes, BamHⅠand EcoRⅠ. Plasmids were transfected into cells with Lipofectamine3000 Transfection Reagent. Using the cDNA and primer F/G to clone fragment DPP9 and set it into plasmid pCMV3-Flag with KpnⅠand XbaⅠ.
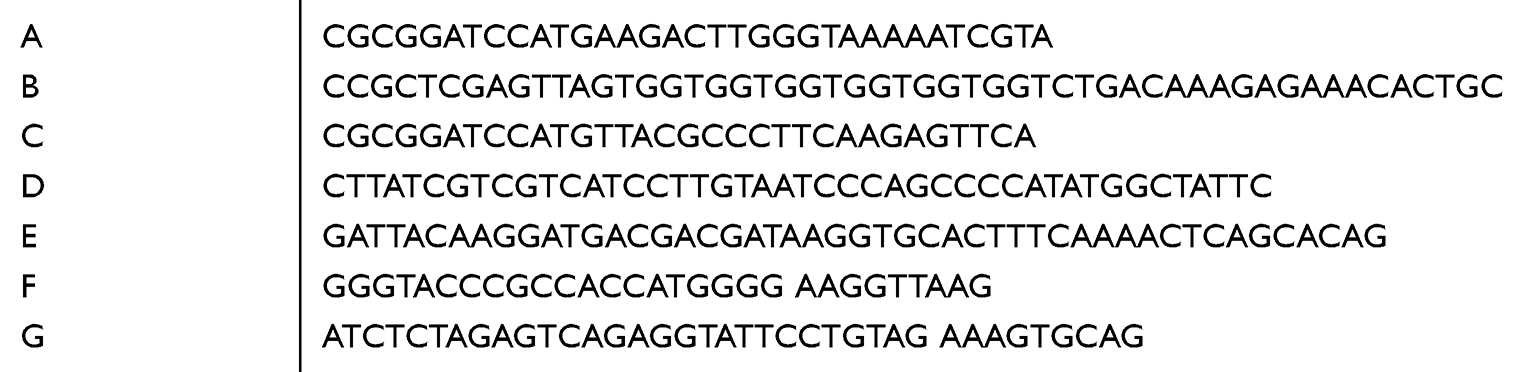 |
Table 1 Primer Used in Plasmid Construction (5ʹ-3ʹ) |
Western Blotting
Western blotting was performed using an SDS-PAGE Electrophoresis System with rabbit polyclonal anti-FAP antibody (1:1000; Santa Cruz Biotechnology Inc., Seattle, USA), mouse monoclonal anti-DPPR2 antibody (1:2000; Santa Cruz Biotechnology Inc., Seattle, USA). Other rabbit polyclonal antibodies against HIS-tag, GAPDH, E-cadherin, N-cadherin, Vimentin, MMP2, P53, SNAIL, SLUG (1:2000; Proteintech Inc., USA) and pan-ERK1/2 and phosphate-ERK1/2(1:2000; CST, USA) were also utilized.
IP Assay and MS Assay
The total protein was extracted from SCC9-FAP+ cells and IP was performed using a Protein A/G Magnetic Beads (Bimake, USA). Briefly, 6.0×105 SCC9-FAP+ cells per reaction were washed with phosphate-buffered saline (PBS), lysed in cold IP lysate buffer, and then centrifuged. Next, the cell lysates were immunoprecipitated with the monoclonal anti-HIS-tag antibody (Cell Signaling Technology, Inc.; USA) and incubated overnight at 4°C. SDS-PAGE was then conducted to examine the protein levels. The lysate including HIS-FAP-interacting peptides were collected and submit to an MS facility (Thermo Fisher, Inc.; USA) for analysis. The experiments were repeated three times and the detected peptides were intersected.
Immunohistochemistry (IHC) Assay
Examination of DPP9 expression in tumor (TUM) and matched normal tissue (MNT) by IHC was processed with the DPP9 antibody (1:50, Abcam Inc., USA). For statistical analysis of DPP9 expression in MNT against TUM, staining scores of 0–5 and 6–10 were, respectively, grouped into low and high levels of expression. IHC score is equal to the percentage positive for each slide ×100. (No digits after decimal point).
CCK-8 Assay
Cell Counting Kit-8 assay was used to assess the rate of cell proliferation. Cells were seeded in 96-well plates at a density of 1000 cells/well and, respectively, incubated for 12, 24, 48, 70 hrs. 10 μL of CCK-8 (DOJINDO LABORATORIES Ltd., China) was added to each well (with 90 μL medium without FBS) and incubated for 2 h. The absorbance value (OD) was measured at 450 nm. Experiments were carried out three times.
Colony Formation Assay
We placed 1×105 treated cells per well in 6-well plates, and after their attachment, there was no contact between cells. The cells were incubated in a CO2 incubator at 37°C for 10–15 days until the cells formed substantially large colonies (50 cells per colony). Media was removed and cells were rinsed twice with 1X PBS. 1 mL of methanol was added per well and incubated at room temperature (RT) for 5 min. Methanol was removed and 0.5% crystal violet was added carefully then incubated at RT for 2 hrs. Crystal violet was removed and the plates were rinsed in tap water followed by drying at RT for 1 day. Plates were scanned and the number of colonies was counted. Plating efficiency (PE) was calculated by the following equation:
PE = no. of colonies formed/no. of cells seeded × 100%.
Invasion and Migration Assays
For the invasion assay, cells were seeded in 100 mL DMEM/F12 media on top of polyethylene terephthalate (PET) membranes coated with Matrigel TM (1.5 mg/mL, BD Biosciences Inc.) within transwell cell culture inserts (24-well inserts, 8 mm pore size; Corning Life Sciences, Corning, NY, USA). The bottom chamber was filled with 600 mL of DMEM/F12 media containing 20% FBS. The cells were incubated for 12 h at 37°C with 5% CO2. Subsequently, the cells were fixed in 2.5% (v/v) glutaraldehyde and stained with 0.1% crystal violet. For the migration assay, the same conditions were followed as the invasion assay without the addition of Matrigel TM. Both groups of cells on the membranes bottom were visualized under a microscope (Zeiss Ltd., China) and quantified by counting the number of cells in three randomly chosen fields at 200-fold magnification.
In vivo Tumor Growth Assay
BALB/c-nude mice (4 weeks old, 18–20 g) were purchased from The Laboratory Animal Centre, Southern Medical University. The Institutional Animal Care and Use Committee of Southern Medical University approved all experimental procedures. Cells were dispersedly suspended, washed twice with cold serum-free medium, and resuspended with serum-free medium. 2 × 106 treated cells were injected subcutaneously into the left axilla in 5 nude mice each group. Every 5 days, the length (L) and width (W) of tumors were measured and their volumes were calculated using the equation (L ×W2)/2. The animals were euthanized on the 20th day after injection, and the tumors were excised, weighed, serially sectioned and stained with hematoxylin and eosin (H&E). Animal studies were conducted in accordance with the guidelines as published in the Guide for the Care and Use of Laboratory Animals.18
Statistical Analysis
SPSS 22.0 software (SPSS Inc., Chicago, IL, USA) and GraphPad software (GraphPad Software, Inc., La Jolla, CA, USA) were used to analyze all data for statistical significance. Student's t-test was used for comparisons of two independent groups. One-way ANOVA was used to determine differences between groups for all in vitro analyses. Spearman correlation analysis and Cox-regression analysis were used to determine the prognostic role of DPP9 in clinical data. A p-value ≤ 0.05 was considered statistically significant.
Results
FAP Negatively Regulates DPP9 in Its Upstream
Current research has been focused primarily on the enzymatic activity of FAP and its substrates. Clinical studies involving Talabostat, which inhibits the enzymatic functions of FAP showed less than promising results in patients, necessitating the exploration of the non-enzymatic activities of FAP. We overexpressed FAP with a HIS-tag in the SCC9 tumor cell line and conducted IP using anti-HIS antibody to find possible interaction proteins with FAP. The lysates from the HIS-tag antibody group and IgG antibody group were analyzed by MS, and 13 proteins showed significant difference compared with the IgG group. We focused on DPP9 with three filter conditions: subcellular localization, GO function and protein domain (Figure 1A). We verified the association between FAP and DPP9 in the antibody group lysates by Western blotting (Figure 1B). In SCC15 cell line transiently silence FAP showed higher DPP9 expression. In both SCC9/SCC25 FAP+ cell lines, DPP9 was downregulated (Figure 1C. p<0.05). We next asked which site on FAP may relate with DPP9. To answer this question, wild type FAP, intracellular segment deletion type FAP (tFAP) and extracellular segment mutation type FAP (mFAP) were each cloned into the pcDNA3.1+ plasmid (Fig. S1A) and were transiently transfected into SCC9 cell lines. DPP9 antibody was used to execute conditioned-IP, and no FAP staining in tFAP group. We compared the DPP9 expression in SCC9 cells which were transfected by wFAP or tFAP and found that losing the extracellular segment of FAP will not affect the expression of DPP9 (Figure 1D). This suggested that the intracellular part of FAP may be the cooperating site with DPP9. These results show that FAP can reduce DPP9 and the functional site may be inside the cell.
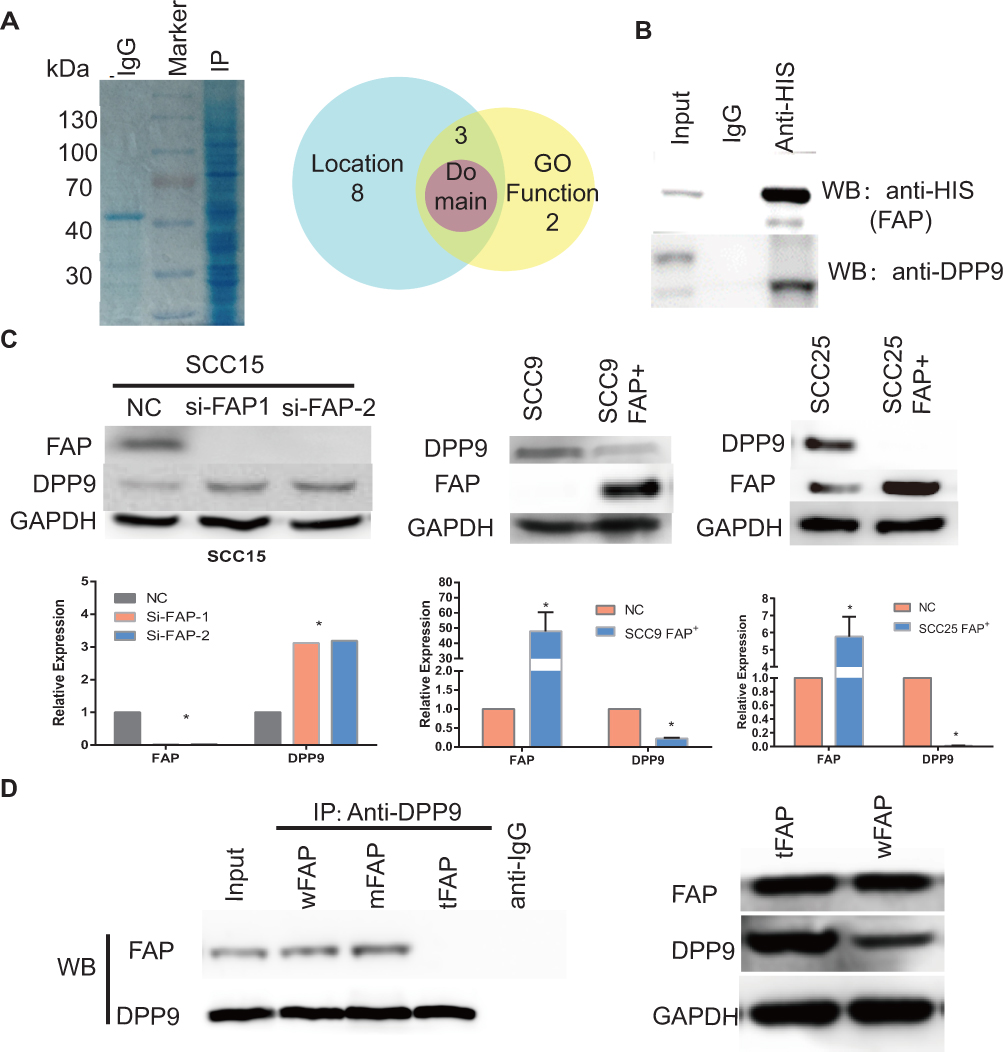 |
Figure 1 Negative correlation between FAP and DPP9. (A) Coomassie blue staining shows IP lysate with His-antibody. Venn diagram shows the filter condition of 13 peptides from IP-MS. (B) Verify DPP9 in IP lysate using DPP9 antibody. (C) In FAP-low-expression SCC9 and SCC25, overexpressing FAP and reduced DPP9 can be detected. And in FAP-high-expression SCC15, knockdown FAP shows higher DPP9. All data are represented as mean ± SD; *p < 0.05. (D) SCC9 cells transfected with either pcDNA-wFAP/mFAP/tFAP then immunoprecipitated with DPP9 antibody showed no DPP9 reactivity when the intracellular section of FAP was deleted (tFAP) compared with wFAP or mFAP groups. Transfecting wFAP and tFAP plasmid showed different DPP9 expression. |
DPP9 Is Downregulated in OSCC
Through analysis of DPP9 expression from TCGA head and neck cancer patients in Oncomine, the log2 copy number unit of DPP9 was downregulated in head and neck carcinoma samples (290 cases) compared with head and neck part (74 cases) and blood samples (264 cases) (Fig. S1B). DPP9 mRNA in TUM is relatively lower in MNT (p< 0.0001, n=90, Figure 2A). Three randomized-picked paired OSCC tumor tissues (TUM) and the matched normal tissues (MNT) were analyzed, and immunoblotting showed that DPP9 protein exhibited markedly lower expression levels in the TUM tissues compared to the MNT. (Figure 2B). Also, protein levels of DPP9 were measured in samples of 118 pairs TUM and MNT using IHC (p< 0.005, n=45) (Figure 2C). Also, the FAP mRNA level in 90 pairs of specimens were tested and the regression analysis showed that DPP9 is negatively associated with FAP (R2=0.121, p=0.001, Figure 2D and E). Taken together, these results strongly indicate that DPP9 is downregulated in human OSCC.
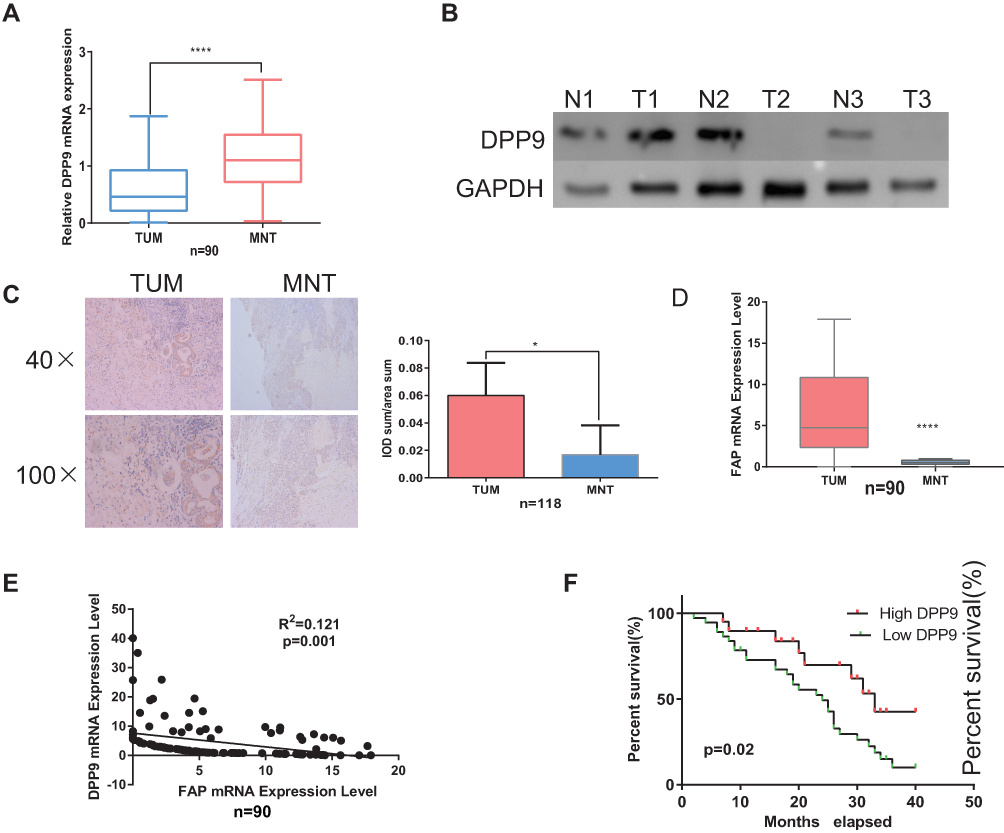 |
Figure 2 Decreased expression of DPP9 is unfavorable for OSCC prognosis. (A) mRNA level of DPP9 in 90 pairs of clinical samples. (B) Three random pairs of samples detecting the DPP9 protein expression. (C) By IHC and statistical analysis show DPP9 protein level is less in TUM compared with MNT (*p<0.05,****p<0.0001). (D, E) mRNA level of FAP in 90 pairs of clinical samples and regression analysis showed that DPP9 is negatively associated with FAP in OSCC tissues (R2=0.121, p=0.001). Regression analysis compared the mRNA level of FAP and DPP9 in both 90 pairs samples. (F) Survival curves (Kaplan–Meier plots) show low-DPP9 is related to lower survival rates (p=0.02). |
Decreased Expression of DPP9 Is Unfavorable for OSCC Prognosis
Kaplan–Meier survival analysis revealed a correlation between DPP9 expression level and overall survival duration (p=0.02, Figure 2F). To explore the prognostic value of DPP9 expression for OSCC, 118 OSCC patients for up to 40 months were stained by IHC assay, scored, and divided into low or high expression groups (summarized in Table 2). We analyzed the association between DPP9 and the clinicopathological features of OSCC. As showed in Table 2, strong associations were observed between DPP9 expression and clinical stage (p=0.036), T classification (p=0.017), N classification (p=0.041). However, the expression of DPP9 was not associated with age, gender and lymphatic metastasis. Cox-regression analysis was used to determine whether DPP9 could serve as a risk factor. As shown in Table 3, low expression of DPP9 was associated with a significantly increased risk of death in OSCC patients (P =0.045) compared to those with high DPP9 expression by univariate Cox-regression analyses. Multivariate Cox-regression analysis found that DPP9 could be a biomarker of OSCC which predicting unfavorable survival rate (Table 3). These results indicate a significant correlation of the expression of DPP9 with the prognosis of OSCC.
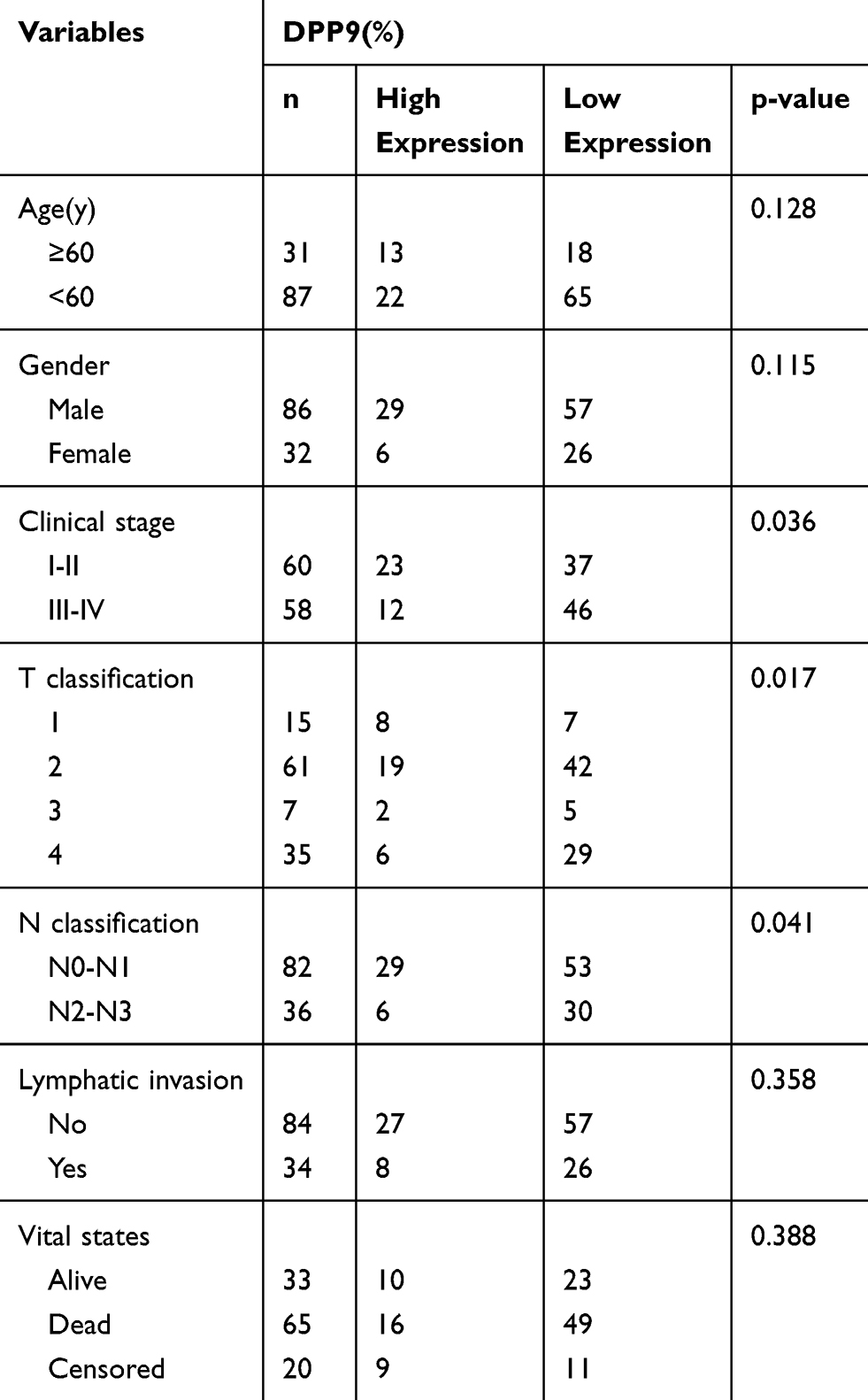 |
Table 2 Clinicopathological Characteristics of Patient Samples and Expression of DPP9 in OSCC |
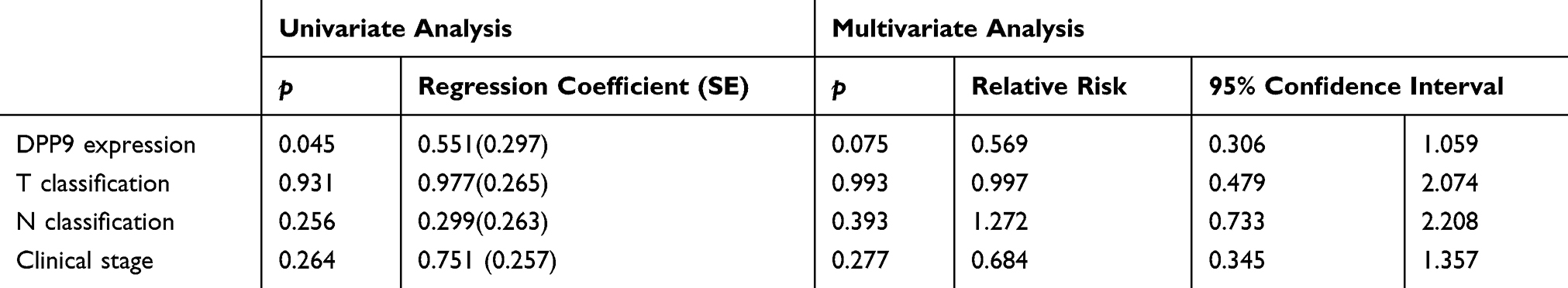 |
Table 3 Multivariate Analyses of Various Prognostic Parameters in Patients with OSCC Cox-Regression Analysis |
DPP9 Modulates Cell Growth of OSCC Cells
To evaluate the function of DPP9 on cancer cell proliferation, siRNA was transfected into SCC9 and SCC25 cell lines to specifically salience DPP9 expression and observed no change of FAP (Figure 3A, **p<0.01). Depleted DPP9 promoted cell growth by Colony formation assay and CCK-8 assay (Figure 3B and C). Taken together, these results suggested that DPP9 significantly attenuated cell growth in vitro.
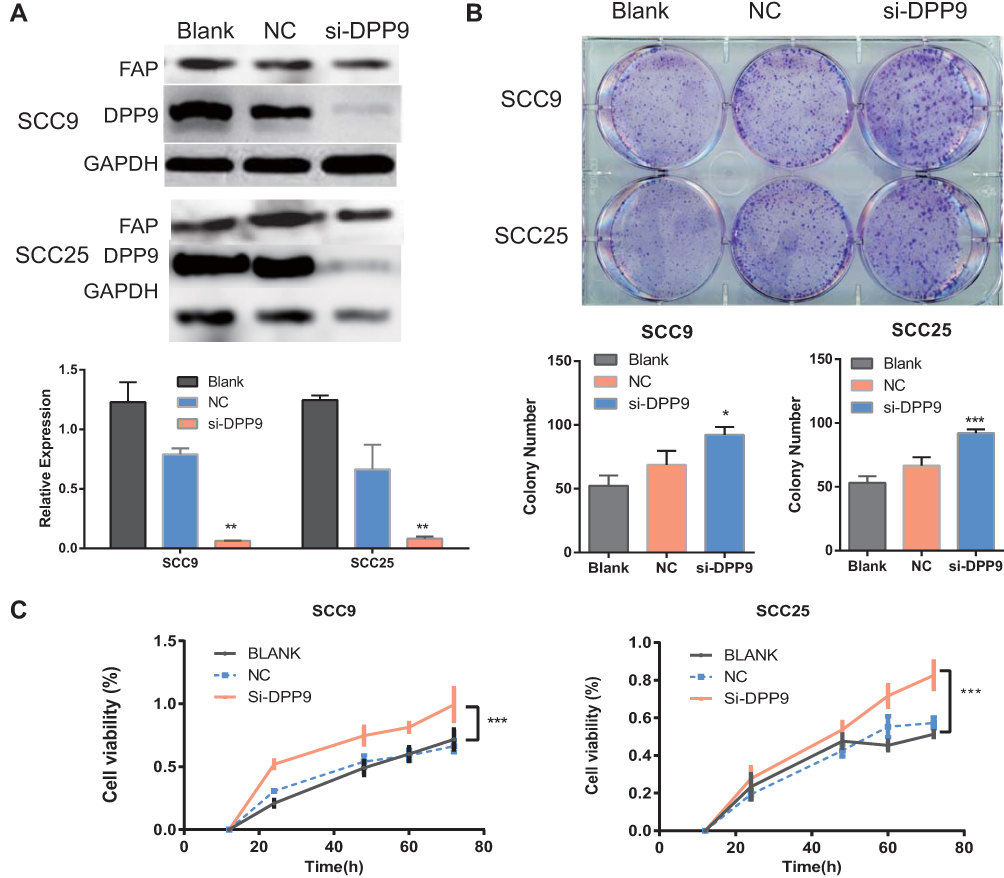 |
Figure 3 Knockdown of DPP9 causes an anti-tumor effect on OSCC cells. (A) Western blot analysis of DPP9 following DPP9 knockdown for 72 hrs. GAPDH was used as an internal control. (B) Colony formation assay after knockdown of DPP9 in SCC9 and SCC25 cells for 10 days (Top). The mean number of colonies for each well was determined from three independent assays (Bottom). (C) Growth rates of SCC9 and SCC25 cells measured by CCK-8 assay after DPP9 knockdown. All data are presented as mean ± SD; *p < 0.05; **p < 0.01; ***p < 0.001 versus control group. |
DPP9 Depletion Promoted Cell Migration/Invasion and Induced EMT in vitro
To determine the effects of DPP9 on cancer cell metastatic ability, DPP9 repression in SCC9 and SCC25 cells was conducted to examine the cellular function of invasion and migration. Matrigel invasion assays showed SCC9 and SCC25 cells with downregulated DPP9 were much more invasive than controls and exhibited a similar tendency in migration assays (Figure 4A, *p<0.05, **p<0.01, ***p<0.005). Immunoblotting showed DPP9-depletion can reduce E-cadherin, P53, phosphorylated-ERK 1/2, with increasing N-cadherin, MMP2, SAIL1 and SLUG, compared with no change of Vimentin and pan-ERK1/2 (Figure 4B).
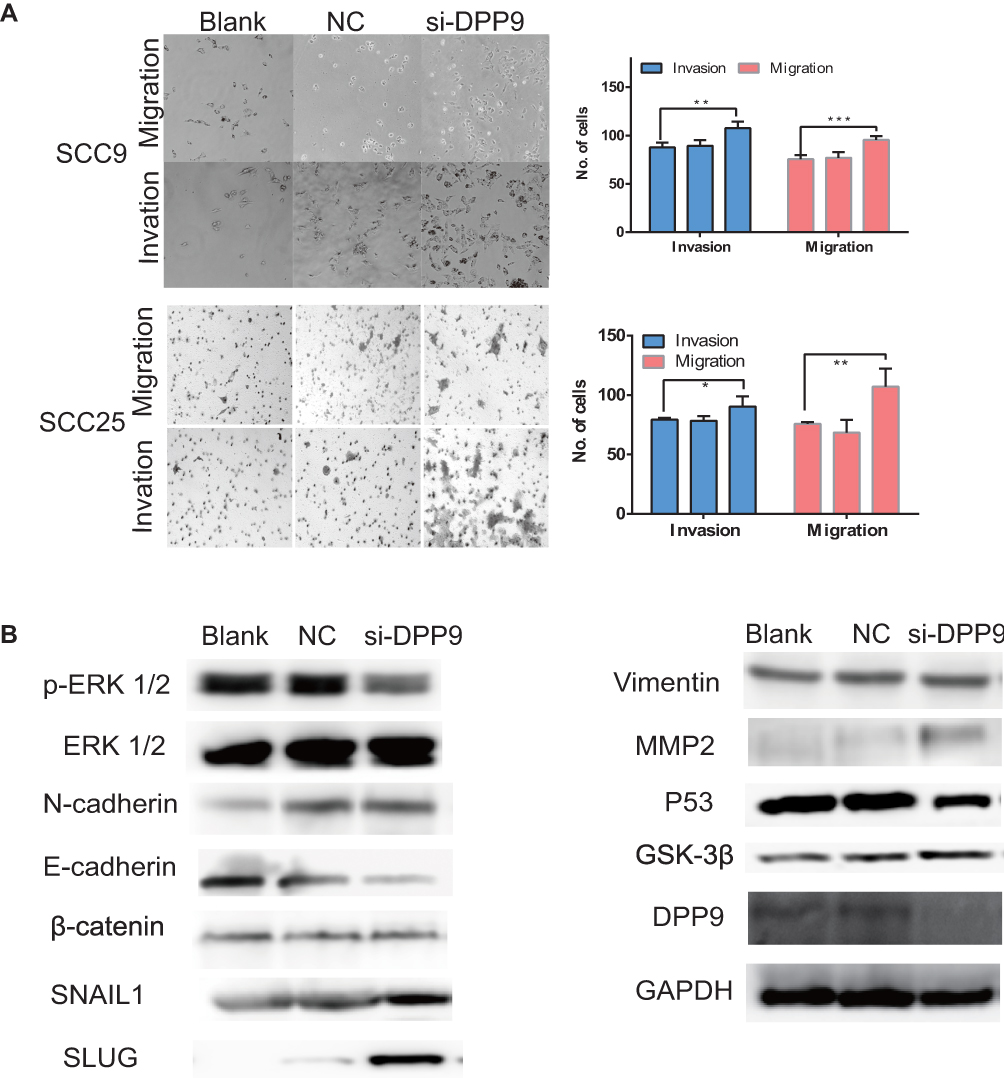 |
Figure 4 DPP9 depletion promoted cell migration/invasion and induced EMT in vitro. (A) Transwell migration and invasion assay after DPP9 knockdown for 48 hrs. (All data are presented as mean ± SD; *p < 0.05; **p < 0.01; ***p < 0.001.) (B) Western blot analysis of p-ERK1/2, ERK, EMT-associated proteins and P53, DPP9, Cyclin D1 following knockdown for 48 hrs. GAPDH was used as an internal control. |
DPP9 Modulates Cell Growth of OSCC Cells in vivo
To confirm whether DPP9 repression effects the OSCC growth, DPP9-depleted SCC9 cells, or control cells were injected into nude mice subcutaneously, and tumor size was observed. Tumor generation speed of si-DPP9 cells was significantly faster than in control cells, and final volume/weight (20 days) of si-DPP9 group is significantly larger (Figure 5A). The EMT-associated protein in si-DPP9 group change similarly with treated cells in vitro compared with the untreated group by WB (Figure 5B). All tumors were stained with H&E and IHC stained using DPP9, CD44, and Ki67 antibodies (Figure 5C).
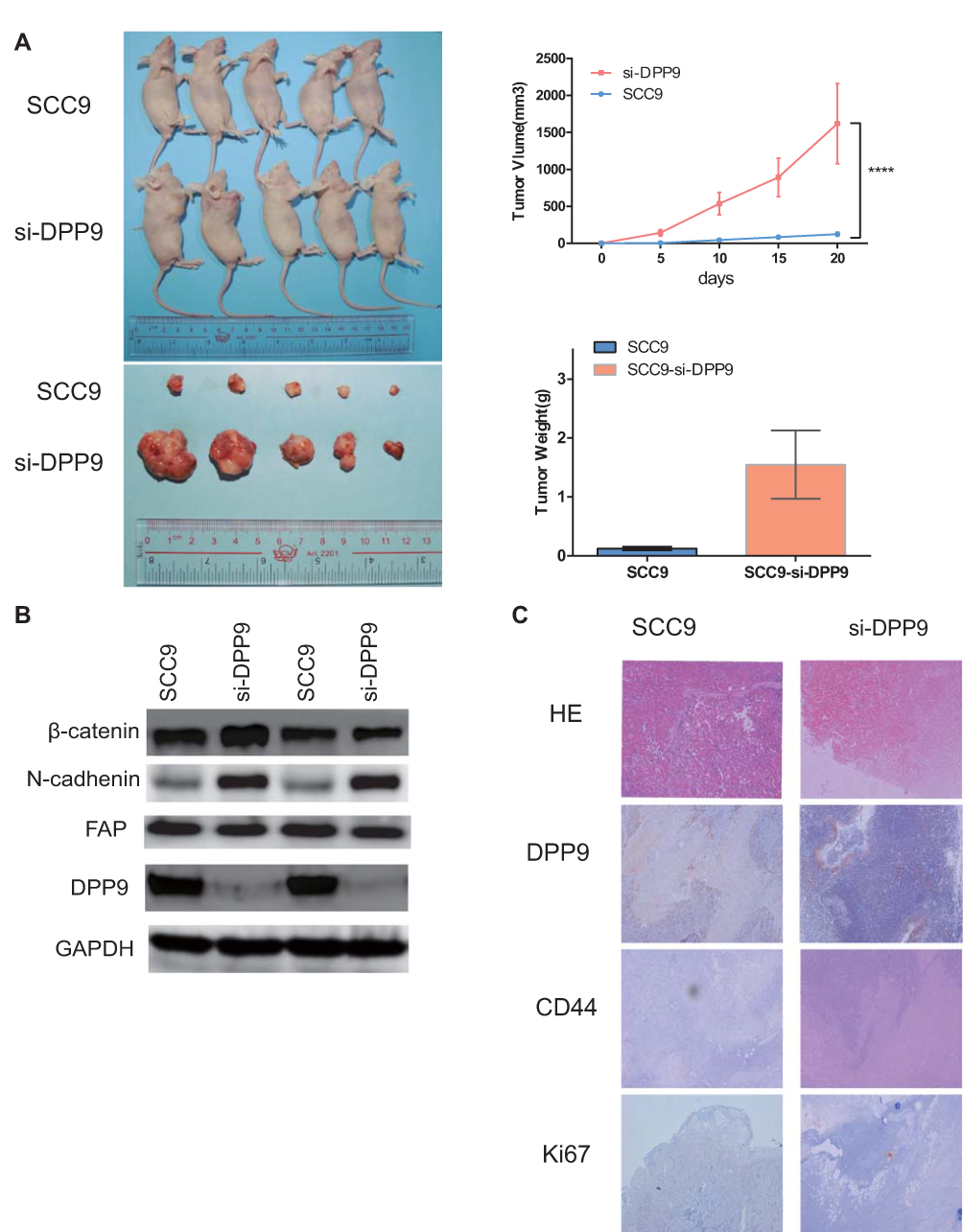 |
Figure 5 DPP9 modulates cell growth of OSCC cells in vivo. (A) Tumor formation for 20 days after injecting cells to BALBc/nude right armpit. The weight of the tumors and consequent volume change were observed every 5 days, and the mean weight of the tumors observed for each group is shown in the bar graph. (****p<0.0001) (B) Western blot analysis of DPP9, FAP, EMT-associated proteins in two tumor tissues each group. (C) IHC of CD44, Ki-67 and DPP9 verification and H&E staining of each group. |
FAP/DPP9 Regulated EMT to Regulate Migration and EMT in OSCC Cells
To further delineate the role of DPP9 in the proliferation and migration/invasive regulated by FAP, DPP9 was restored by transfecting pCMV-DPP9 plasmid in FAP-overexpressing SCC9 cells (Figure 6A). CCK8 assay and colony formation assay demonstrated that the DPP9 plasmid decreased the cell proliferation and tumorigenesis (Figure 6B and D). Consistently, cells numbers of DPP9 plasmid treated cells of migration and invasion was reduced (Figure 6C). The increased vimentin, E-cadherin and N-cadherin expression levels were reversed to wildtype levels (Figure 6E). And Adhesion assay showed DPP9 transfection can furtherly reduce the adhesion ability of cells. (Fig. S1C) These findings suggest that proliferation, tumorigenesis by overexpressing FAP can be reversed by restoring DPP9 in OSCC cells.
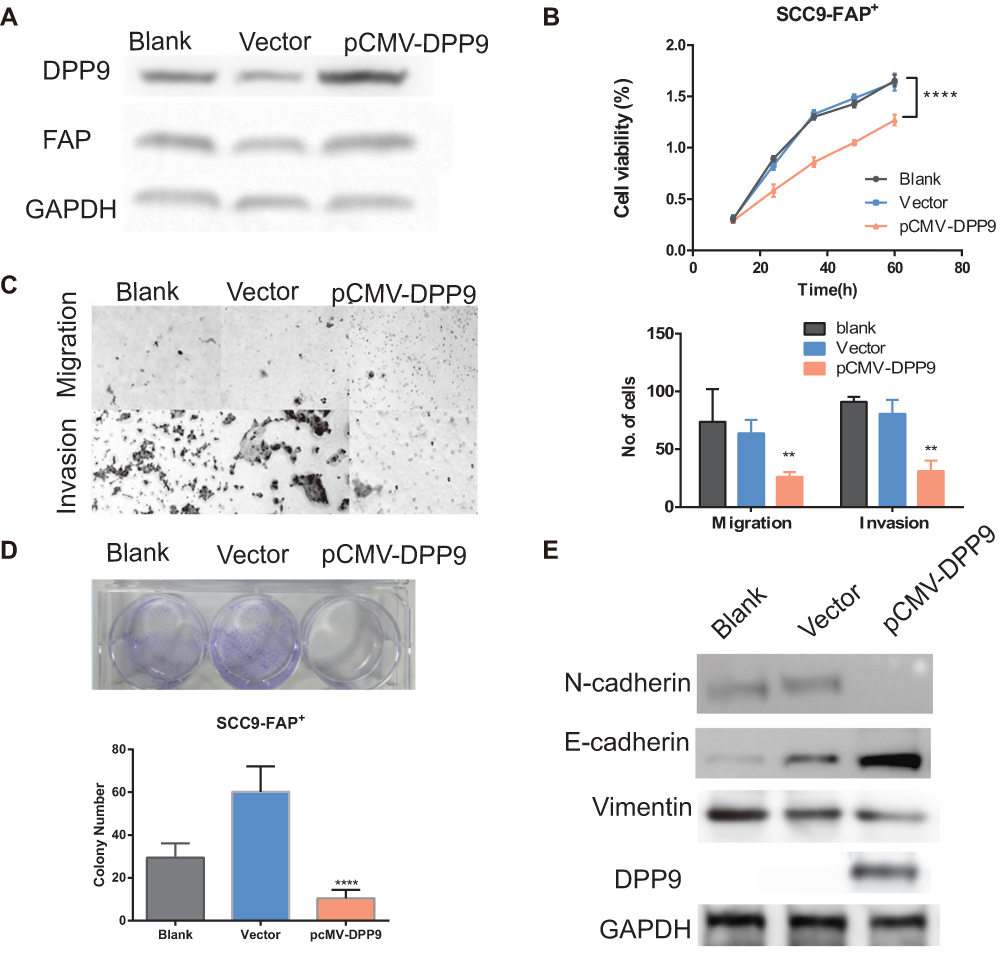 |
Figure 6 FAP/DPP9 regulated EMT so as to regulate migration and EMT in OSCC cells. (A) Verification of DPP9 overexpression in SCC9-FAP+ cells by Western Blotting. (B) CCK-8 shows a decreased growth rate of treated cells. (C) Transwell migration and invasion assay after DPP9 overexpressing 48 hrs. (D) Top: Colony formation assay after DPP9 plasmid transfecting for 10 days. Bottom: The mean number of colonies for each well was determined from three independent assays. (E) Western blot analysis of N-cadherin, E-cadherin and Vimentin level in SCC9-FAP+ cells after treatment with pCMV-DPP9 for 48 hrs. (All data are presented as mean ± SD; **p < 0.01; ***p < 0.001 versus control). |
Discussion
FAP has been considered as a potential immunotherapeutic target,19 and it plays a key role in cancer promotion both by enzymatic effects and non-enzymatic effects. Its enzymatic effects have attracted more attention so far. FAP shares substrates with DPP4.20,21 However, Talabostat, the first inhibitor of enzymatic activity of FAP, showed no significant response, or improvement in patient outcomes, in clinical trials.23,24,22 Emerging evidence support the idea that FAP might affect tumorigenicity independent of its enzymatic activity. Overexpression of FAP and expression of mutant FAP, both displayed enhanced cancer phenotypes with no significant differences between the groups.13 Chung suggested FAP leads cellular migration and its peptidase activity is not essential for such migration.26 Yang et al14 inhibited FAP with Talabostat does not reduce levels of its downstream CCL-2 secretion. Knopf et al25 suggested that FAP directly binds to erlin-2, stomatin, and caveolin-1. However, it is not clear whether this binding is due to FAP enzyme activity pattern or non-enzyme activity pattern. FAP may bind to erlin-2 through a non-enzymatic pattern for erlin-2 is located on the endoplasmic reticulum. Therefore, we are committed to exploring the closest downstream effectors on/inside the cell that may mediate the regulation of proliferation and metastasis of FAP, and in the 13 proteins isolated by our IP-MS experiment. Two of the 13 proteins are located on the cell membrane and the other 11 are distributed separately on the nucleus, cytoplasm, mitochondrion, cytosol, and cytoskeleton. We finally focused on DPP9 because of its similar location, likely interaction domain, and analogous GO function compared to FAP.
In our study, we verified that DPP9 expresses in lower than normal levels in OSCC samples both from TCGA database and in our sample base and the lower expression is associated with worse prognosis, including clinical stage, T, N classification and lymph node status. Survival analysis showed that downregulated DPP9 was an independent prognostic factor for poor 3-year overall survival in OSCC patients. DPP9 knockdown fostered the cell proliferation speed, migration, and EMT of OSCC cells. All these results showed that DPP9 could play an oncogenic role in OSCC and be a petanial. To the best of our knowledge, this is the first study to explore the role of DPP9 in OSCC carcinogenesis. DPP9 has two distinct biological effects on various types of cancer. On the one hand, Lu et al26 found DPP9 may act as a survival factor for cells from the Ewing’s sarcoma family of tumors cells. Yao et al27 reported that DPP9 overexpression can inhibit PI3K/AKT signaling, attenuate cell proliferation, and promote apoptosis in human hepatoma cells.28 On the other hand, DPP9 expression is significantly increased in testicular cancer.17 Thence the pro- or anti-tumor activity of DPP9 may depend on the cell type and the molecular context within the tumor microenvironment.
EMT has been confirmed to play a significant role in promoting metastasis in epithelium-derived carcinoma. Accumulating evidence has shown that EMT confers adhesion, migration, invasion and metastasis capacity, stemness, and multidrug resistance in tumor cells.30,29,10,31 There are many existing studies on how DPP432 and FAP33 affect EMT. Our data and results indicate that the underlying mechanism of OSCC tumorigenesis may be dysregulation of EMT properties through the DPP9 pathway that requires further experimentation to elucidate. Currently, there is not much research on whether DPP9 affects EMT, while Tang34 found upregulated DPP9 promotes tumorgenicity and EMT in non-small cell lung cancer (NSCLC). This difference may be because NSCLC samples in Tang’s study included adenocarcinoma and squamous cell carcinoma, which may have a certain impact on the study. At present, the relationship between FAP and histones such as DPP4 is limited. Augoff35 found FAP and DPP4 mRNA expression were upregulated in patients with esophageal squamous cell carcinoma while only DPP4 protein was increased. Further studies are needed to clarify the relationship between FAP and histones.
It can be observed that enforcing DPP9 expression in SCC9-FAP+ cells can reverse tumor growth, migration, invasion and EMT-biomarkers. The intracellular part of FAP is the possible site interaction with DPP9 observed by conditioned IP assay. We reach the conclusion that FAP facilitates carcinogenesis and EMT by down-regulating DPP9 inside OSCC cells. Results innovatively indicate that FAP may induce pro-tumorigenic effects in a non-enzymatic manner. The intracellular part of FAP consists of only 7 amino; currently, we infer that the intracellular FAP may repress DPP9 by ubiquitination. The specific mechanism still needs to be further explored.
Conclusion
Overall, our study finds that FAP promotes EMT of OSCC by down-regulating DPP9 in a non-enzymatic manner. FAP-DPP9 pathway could be a potential therapeutic target of OSCC.
Acknowledgments
We would like to acknowledge Stacks, Delores A and Xiaoxiao Hao for their help and support in this work. This study was supported by National Natural Science Foundation of China (No: 81472536), Science and Technology Planning Project of Guangdong Province of China (No:2017A020215181), the Southern Medical University Scientific Research Fund (CX2018N016), Project of Educational Commission of Guangdong Province of China (2018KTSCX026), and the Presidential Foundation of the Nanfang Hospital (2014027, 2019Z030).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
2. Pannone G, Santoro A, Papagerakis S, Lo Muzio L, De Rosa G, Bufo P. The role of human papillomavirus in the pathogenesis of head & neck squamous cell carcinoma: an overview. Infect Agent Cancer. 2011;6:4. doi:10.1186/1750-9378-6-4
3. Nosratzehi T. Salivary Chemical Factors in Relation with Oral Cancer in Smokers and Non-Smokers: a Literature Review. J Dent (Shiraz). 2017;18(4):237–243.
4. Shao T, Huang J, Zheng Z, Wu Q, Liu T, Lv X. SCCA, TSGF, and the Long Non-Coding RNA AC007271.3 are Effective Biomarkers for Diagnosing Oral Squamous Cell Carcinoma. Cell Physiol Biochem. 2018;47(1):26–38. doi:10.1159/000489741
5. Scanlan MJ, Raj BK, Calvo B, et al. Molecular cloning of fibroblast activation protein alpha, a member of the serine protease family selectively expressed in stromal fibroblasts of epithelial cancers. Proc Natl Acad Sci USA. 1994;91(12):5657–5661. doi:10.1073/pnas.91.12.5657
6. Hamson EJ, Keane FM, Tholen S, Schilling O, Gorrell MD. Understanding fibroblast activation protein (FAP): substrates, activities, expression and targeting for cancer therapy. Proteomics Clin Appl. 2014; 8 (5–6): 454–463. doi:10.1002/prca.201300095
7. Yang W, Han W, Ye S, et al. Fibroblast activation protein-α promotes ovarian cancer cell proliferation and invasion via extracellular and intracellular signaling mechanisms. Exp Mol Pathol. 2013;95(1):105–110. doi:10.1016/j.yexmp.2013.06.007
8. Errarte P, Guarch R, Pulido R, et al. The Expression of Fibroblast Activation Protein in Clear Cell Renal Cell Carcinomas Is Associated with Synchronous Lymph Node Metastases. PLoS ONE. 2016;11(12):e0169105. doi:10.1371/journal.pone.0169105
9. Wang H, Wu Q, Liu Z, et al. Downregulation of FAP suppresses cell proliferation and metastasis through PTEN/PI3K/AKT and Ras-ERK signaling in oral squamous cell carcinoma. Cell Death Dis. 2014;5:e1155. doi:10.1038/cddis.2014.122
10. Lu W, Kang Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev Cell. 2019;49(3):361–374. doi:10.1016/j.devcel.2019.04.010
11. Park JE, Lenter MC, Zimmermann RN, Garin-Chesa P, Old LJ, Rettig WJ. Fibroblast activation protein, a dual specificity serine protease expressed in reactive human tumor stromal fibroblasts. J Biol Chem. 1999;274(51):36505–36512.
12. Puré E, Blomberg R. Pro-tumorigenic roles of fibroblast activation protein in cancer: back to the basics. Oncogene. 2018;37(32):4343–4357. doi:10.1038/s41388-018-0275-3
13. Lv B, Xie F, Zhao P, et al. Promotion of Cellular Growth and Motility Is Independent of Enzymatic Activity of Fibroblast Activation Protein-α. Cancer Genomics Proteomics. 2016;13(3):201–208.
14. Yang X, Lin Y, Shi Y, et al. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3-CCL2 Signaling. Cancer Res. 2016;76(14):4124–4135. doi:10.1158/0008-5472.CAN-15-2973
15. Chung K-M, Hsu S-C, Chu Y-R, et al. Fibroblast activation protein (FAP) is essential for the migration of bone marrow mesenchymal stem cells through RhoA activation. PLoS ONE. 2014;9(2):e88772. doi:10.1371/journal.pone.0088772
16. Liu J, Huan Y, Li C, Liu M, Shen Z. Establishment of a selective evaluation method for DPP4 inhibitors based on recombinant human DPP8 and DPP9 proteins. Acta Pharm Sin B. 2014;4(2):135–140. doi:10.1016/j.apsb.2013.12.007
17. Yu DMT, Ajami K, Gall MG, et al. The in vivo expression of dipeptidyl peptidases 8 and 9. J Histochem Cytochem. 2009;57(11):1025–1040. doi:10.1369/jhc.2009.953760
18. Workman P, Aboagye EO, Balkwill F, et al. Guidelines for the welfare and use of animals in cancer research. Br J Cancer. 2010;102(11):1555–1577. doi:10.1038/sj.bjc.6605642
19. Jiang G-M, Xu W, Du J, et al. The application of the fibroblast activation protein α-targeted immunotherapy strategy. Oncotarget. 2016;7(22):33472–33482. doi:10.18632/oncotarget.8098
20. Keane FM, Nadvi NA, Yao T-W, Gorrell MD. Neuropeptide Y, B-type natriuretic peptide, substance P and peptide YY are novel substrates of fibroblast activation protein-α. FEBS J. 2011;278(8):1316–1332. doi:10.1111/j.1742-4658.2011.08051.x
21. Koczorowska MM, Tholen S, Bucher F, et al. Fibroblast activation protein-α, a stromal cell surface protease, shapes key features of cancer associated fibroblasts through proteome and degradome alterations. Mol Oncol. 2016;10(1):40–58. doi:10.1016/j.molonc.2015.08.001
22. Narra K, Mullins SR, Lee H-O, et al. Phase II trial of single agent Val-boroPro (Talabostat) inhibiting Fibroblast Activation Protein in patients with metastatic colorectal cancer. Cancer Biol Ther. 2007;6(11):1691–1699. doi:10.4161/cbt.6.11.4874
23. Eager RM, Cunningham CC, Senzer N, et al. Phase II trial of talabostat and docetaxel in advanced non-small cell lung cancer. Clin Oncol (R Coll Radiol). 2009;21(6):464–472. doi:10.1016/j.clon.2009.04.007
24. Eager RM, Cunningham CC, Senzer NN, et al. Phase II assessment of talabostat and cisplatin in second-line stage IV melanoma. BMC Cancer. 2009;9:263. doi:10.1186/1471-2407-9-263
25. Knopf JD, Tholen S, Koczorowska MM, De Wever O, Biniossek ML, Schilling O. The stromal cell-surface protease fibroblast activation protein-α localizes to lipid rafts and is recruited to invadopodia. Biochim Biophys Acta. 2015;1853(10 Pt A):2515–2525. doi:10.1016/j.bbamcr.2015.07.013
26. Lu C, Tilan JU, Everhart L, et al. Dipeptidyl peptidases as survival factors in Ewing sarcoma family of tumors: implications for tumor biology and therapy. J Biol Chem. 2011;286(31):27494–27505. doi:10.1074/jbc.M111.224089
27. Yao T-W, Kim W-S, Yu DMT, et al. A novel role of dipeptidyl peptidase 9 in epidermal growth factor signaling. Mol Cancer Res. 2011;9(7):948–959. doi:10.1158/1541-7786.MCR-10-0272
28. Spagnuolo PA, Hurren R, Gronda M, et al. Inhibition of intracellular dipeptidyl peptidases 8 and 9 enhances parthenolide’s anti-leukemic activity. Leukemia. 2013;27(6):1236–1244. doi:10.1038/leu.2013.9
29. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. doi:10.1172/JCI39104
30. Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119(6):1438–1449. doi:10.1172/JCI38019
31. van Zijl F, Mall S, Machat G, et al. A human model of epithelial to mesenchymal transition to monitor drug efficacy in hepatocellular carcinoma progression. Mol Cancer Ther. 2011;10(5):850–860. doi:10.1158/1535-7163.MCT-10-0917
32. Yang F, Takagaki Y, Yoshitomi Y, et al. Inhibition of Dipeptidyl Peptidase-4 Accelerates Epithelial-Mesenchymal Transition and Breast Cancer Metastasis via the CXCL12/CXCR4/mTOR Axis. Cancer Res. 2019;79(4):735–746. doi:10.1158/0008-5472.CAN-18-0620
33. Liu J, Huang C, Peng C, et al. Stromal fibroblast activation protein alpha promotes gastric cancer progression via epithelial-mesenchymal transition through Wnt/ β-catenin pathway. BMC Cancer. 2018;18(1):1099. doi:10.1186/s12885-018-5035-9
34. Tang Z, Li J, Shen Q, et al. Contribution of upregulated dipeptidyl peptidase 9 (DPP9) in promoting tumoregenicity, metastasis and the prediction of poor prognosis in non-small cell lung cancer (NSCLC). Int J Cancer. 2017;140(7):1620–1632. doi:10.1002/ijc.30571
35. Augoff K, Hryniewicz-Jankowska A, Tabola R, et al. Upregulated expression and activation of membrane‑associated proteases in esophageal squamous cell carcinoma. Oncol Rep. 2014;31(6):2820–2826. doi:10.3892/or.2014.3162
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.