Back to Journals » Journal of Inflammation Research » Volume 19
FGF21 Exacerbates Obesity-Induced Airway Hyperresponsiveness and FGFR1-Dependent Mast Cell Activation in Mice
Authors Ren L, Xuan L, Zhang J, Zhang W, An Z
Received 10 October 2025
Accepted for publication 17 January 2026
Published 24 January 2026 Volume 2026:19 570000
DOI https://doi.org/10.2147/JIR.S570000
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Tara Strutt
Lulu Ren,1 Lingling Xuan,1 Jie Zhang,1 Wen Zhang,2 Zhuoling An2
1Medical Research Center, Beijing Institute of Respiratory Medicine and Beijing Chao-Yang Hospital, Capital Medical University, Beijing, People’s Republic of China; 2Department of Pharmacy, Beijing Chao-Yang Hospital, Capital Medical University, Beijing, 100020, People’s Republic of China
Correspondence: Lulu Ren, Medical Research Center, Beijing Institute of Respiratory Medicine and Beijing Chao-Yang Hospital, Capital Medical University, 8 Gongren Tiyuchang Nanlu, Beijing, 100020, People’s Republic of China, Tel +86-010-85231720, Email [email protected] Zhuoling An, Department of Pharmacy, Beijing Chao-Yang Hospital, Capital Medical University, 8 Gongren Tiyuchang Nanlu, Beijing, 100020, People’s Republic of China, Tel +86-010-85231464, Email [email protected]
Introduction: Obesity is a well-established risk factor for asthma pathogenesis. However, the underlying mechanisms remain incompletely understood, and effective therapeutic interventions are currently lacking, making asthma management in obese individuals particularly challenging. Asthma is characterized by chronic airway inflammation, eosinophilic infiltration, and airway hyperresponsiveness (AHR). In this study, we investigated the novel role of fibroblast growth factor 21 (FGF21), a stress-inducible hepatokine with pleiotropic metabolic regulatory functions, in obesity-associated AHR using a diet-induced obesity mouse model (n = 10).
Material and Methods: Serum samples were collected from obese and lean asthma patients, along with relevant clinical indicators, including body mass index (BMI), forced expiratory volume in 1 second (FEV1%), and the FEV1/forced vital capacity (FVC) ratio, to facilitate the investigation. Moreover, diet-induced obese mice with innate AHR (male, n = 10) were employed to clarify the effects of FGF21 and FGF21-neutralizing antibody on obesity induced AHR. In vitro, LAD2 human mast cells and P815 murine mast cells activated by compound 48/80 were used to elucidate the underlying mechanisms.
Results: Our findings demonstrate that serum FGF21 levels exhibit reportedly elevated in participants with obesity and are associated with impaired pulmonary function. In diet-induced obese (DIO) mice, FGF21 levels were increased in both serum and bronchoalveolar lavage fluid (BALF). In vivo investigations demonstrate that administration of recombinant FGF21 exacerbated AHR in DIO mice, whereas FGF21-neutralizing antibody treatment ameliorated obesity-induced AHR and suppressed mast cell infiltration. Mechanistically, FGF21 was found to potentiate mast cell activation through cholesterol biosynthesis modulation. Crucially, pharmacological inhibition of FGFR1 abrogated FGF21-induced mast cell hyperactivity and cholesterol synthesis, indicating FGFR1-dependent signaling in this process.
Conclusion: These findings may represent the FGF21/FGFR1 axis as a potential therapeutic target for obesity-related AHR and asthma.
Keywords: obesity, airway hyperresponsiveness, FGF21, mast cell
Introduction
Obesity constitutes a significant independent risk factor for asthma development.1 The escalating global prevalence of obesity has led to a parallel surge in asthma cases among obese populations, prompting recognition of “obesity-associated asthma” as a distinct clinical phenotype characterized by heightened symptom severity and increased exacerbation frequency.2 Current therapeutic strategies exhibit limited efficacy in this patient subgroup, with clinical studies demonstrating reduced responsiveness to corticosteroids and β-agonists in obese asthmatics.3,4 Although substantial evidence links obesity to asthma pathogenesis and progression, the precise molecular mechanisms underlying this association remain poorly understood.
Fibroblast growth factor 21 (FGF21), a hepatokine induced by metabolic stress, serves as a central regulator of glucose and lipid homeostasis.5,6 This pleiotropic hormone modulates fatty acid oxidation in hepatic tissue while enhancing glucose metabolism in adipose depots.7 Notably, emerging evidence suggests FGF21 promotes white adipose tissue browning, a process critical for systemic energy balance regulation.8 These metabolic properties have positioned FGF21 and its analogs as promising therapeutic candidates for obesity-related comorbidities, including hyperglycemia, insulin resistance, and non-alcoholic steatohepatitis.5,9 Despite its metabolic benefits, the potential roles of FGF21 in respiratory pathologies remain insufficiently explored. FGF21 has been reported to exert immunomodulatory effects by regulating immune cell metabolism, thereby influencing immune cell activation and effector function under stress conditions. For example, FGF21 in the tumor microenvironment has been shown to promote T cell exhaustion and impair CD8⁺ T cell cytotoxicity. These observations suggest that stress-induced FGF21 upregulation may remodel local immune microenvironments through metabolic reprogramming of immune cells, although the precise mechanisms remain to be clarified.10 Previous studies have shown that in patients with allergic asthma, fibroblast growth factor 21 (FGF21) levels are negatively correlated with pulmonary function.11 Clinically relevant elevations in circulating FGF21 observed in obese individuals12,13 raise intriguing questions about its potential immune regulatory effects on airway pathophysiology.
Airway hyperresponsiveness (AHR), recognized as a defining clinical characteristic of asthma, is described as an abnormal tendency of the airways to undergo bronchoconstriction when exposed to stimuli that are typically innocuous to healthy individuals.14 The pathogenesis of AHR involves multifactorial mechanisms, including dysregulated inflammatory mediators and environmental triggers that potentiate airway smooth muscle hypercontractility, ultimately contributing to asthma exacerbations.15 Preclinical studies have established that obesity induces innate AHR,16 positioning DIO mice as a validated experimental model for investigating obesity-related asthma pathophysiology. Preclinical studies have established that obesity induces innate AHR,16 positioning diet-induced obese (DIO) mice as a validated experimental model for investigating obesity-related asthma pathophysiology. Building on these observations, we employed a model of obesity-induced AHR in mice in the absence of any allergen or antigen challenge, allowing us to examine the direct effects of obesity on airway physiology independently of classical allergic sensitization.
Mast cells are important immune defense cells that can rapidly respond to a variety of stimuli and participate in inflammatory responses and tissue repair.17,18 However, the activation of mast cells in many diseases is potentially deleterious, including asthma and systemic mastocytosis.19,20 Mounting research reports that the release of mediators by activated mast cells plays an important role in promoting asthma.19,21 The activation of mast cells can be induced by a variety of stimuli, including IgE, cytokines, complement, pollutants, and drugs.19,22 Emerging evidence suggests that mast cells may serve as a mechanistic link between obesity and asthma. Obesity-related factors, including elevated adipokines, IL-33, IL-9, and neuropeptides, can activate pulmonary mast cells, leading to airway inflammation, hyperresponsiveness, and remodeling, supporting a potential role for mast cells in mediating obesity-associated airway dysfunction.23 Nevertheless, the precise mechanisms require further investigation. In the previous study, bariatric surgery could improve pulmonary function and reduce the number of mast cell in the airways.24 However, the role of mast cell activation in obesity-associated AHR is not yet illustrated.
In this study, we elucidate a previously unrecognized role of FGF21 in obesity-associated AHR using a DIO murine model. Our findings demonstrate that elevated FGF21 levels in both serum and bronchoalveolar lavage fluid (BALF) inversely correlate with impaired pulmonary function parameters, including FEV1% and the FEV1/FVC ratio. Through in vivo experimentation, we observed that exogenous recombinant FGF21 administration exacerbated AHR in obese mice, whereas FGF21-neutralizing antibody treatment attenuated obesity-induced AHR and suppressed mast cell activation. Mechanistic investigations revealed that FGF21 potentiates AHR through FGFR1-dependent upregulation of cholesterol biosynthesis pathways. Collectively, these findings provide compelling preclinical evidence supporting the therapeutic potential of targeting the FGF21/FGFR1 axis for asthma management in obese populations.
Methods
Study Subjects
This study was approved by the Research Ethics Committee of Beijing Chao-Yang Hospital (No. 2020-ke-415). All participants provided written informed consent after being fully informed of the study objectives, procedures, potential risks and benefits, and the voluntary nature of participation. Lean control participants (n=26, BMI 18.5–24 kg/m2) were recruited from routine health check-up attendees, and obese control participants (n=17, BMI ≥ 28 kg/m2) were recruited as individuals with obesity. All control participants had no history of asthma or other chronic respiratory diseases. Exclusion criteria were applied as described for the patient groups. Fasting peripheral blood samples were collected from all participants for subsequent analyses. Adult patients with asthma were recruited between July 2020 and December 2020, including lean patients (n = 23) and obese patients (n = 20). Obesity (BMI ≥ 28 kg/m2) and leanness (18.5 ≤ BMI < 24 kg/m2) were defined according to body mass index (BMI) criteria in Chinese Guidelines for the Prevention and Control of Overweight and Obesity in Adults (2021 Revision).25 The diagnosis of asthma was established based on Global Initiative for Asthma (GINA, 2023).26 The exclusion criteria include: (1) Patients with chronic obstructive pulmonary disease (COPD), bronchiectasis, cystic fibrosis, interstitial lung disease (ILD), infectious lung disease, pulmonary embolism, pulmonary hypertension, acute respiratory distress syndrome (ARDS), and pneumoconiosis; (2) Patients who receive systemic steroids therapy or biologic therapy, including omalizumab; (3) Pregnant and lactating females; (4) Cancer patients. Detailed demographic and clinical information was collected at enrollment. Asthma treatment status was recorded according to the GINA recommendations. Asthma-related medications, particularly inhaled corticosteroids (ICS), systemic corticosteroids, and biologic therapies (eg, omalizumab), were carefully recorded due to their potential effects on FGF21 levels and pulmonary function. At the time of enrollment, some patients (n=23) were receiving inhaled corticosteroid (ICS)–based therapy, while others (n=20) had not yet initiated controller medication. Patients receiving systemic corticosteroids or biologic therapies were excluded from the study in accordance with the predefined exclusion criteria. Clinical characteristics of the study participants are summarized in Table 1. Fasting peripheral venous blood was collected from all patients, and 1 mL of residual serum after routine clinical tests was used for analysis. Samples were immediately aliquoted and stored at −80°C, with a maximum of one freeze-thaw cycle prior to analysis. The time from blood draw to freezing did not exceed 2 hours. Pulmonary function tests were performed at the same visit as fasting blood collection, ensuring temporal alignment with the serum measurements. The study adhered to the Declaration of Helsinki.
 |
Table 1 Characteristics of the Subjects |
Animals
Mouse models of obesity-induced AHR were used in this study. C57BL/6J mice (6-week-old, male) were provided by Vital River Laboratory Animal Technology Company (Beijing, China). Only male C57BL/6J mice were used in this study, which may limit the generalizability of the findings to females. After a 1-week adaptation period, the mice were fed with a high fat diet (HFD, 60% fat, D12492, Research Diets) or a regular chow diet (CD, 10% fat, D12450J, Research Diets) for 16 weeks. Each experimental group consisted of 10 mice. A predefined exclusion criterion was a body weight of less than 35 g; however, all mice in the diet-induced obesity model exceeded this threshold, and no animals were excluded from the analysis. Obese mice fed with HFD at 21–24 weeks of age exhibited marked innate AHR when contrasted with lean mice.27 Therefore, these obese mice were used as models for obesity-induced AHR in this study. Mice were randomly assigned to the control and treatment groups using a random number method. To clarify the effect of FGF21 in obesity-induced AHR, the mice were intraperitoneally injected with recombinant FGF21 (i.p, 1.5 mg/kg). Recombinant mouse FGF21 was purchased from MedChemExpress (HY-P72651). For all in vivo experiments, vehicle-treated controls were included to control for non-specific effects. Moreover, the mice were treated with anti-FGF21 neutralizing antibody or control IgG (i.p, 10 μg per mouse) every four days. Mice were divided into four groups: lean + IgG, obese + IgG, lean + anti-FGF21, and obese + anti-FGF21. Anti-FGF21 antibody (AF3057) and control IgG (AB-108-C) were purchased from Bio-Techne. All mice were housed in static cages (5 mice/cage) at 22–24°C under a cycle with 12 h/12 h of darkness and light. Water and food were available ad libitum. At the end of the experiment, mice were euthanized under anaesthesia via pentobarbital sodium (0.3%), and serum and tissues were obtained for subsequent experiments. Mice were fasted for 6 h before blood sampling. BALF was collected by euthanizing the mice, cannulating the trachea, and gently lavaging the lungs three times with 0.5 mL of cold sterile PBS. The recovered lavage fluid was pooled for subsequent analyses. A total of 40 mice were used in this study. The sample size for the formal experiment was estimated based on the results of the preliminary study. All animal procedures were performed in compliance with the Health Guidelines established by Capital Medical University (No. AEEI-2024-102) and in accordance with the ARRIVE guidelines.
Enzyme-Linked Immunosorbent Assay (ELISA)
FGF21 concentration in serum collected from patients was measured by ELISA kits (Human FGF21 ELISA Kit, Sangon Biotech) with a detection range of 31.25–2000 pg/mL. And ELISA kits (Mouse FGF21 ELISA Kit, Sangon Biotech) were used to measure FGF21 concentration in mouse serum and BALF according to the manufacturer’s protocols, which had a detection range of 31.25–2000 pg/mL.
AHR Measurement
The specific airway resistance (sRaw) of 22 week age of mice was measured using non-invasive airway mechanics (Buxco FinePointe, DSI). In the test, methacholine (0, 12.5, 25, 50 mg/mL) was used as a stimulant. The methacholine solution was nebulized via an atomizing pump and the mice were subjected to methacholine exposure. The adaptation period lasted for 5 minutes, the nebulization time was 1 minute, the reaction time was 5 minutes, and the recovery time was 2 minutes. The sRaw values within the reaction time range were recorded. To investigate the role of Anti-FGF21 in obesity induced AHR, changes in pulmonary resistance (RL) was assessed using the animal pulmonary function analysis system (AniRes2005, Bioon, China). In the test, methacholine (0, 0.2, 0.4, 0.8 mg per mouse) was used as a stimulant. The mice were injected intravenously with methacholine. The adaptation period lasted for 5 minutes, the reaction time was 5 minutes, and the recovery time was 2 minutes. The RL values within the reaction time range were recorded. Average RL values were calculated. A different respiratory measurement platform was used in the anti-FGF21 intervention experiments due to equipment availability; however, all measurements were performed under standardized conditions and using established methacholine challenge protocols. Comparisons were made within each experimental system rather than across platforms.
Immunohistochemistry
Immunohistochemical staining was performed to detect chymase expression in lung tissues. Lung samples were fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned at 5 μm thickness. After deparaffinization, rehydration, and antigen retrieval, tissue sections were incubated with a primary antibody against chymase (Santa Cruz Biotechnology, sc-71540; dilution 1:100) overnight at 4 °C. A polymer-based horseradish peroxidase–conjugated secondary antibody was then applied according to the manufacturer’s instructions. Images were acquired using a fluorescence microscope and quantified with ImageJ.
Cell Culture and Treatment
The P815 cells were purchased from Oricell (Cyagen Biosciences, China). It was cultured in complete DMEM (Gibco, USA) supplemented with 10% fetal bovine serum (FBS). The LAD2 (laboratory of allergic diseases 2) cells used in this study were generously provided by professor Zhao Hongmei from Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences. LAD2 cells were cultured in complete RPMI1640 medium (Gibco, USA). To investigate the effect of FGF21 at various dosages on mast cell activation, P815 and LAD2 cells were pretreated with FGF21 (0, 100, 200, 400 ng/mL) for 24 h. For the in vitro mast cell activation assay, the cells were incubated with Compound 48/80 (40 μg/mL) dissolved in TM buffer for 1 hour before the release detection of β-hexosaminidase and histamine. For the knockdown of sterol regulatory element binding transcription factor 1 (SREBF1), cells were transfected with siRNA using Lipofectamine™ RNAiMAX (Thermo Fisher Scientific, No. 13778075) according to the manufacturer’s instructions. The sequence of SREBF1-siRNA was 5′CGGAGAAGCUGCCUAUCAATT3′, and the sequence of siRNA-NC was 5′UUCUCCGAACGUGUCACGUTT3’. To inhibit the activation of FGFR1, we used the specific antagonist PD173074 at a concentration of 200 nM. The cells were pretreated with PD173074 for 24 hours prior to FGF21 administration. In the in vitro experiment, three independent experiments were performed and three replicas were analyzed in each experiment.
Quantitative RT-PCR
The relative expression levels of mRNA were examined by quantitative real-time polymerase chain reaction (qRT-PCR) method. The primers used in the experiment were synthesized by BGI Genomics and the sequences are listed inTable S1. Total RNA was extracted using RNAiso Plus (Takara, Shiga, Japan) and reverse transcripted using the FastKing RT kit (Tiangen, China). And PCR was conducted with the qPCR PreMix (SYBR Green) kit (Tiangen, China) Gene expression levels was normalised to GAPDH (2–ΔΔCT).
Western Blotting
Total protein was extracted using RIPA buffer supplemented with protease and phosphatase inhibitors and quantified using a Bradford assay. Equal amounts of protein were separated by 8% SDS–PAGE and transferred to PVDF membranes. Membranes were blocked with 5% skim milk and incubated with primary antibodies followed by fluorescently labeled secondary antibodies. Protein bands were visualized using an LI-COR Odyssey imaging system. Primary antibodies included anti-GAPDH (ab8245, Abcam), anti-GABABR1 (CST #3835), and anti-GABAARα (sc-376282, Santa Cruz Biotechnology). Primary antibodies included anti-GAPDH (ab8245, Abcam) and anti-FGFR1 (ab63601, Abcam).
β-Hexosaminidase Release Assay
After incubation with Compound 48/80, the cells were centrifuged (1500 rpm, 4 °C) for 10 min. Then the culture supernatant was obtained. Triton X- 100 dissolved in TM buffer (1.0%) was applied for cell lysis. Then the cell lysis mixture was centrifuged (1500 rpm, 4 °C) for 10 min. And the lysate supernatant was obtained. The culture supernatant or lysate supernatant was mixed with the equal volume of N-Acetyl-D-glucosamine (NAG, 5 μM) dissolved in citrate buffer (pH4.5) and incubate at 37°C for 1 h. After that, NaHCO3/Na2CO3 buffer (0.1 M, pH10.0) was used for terminating the reaction. At the end, a microplate reader was used for measuring the OD value at 405 nm. The β-hexosaminidase release rate was calculated as the ratio of supernatant activity to total activity (supernatant + cell lysate).
Histamine Release Assay
Histamine concentration in the culture supernatant released from mast cells was measured using His ELISA kit (D751012, Sangon Biotech, China).
Measurement of Cellular Calcium Concentration
Cellular calcium concentration was measured using Fluo-4 Calcium Assay Kit (S1061S, Beyotime, China). At the end, the cells were analyzed by fluorescent microscope (Olympus, Japan). The quantitative data was analyzed using Image J.
Measurement of Cellular Cholesterol Content
The Cholesterol Assay Kit (Cell-Based) (ab133116) was used for the measurement of cellular cholesterol content. Cells were fixed with 4% paraformaldehyde at room temperature for 15 min, followed by washing with phosphate-buffered saline. Filipin staining was performed for 1 hour using filipin III (50 μg/mL) according to the manufacturer’s instructions. The mast cells were pictured by a fluorescent microscope and the quantitative data was analyzed using Image J. Fluorescence images were acquired using a fluorescence microscope equipped with appropriate filter sets. Filipin fluorescence was detected using an excitation wavelength of 360 nm and an emission filter of 460 nm. All images were captured using identical exposure times, gain, and acquisition settings across experimental groups.
Statistical Analysis
Data conforming to a normal distribution are presented as means ± SEM, whereas data not conforming to a normal distribution presented as median (interquartile range). The data were analyzed using GraphPad Prism 8.00. An unpaired two-tailed t-test was employed to assess the differences between two groups when the data passed the equal variance test. For comparisons involving more than two groups, we used ANOVA followed by Bonferroni’s post hoc multiple comparisons tests. Mann–Whitney test was employed to assess the differences between two groups when the data did not follow a normal distribution. No data points were excluded. The experimenters and data analysts were blinded to the experimental group allocations. Statistically significant was set at p < 0.05.
Results
FGF21 Elevation in Obese Mice Correlates with Airway Hyperresponsiveness
HFD-induced obese mice develop intrinsic airway hyperresponsiveness (AHR), with significantly exacerbated respiratory resistance observed at 21–24 weeks compared to age-matched lean controls.27 To clarify FGF21’s role in obesity-associated AHR, we employed a DIO model (Figure 1A and B). Biochemical analysis revealed substantial upregulation of FGF21 in both serum (456.7 ± 131.7 pg/mL vs 42.4 ± 6.4 pg/mL, unpaired two-tailed t test, P=0.0056) and BALF (166.5 ± 14.2 pg/mL vs 76.9 ± 5.0 pg/mL, unpaired two-tailed t test, P<0.0001) in HFD-fed obese mice (n=10) compared to chow-fed lean controls (n=10) (Figure 1C and D). Consistent with previous reports, DIO mice exhibited progressive AHR development, demonstrated by dose-dependent increases in airway resistance following methacholine challenge (0–50 mg/mL; Figure 1E). Strikingly, Pearson correlation analysis revealed strong positive associations between FGF21 levels (both serum: r=0.87, p<0.05; BALF: r=0.82, p<0.05) and methacholine-induced airway resistance (Figure 1F and G). These findings implicate FGF21 as a potential mediator in obesity-related AHR pathogenesis.
 |
Figure 1 FGF21 is upregulated in serum and BALF of obese mice, and positively correlated with airway resistance. Mice were fed either a standard chow diet (lean) or a high-fat diet (obese) for 16 weeks prior to analysis. (A) Measurements of body weight from the lean and obese group mice. (B) Representative pictures of lean and obese mice. (C) Levels of serum FGF21 in lean and obese mice were measured by ELISA (n=10). (D) Levels of BALF FGF21 in lean and obese mice were measured by ELISA (n=10). (E) DIO mice exhibit pronounced AHR. Results showed the changes in specific airway resistance (sRaw) as a measure of AHR. (F) Pearson’s correlation tests results represent FGF21 level in serum of obese mice was positively correlated with sRaw (Cmethacholine:50.0 mg/mL). (G) Pearson’s correlation tests results represent FGF21 level in BALF of obese mice was positively correlated with sRaw (Cmethacholine:50.0 mg/mL). Data are mean ± SEM, **p < 0.01, ***p < 0.001. |
Circulating FGF21 Elevation Correlates with Pulmonary Dysfunction in Asthma Patients
To investigate the clinical relevance of FGF21 in obesity-associated AHR, we conducted a cross-sectional analysis comparing serum FGF21 levels between asthma patients with obesity and lean asthmatics. Quantification via ELISA revealed significantly elevated FGF21 concentrations in obese asthmatics (309.5 ± 21.7 pg/mL) compared to their lean counterparts (215.8 ±11.6 pg/mL) (Figure 2A). Serum FGF21 levels in asthma patients were significantly positively correlated with BMI (Pearson r = 0.49, P < 0.05), consistent with previous reports linking FGF21 to adiposity (Figure S1). To establish the functional relationship between FGF21 and respiratory pathophysiology, we performed correlation analyses of FGF21 levels against spirometric parameters. Multivariable linear regression analysis adjusting for age demonstrated that serum FGF21 levels were independently associated with FEV1% (β= −0.65, P=0.028) and FEV1/FVC % (β=−0.65, P=0.037) (Figure 2B and C). These clinical observations suggest that FGF21 upregulation may contribute to airway dysfunction in obese asthma patients.
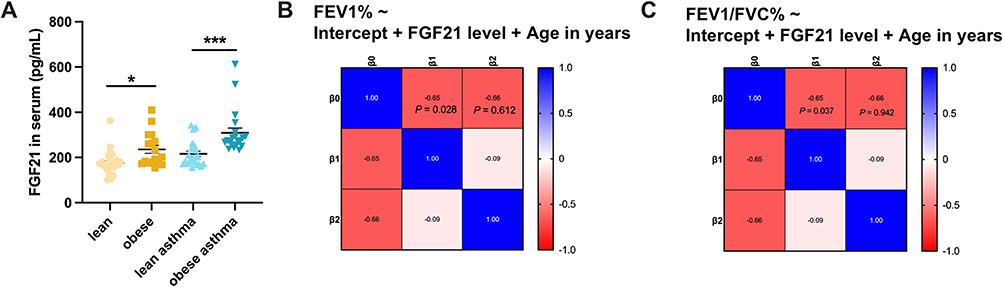 |
Figure 2 FGF21 level is increased in serum from obese patients with asthma and positively correlated with reduced pulmonary function. (A) Serum FGF21 levels in lean patients with asthma and obese patients with asthma were measured by ELISA (n=20-23). Data are mean ± SEM, *p < 0.05, ***p < 0.001 (B) Multivariable linear regression results represent serum FGF21 level was negatively correlated with FEV1%. (C) Multivariable linear regression results represent serum FGF21 level was negatively correlated with FEV1/FVC%, n=43. |
FGF21 Neutralization Ameliorates Obesity-Induced AHR and Inhibits Mast Cell Infiltration
To establish causal relationships, we administered recombinant FGF21 (1.5 mg/kg, i.p.) or saline to DIO mice during methacholine challenge protocols (Figure 3A). Specific airway resistance (sRaw) measurements confirmed baseline AHR in DIO mice, consistent with previous reports.28 Notably, FGF21 administration exacerbated methacholine-induced airway resistance compared to saline controls (Figure 3B). In therapeutic intervention studies, anti-FGF21 neutralizing antibody or isotype control IgG was administered to lean and DIO mice (Figure 3E). While anti-FGF21 showed no significant effects on airway reactivity in lean mice, it markedly reduced methacholine-evoked lung resistance (RL) in DIO mice compared to IgG controls (Figure 3F).
 |
Figure 3 Recombinant FGF21 aggravates AHR in obese mice, while Anti-FGF21 ameliorates obesity-induced AHR and inhibits mast cell infiltration. (A) The schematic diagram for recombinant FGF21 treatment in DIO mice. (B) Changes of airway resistance in DIO mice treated with recombinant FGF21. (C) Representative immunohistochemical images (× 400) for lung tissue sections staining of chymase. (D) Quantitative analysis of chymase staining. (E) The schematic diagram for Anti-FGF21 treatment in lean mice or DIO mice. (F) Changes of lung resistance (RL) in DIO mice treated with the Anti-FGF21. (G) Representative immunohistochemical images (× 400) for lung tissue sections staining of chymase. (H) Quantitative analysis of chymase staining. n=5. Data are mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001. IgG: the normal IgG control antibody. |
Given the dual roles of mast cells in immune surveillance and pathological inflammation,17–20 we assessed their number and spatial distribution in the airway using chymase immunohistochemistry. DIO mice exhibited higher mast cell density in airway compared to lean controls (Figure 3C and D). FGF21 treatment further amplified mast cell infiltration in obese mice (Figure 3C and D), whereas anti-FGF21 reduced mast cell infiltration in the airway of obese mice (Figure 3G and H).
Considering FGF21’s metabolic benefits, we monitored the body weight, fasting blood glucose (FBG), serum insulin level and total cholesterol of mice following 12-day anti-FGF21 treatment. No significant differences were observed in these parameters between anti-FGF21 and IgG groups (Figure S2A–D). These findings demonstrate that mast cell infiltration and activation associated with FGF21 may contribute to obesity-associated AHR, and the pharmacological inhibition of FGF21 achieves airway benefits without disrupting metabolic homeostasis in a 12-day treatment.
FGF21 Potentiates Mast Cell Activation via Modulating Cholesterol Biosynthesis
To mechanistically interrogate FGF21’s effects on mast cell, we employed LAD2 human mast cells and P815 murine mast cells activated by compound 48/80. Quantitative assessment of degranulation markers revealed that FGF21 dose-dependently enhanced β-hexosaminidase release and histamine secretion compared to vehicle controls (Figure 4A and B). An elevated intracellular calcium concentration is associated with mast cell activation and plays a key role in both initiating mast cell activation and facilitating the release of mast cell-derived mediators. Therefore, we detected the intracellular calcium concentration of mast cells treated with FGF21 or vehicle using the fluorescent probe Fluo-4 AM. Calcium imaging using Fluo-4 AM demonstrated that FGF21 treatment elevated intracellular Ca2⁺ levels in LAD2 and P815 cells versus vehicle (Figure 4C and D), confirming enhanced mast cell activation.
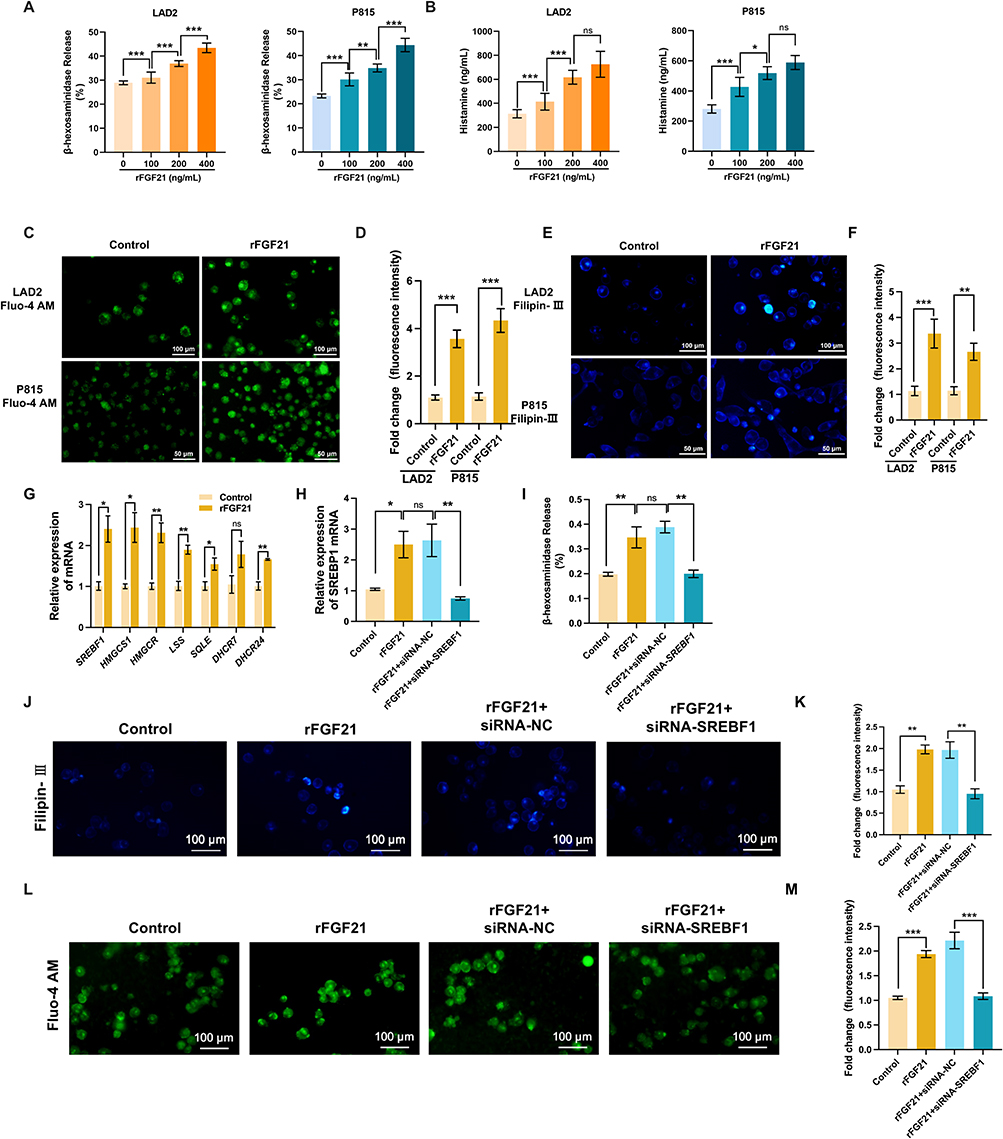 |
Figure 4 FGF21 facilitates mast cell activation through up-regulating cholesterol biosynthesis. (A) Release of β-hexosaminidase from LAD2 and P815 cells pre-activated with compound 48/80 following 24 h treatment with recombinant FGF21 (100–400 ng/mL). (B) Release of histamine from LAD2 and P815 cells pre-activated with compound 48/80 following 24 h treatment with recombinant FGF21 (100–400 ng/mL). (C) Measurement of cellular calcium concentration in mast cells pre-activated with compound 48/80 using fluorescent probe Fluo-4 AM following 24 h treatment with recombinant FGF21 (200 ng/mL). (D) Quantitative analysis of fluorescence intensity of Fluo-4. (E) Filipin III staining of cholesterol in mast cells pre-activated with compound 48/80 following 24 h treatment with recombinant FGF21 (200 ng/mL). (F) Quantitative analysis of fluorescence intensity of Filipin III. (G) Expression levels of cholesterol biosynthesis genes in LAD2 cells. (H) Expression level of SREBF1. (I) Quantitative analysis of fluorescence intensity of Filipin III in siRNA pretreated cells. (J) Representative images of Filipin III staining in siRNA-NC or siRNA-SREBF1 pretreated cells. (K) Release rate of β-hexosaminidase from LAD2 cells treated with siRNA and FGF21. (L) Measurement of cellular calcium concentration in LAD2 cells using fluorescent probe Fluo-4 AM. (M) Quantitative analysis of fluorescence intensity of Fluo-4 in LAD2 cells treated with siRNA and FGF21. n=3. Data are mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant. |
Given FGF21’s established metabolic regulatory roles, we hypothesized cholesterol biosynthesis modulation as a potential mechanism. Filipin III fluorescence quantification showed higher cholesterol in FGF21-treated mast cells (Figure 4E and F), corroborated by qRT-PCR showing upregulation of cholesterol biosynthesis genes in LAD2 cells (Figure 4G). Sterol regulatory element-binding protein 1 (SREBP1), which is a central transcription factor for lipid metabolism, was encoded by SREBF1. To verify that FGF21 promotes mast cell activation via up-regulating cholesterol biosynthesis, we silenced SREBF1 in LAD2 cells using siRNA (Figure 4H), which abrogated FGF21-induced cholesterol biosynthesis (Figure 4J and K). Crucially, restricted the release of β-hexosaminidase (Figure 4I) and the cellular calcium accumulation (Figure 4L and M) induced by FGF21. These data demonstrated that FGF21 functions as a positive regulator of mast cell activation via regulating cholesterol synthesis.
Enhancement of Cholesterol Synthesis and Mast Cell Activation in the Airway Induced by FGF21 Is FGFR1-Dependent
It has been reported that FGF21 could activate the FGFR1, FGFR2 and FGFR3 receptors. The public data in the Human Protein Atlas implies that mast cells within the airway might express FGFR1 at a high level (Figure 5A). To confirm potential receptors activated by FGF21 in airway mast cells, the expression of FGF21 receptors was measured. The result suggested that FGFR1 was abundantly expressed in mast cells, while FGFR2 and FGFR3 were barely expressed in mast cells (Figure 5B). Western blotting analysis confirmed the expression of FGFR1 protein in mast cells (Figure S3). These data indicate that FGF21 might enhance the biosynthesis of cholesterol and activate mast cell in airway by binding FGFR1. To verify this speculation, the FGFR1-specific antagonist PD173074 was used. Release rate of β-hexosaminidase from LAD2 cells pretreated with PD173074 was measured. The result indicated that FGF21 failed to increase the release rate of β-hexosaminidase from cells pretreated with PD173074 (Figure 5C). Moreover, the result suggested that PD173074 restricted the cellular calcium accumulation induced by FGF21 (Figure 5D and E). These data indicated that FGF21 activated mast cells was FGFR1-dependent. To further investigate the effect of PD173074 on the cholesterol synthesis in mast cells, the cholesterol biosynthesis-related genes expression were measured. And the results of qPCR analysis demonstrated FGF21 failed to up-regulate cholesterol biosynthesis-related genes in cells pretreated with PD173074 (Figure 5F). The cellular cholesterol content was measured using Filipin III staining assay and results indicated that the cholesterol content in mast cells treated with PD173074 and FGF21 was decreased compared with the cells treated with vehicle and FGF21 (Figure 5G and H). These data demonstrated that FGF21 promoted AHR in obese mice through increasing cholesterol synthesis and facilitating mast cell activation, which is FGFR1-dependent (Figure 6).
 |
Figure 5 FGF21 promoted cholesterol synthesis and mast cell activation in a FGFR1-dependent manner. (A) Expression of FGFR1, FGFR2 and FGFR3 in lung tissues. (Data from Human Protein Atlas, http://www.proteinatlas.org/). (B) The mRNA expression of FGFR1, FGFR2, and FGFR3 in LAD2 cells. Relative expression levels were normalized to GAPDH. (C) Release rate of β-hexosaminidase from LAD2 cells treated with FGFR1 inhibitor PD173074. (D) Measurement of cellular calcium concentration in PD173074 treated LAD2 cells using fluorescent probe Fluo-4 AM. (E) Quantitative analysis of fluorescence intensity of Fluo-4 in LAD2 cells. (F) Expression levels of cholesterol biosynthesis genes in PD173074 treated LAD2 cells. (G) Representative images of Filipin III staining in PD173074 treated LAD2 cells. (H) Quantitative analysis of fluorescence intensity of Filipin III in PD173074 treated cells. n=3. Data are mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001. |
 |
Figure 6 Schematic of the role of FGF21 in obesity induced AHR. FGF21 promotes AHR in obese mice through increasing cholesterol synthesis and facilitating mast cell activation in a FGFR1-dependent manner. |
Discussion
Obesity constitutes an independent risk factor for the initiation or aggravation of asthma.29 Obesity-associated asthma represents a unique phenotype with specific immunological and pathological traits and demonstrates insensitivity to glucocorticoids and β-agonists.28 Research has failed to clarify the pathogenic mechanism of obese asthma, recognized as a metabolic-immune crosstalk-related disorder, resulting in the absence of potential drug targets.30 Our study identified a novel role of FGF21 in the airways of obese individuals. Contrary to the previous perception regarding the beneficial effects of FGF21, we found that elevated level of FGF21 in obese patients might be one of the crucial factors inducing AHR, and its role is associated with the activation of mast cells in the airway. Hence, FGF21 could serve as a potential novel target for the treatment of obesity induced AHR and asthma.
FGF21 serves as a crucial regulator of metabolism, particularly for lipid and glucose. Although previous studies have elucidated the physiological functions and mechanisms of FGF21 in obesity and other metabolic diseases, its role in obesity-induced respiratory disorders remains largely unknown. A study related to obesity-associated asthma showed that FGF21 levels in serum were negatively correlated with pulmonary function and positively correlated with the number of mast cell progenitors.11 Another study demonstrated that serum FGF21 levels were higher in obese asthma patients than in normal-weight asthma patients, suggesting that FGF21 could serve as a specific serum biomarker for obesity-associated asthma.31 However, these analyses were largely based on unadjusted correlation approaches. In contrast, our study applied multivariable linear regression to evaluate the association between serum FGF21 and pulmonary function while controlling for relevant covariates, thereby providing stronger evidence that FGF21 is independently associated with lung function impairment, though causality cannot be inferred from this cross-sectional analysis. Moreover, our work focuses on obesity-associated airway dysfunction rather than classical allergic asthma, highlighting a distinct pathophysiological context in which FGF21–mast cell interactions may contribute to airway hyperresponsiveness. In an asthma study, high serum FGF21 levels were strongly associated with poor asthma control,32 indicating a potential causal relationship between FGF21 and asthma development, highlighting the urgent need for in-depth studies to clarify its physiological and pathological roles in the airway. Preclinical studies have shown that FGF21 can prevent asthma development in mice by inhibiting NLRP3 inflammasome activation.33 It should be noted that previous mechanistic studies, such as those examining NLRP3 inflammasome regulation, were performed in lean mice, representing a different baseline physiological state. In contrast, our study focuses on obesity-induced airway dysfunction, where elevated FGF21 may interact with obesity-associated metabolic and inflammatory changes to exacerbate airway hyperresponsiveness. Consequently, the effects and mechanisms of FGF21 may differ between obesity alone, classical asthma, or the combination of obesity and asthma, which should be considered when extrapolating these findings to human disease.
In this study, we found that FGF21 increased cholesterol synthesis in mast cells, thereby promoting mast cell activation. It is worth noting that FGF21 alone did not activate mast cells in our in vitro assays (not reported), suggesting that FGF21 enhances rather than initiates mast cell degranulation. In the obese microenvironment, elevated adipokines (eg, leptin, adiponectin) and innate immune alarmins such as IL-33 and IL-9 have been shown to promote mast cell recruitment and activity, potentially synergizing with metabolic stress to drive mast cell activation in vivo.23,34 Activated mast cells release pro-inflammatory mediators, inducing abnormal contraction of the airway smooth muscle and ultimately causing AHR.35 The effects of FGF21 in different diseases appear to be tissue-specific. In energy-metabolic organs, including the liver, adipose tissue, and skeletal muscle, basal FGF21 levels are relatively high, serving to counteract lipid overload and maintain glucose homeostasis.36,37 In contrast, other organs exhibit low basal FGF21 levels that are highly responsive to the organism’s metabolic state. For example, circulating FGF21 levels in patients with obesity-associated asthma are negatively correlated with pulmonary function.11 Consistently, our data show that FGF21 levels in the airways of obese mice are significantly elevated compared with those in lean controls. We further identified a novel role of FGF21 in regulating immune cell metabolism, thereby modulating their immune function. Similarly, Cegui Hu et al recently reported that FGF21 within the tumor microenvironment promotes T cell exhaustion and impairs the cytotoxic function of CD8⁺T cells.10 These observations suggest that in organs with low basal FGF21, stress-induced upregulation of FGF21 may remodel the tissue immune microenvironment via metabolic reprogramming of immune cells, resulting in either protective or pathological outcomes. However, the specific roles of FGF21 in immune cells deserve further investigations.
Previous studies have shown that FGF21 can activate FGFR1, FGFR2, and FGFR3 receptors expressed in various tissues and cells.38,39 Combining public data and in vitro validation data, we found that mast cells primarily express FGFR1 receptor. Moreover, our data showed that the FGFR1 specific inhibitor can significantly diminish the effects of FGF21 on mast cells. Therefore, these conclusions suggest that inhibiting FGFR1 may be a new effective strategy for preventing asthma in obese individuals. Given the many benefits of the FGF21/FGFR1 pathway in metabolism, the adverse effects of its inhibitors on metabolism deserve our particular attention.40 Our study found that a 12-day injection of FGF21 antibody had no significant impact on blood glucose and serum cholesterol levels in mice. However, if anti-FGF21 or FGFR1 inhibitors are used for a long time, they might cause pathological impacts on the glucose and lipid metabolism. Therefore, in subsequent studies, we will modify anti-FGF21 or FGFR1 inhibitors in terms of drug delivery systems or administration methods to minimize their adverse metabolic effects, such as using novel nanomedicine delivery systems and nebulization methods for administration.
While the present study provides mechanistic insights into the role of FGF21 in obesity-associated airway hyperresponsiveness, several considerations warrant cautious interpretation. First, the relatively small human cohort and cross-sectional design limit statistical power and preclude causal inference between FGF21 levels and pulmonary function. Second, all animal experiments were conducted exclusively in male mice. Although this approach is commonly adopted in obesity-related research to reduce hormonal variability and to facilitate the development of a robust obese phenotype, future studies including female mice are warranted to determine potential sex-specific effects. We did not include a FGF21 administrated lean group; therefore, our conclusions are limited to obesity-associated AHR. Notably, Anti-FGF21 treatment in lean mice did not affect airway responsiveness, suggesting that FGF21 alone may be insufficient to drive AHR in the absence of obesity. Third, in vitro mast cell activation was induced using compound 48/80, a non-physiological stimulus, which may not fully recapitulate endogenous activation pathways in vivo. In addition, although pharmacological inhibition was employed to interrogate FGFR1 signaling, genetic validation using FGFR1-deficient models was not performed, and potential off-target effects of the inhibitors cannot be completely excluded. Importantly, while our data support a role for mast cell activation in FGF21-associated AHR, FGF21 may also modulate additional immune or structural cells within the obese airway microenvironment. Future studies using mast cell–deficient mouse models will be necessary to directly test the FGF21-driven AHR is mast cells-dependent. Finally, the duration of anti-FGF21 treatment was relatively limited, and therefore the long-term efficacy and safety of FGF21 blockade in obesity-associated airway hyperresponsiveness remain to be determined. Future studies incorporating larger cohorts, longitudinal designs, sex-balanced animal models, and genetic approaches will be necessary to further validate and extend our findings.
Obesity has emerged as one of the predominant public health predicaments globally, and numerous studies have clarified the influence of obesity on diverse diseases, such as cancer and cardiovascular disorders.41–43 Over the past few years, in the wake of the eruption of a variety of novel respiratory infectious diseases, the influence of obesity on respiratory system diseases has also attracted increasing attention.44–46 Obesity has been associated with reduced pulmonary function even in individuals without asthma, likely due to mechanical restriction of the thoracic cavity and altered airway physiology.47,48 Of course, other important non-mechanical pathological factors contributing to impaired pulmonary function in obesity remain to be fully elucidated. The unique immune characteristics of obese patients are evoked by the abnormal metabolism, and improving metabolic imbalance might constitute a crucial strategy for addressing immune-related disorders in obese patients.49–51 Consequently, ameliorating the immune function by restoring the metabolic balance of immune cells might potentially constitute a novel breakthrough in the treatment of obesity-associated asthma.
In summary, this study provides a novel insight into the role of FGF21 in the AHR triggered by obesity and delved into the mechanism. We found that anti-FGF21 or FGFR1 antagonists could diminish the activation of mast cells in the airway by suppressing the synthesis of cholesterol, thereby alleviating the AHR induced by obesity. Our research offers a new target and theoretical foundation for the treatment of obesity-associated asthma.
Data Sharing Statement
Data will be made available on reasonable request. For access, please contact the corresponding author.
Author Contributions
Lulu Ren: Writing – review & editing, Writing – original draft, Resources, Methodology, Investigation, Formal analysis, Data curation, Funding acquisition, Conceptualization. Lingling Xuan: Writing – review & editing, Writing – original draft, Resources, Methodology, Investigation, Formal analysis, data curation. Jie Zhang: Resources, data curation, Validation. Wen Zhang: Resources, Methodology, Software. Zhuoling An: Writing – review & editing, Supervision, Resources, Methodology, Conceptualization.
All authors took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was funded by the Beijing Natural Science Foundation (no.7262048).
Disclosure
Dr Jie Zhang reports a patent CN119139471A pending to Beijing Chaoyang Hospital Affiliated to Capital Medical University. The authors declare that they have no other competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Peters U, Dixon AE, Forno E. Obesity and asthma. J Allergy Clin Immunol. 2018;141(4):1169–15. doi:10.1016/j.jaci.2018.02.004
2. Arismendi E, Bantula M, Picado C. Obese asthma syndrome: much work to do. Arch Bronconeumol. 2023;59(8):473–475. doi:10.1016/j.arbres.2023.02.012
3. Hammad H, Lambrecht BN. The basic immunology of asthma. Cell. 2021;184(6):1469–1485. doi:10.1016/j.cell.2021.02.016
4. Bantula M, Tubita V, Roca-Ferrer J, et al. Weight loss and vitamin D improve hyporesponsiveness to corticosteroids in obese asthma. J Investig Allergol Clin Immunol. 2023;33(6):464–473. doi:10.18176/jiaci.0861
5. Geng L, Lam KSL, Xu A. The therapeutic potential of FGF21 in metabolic diseases: from bench to clinic. Nat Rev Endocrinol. 2020;16(11):654–667. doi:10.1038/s41574-020-0386-0
6. Tan H, Yue T, Chen Z, et al. Targeting FGF21 in cardiovascular and metabolic diseases: from mechanism to medicine. Int J Biol Sci. 2023;19(1):66–88. doi:10.7150/ijbs.73936
7. Cui X, Feng J, Wei T, et al. Pancreatic alpha cell glucagon-liver FGF21 axis regulates beta cell regeneration in a mouse model of type 2 diabetes. Diabetologia. 2023;66(3):535–550. doi:10.1007/s00125-022-05822-2
8. Cuevas-Ramos D, Mehta R, Aguilar-Salinas CA. Fibroblast growth factor 21 and browning of white adipose tissue. Front Physiol. 2019;10:37. doi:10.3389/fphys.2019.00037
9. Jin L, Yang R, Geng L, et al. Fibroblast growth factor-based pharmacotherapies for the treatment of obesity-related metabolic complications. Annu Rev Pharmacol Toxicol. 2023;63(1):359–382. doi:10.1146/annurev-pharmtox-032322-093904
10. Hu C, Qiao W, Li X, et al. Tumor-secreted FGF21 acts as an immune suppressor by rewiring cholesterol metabolism of CD8(+)T cells. Cell Metab. 2024;36(3):630–647e8. doi:10.1016/j.cmet.2024.01.005
11. Salomonsson M, Malinovschi A, Kalm‐Stephens P, et al. Circulating mast cell progenitors correlate with reduced lung function in allergic asthma. Clin Experim Allergy. 2019;49(6):874–882. doi:10.1111/cea.13388
12. Nygaard EB, Ørskov C, Almdal T, et al. Fasting decreases plasma FGF21 in obese subjects and the expression of FGF21 receptors in adipose tissue in both lean and obese subjects. J Endocrinol. 2018;239(1):73–80. doi:10.1530/JOE-18-0002
13. Ter Horst KW, Gilijamse PW, Demirkiran A, et al. The FGF21 response to fructose predicts metabolic health and persists after bariatric surgery in obese humans. Mol Metab. 2017;6(11):1493–1502. doi:10.1016/j.molmet.2017.08.014
14. Kim HY, DeKruyff RH, Umetsu DT. The many paths to asthma: phenotype shaped by innate and adaptive immunity. Nat Immunol. 2010;11(7):577–584. doi:10.1038/ni.1892
15. Zhang J, Zou Y, Chen L, et al. Regulatory T cells, a viable target against airway allergic inflammatory responses in asthma. Front Immunol. 2022;13:902318. doi:10.3389/fimmu.2022.902318
16. Shore SA. Obesity, airway hyperresponsiveness, and inflammation. J Appl Physiol. 2010;108(3):735–743. doi:10.1152/japplphysiol.00749.2009
17. Johnson-Weaver B, Choi HW, Abraham SN, et al. Mast cell activators as novel immune regulators. Curr Opin Pharmacol. 2018;41:89–95. doi:10.1016/j.coph.2018.05.004
18. Galli SJ, Gaudenzio N, Tsai M. Mast cells in inflammation and disease: recent progress and ongoing concerns. Annu Rev Immunol. 2020;38(1):49–77. doi:10.1146/annurev-immunol-071719-094903
19. Bradding P. Mechanisms of mast cell activation in severe asthma: beyond IgE. Am J Respir Crit Care Med. 2022;205(4):375–377. doi:10.1164/rccm.202110-2322ED
20. Pardanani A. Systemic mastocytosis in adults: 2021 Update on diagnosis, risk stratification and management. Am J Hematol. 2021;96(4):508–525. doi:10.1002/ajh.26118
21. Pejler G. The emerging role of mast cell proteases in asthma. Eur Respir J. 2019;54(4):1900685. doi:10.1183/13993003.00685-2019
22. Tiotiu A, Badi Y, Kermani NZ, et al. Association of differential mast cell activation with granulocytic inflammation in severe asthma. Am J Respir Crit Care Med. 2022;205(4):397–411. doi:10.1164/rccm.202102-0355OC
23. Sismanopoulos N, Delivanis D-A, Mavrommati D, et al. Do mast cells link obesity and asthma? Allergy. 2013;68(1):8–15. doi:10.1111/all.12043
24. Van Huisstede A, Rudolphus A, Castro Cabezas M, et al. Effect of bariatric surgery on asthma control, lung function and bronchial and systemic inflammation in morbidly obese subjects with asthma. Thorax. 2015;70(7):659–667. doi:10.1136/thoraxjnl-2014-206712
25. Chinese guidelines for the prevention and control of overweight and obesity in adults (2021 Revision). Beijing: Chinese Medical Association; 2021.
26. Global lnitiative for Asthma. Global strategy for asthma management and prevention. 2023. Available from: www.ginasthma.org.
27. Kim HY, Lee HJ, Chang Y-J, et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med. 2014;20(1):54–61. doi:10.1038/nm.3423
28. Miethe S, Karsonova A, Karaulov A, et al. Obesity and asthma. J Allergy Clin Immunol. 2020;146(4):685–693. doi:10.1016/j.jaci.2020.08.011
29. Wong M, Forno E, Celedon JC. Asthma interactions between obesity and other risk factors. Ann Allergy Asthma Immunol. 2022;129(3):301–306. doi:10.1016/j.anai.2022.04.029
30. Boonpiyathad T, Sözener ZC, Satitsuksanoa P, et al. Immunologic mechanisms in asthma. Semin Immunol. 2019;46:101333. doi:10.1016/j.smim.2019.101333
31. Björkander S, Klevebro S, Hernandez‐Pacheco N, et al. Obese asthma phenotypes display distinct plasma biomarker profiles. Clin Transl Allergy. 2023;13(3):e12238. doi:10.1002/clt2.12238
32. Kasaian MT, Lee J, Brennan A, et al. Proteomic analysis of serum and sputum analytes distinguishes controlled and poorly controlled asthmatics. Clin Experim Allergy. 2018;48(7):814–824. doi:10.1111/cea.13151
33. Liu Y, Li J, Wu Z, et al. Fibroblast growth factor 21 confers protection against asthma through inhibition of NLRP3 inflammasome activation. Inflammation. 2024;48(4):2720–2731. doi:10.1007/s10753-024-02222-z
34. Żelechowska P, Brzezińska‐Błaszczyk E, Wiktorska M, et al. Adipocytokines leptin and adiponectin function as mast cell activity modulators. Immunology. 2019;158(1):3–18. doi:10.1111/imm.13090
35. Periyalil HA, Wood LG, Scott HA, et al. Macrophage activation, age and sex effects of immunometabolism in obese asthma. Eur Respir J. 2015;45(2):388–395. doi:10.1183/09031936.00080514
36. Chu Y, Yang S, Chen X, et al. Fibroblast growth factor receptor signaling in metabolic dysfunction-associated fatty liver disease: pathogenesis and therapeutic targets. Pharmacol Ther. 2025;269:108844. doi:10.1016/j.pharmthera.2025.108844
37. Chen L, Gao M, Ong SB, Gong G. Functions of FGF21 and its role in cardiac hypertrophy. J Adv Res. 2025.
38. Yie J, Wang W, Deng L, et al. Understanding the physical interactions in the FGF21/FGFR/beta-Klotho complex: structural requirements and implications in FGF21 signaling. Chem Biol Drug Des. 2012;79(4):398–410. doi:10.1111/j.1747-0285.2012.01325.x
39. Kaess BM, Barnes TA, Stark K, et al. FGF21 signalling pathway and metabolic traits - genetic association analysis. Eur J Hum Genet. 2010;18(12):1344–1348. doi:10.1038/ejhg.2010.130
40. Wu YK, Ren Z-N, Zhu S-L, et al. Sulforaphane ameliorates non-alcoholic fatty liver disease in mice by promoting FGF21/FGFR1 signaling pathway. Acta Pharmacol Sin. 2022;43(6):1473–1483. doi:10.1038/s41401-021-00786-2
41. Singh A, Mayengbam SS, Yaduvanshi H, et al. Obesity programs macrophages to support cancer progression. Cancer Res. 2022;82(23):4303–4312. doi:10.1158/0008-5472.CAN-22-1257
42. Iacobellis G. Epicardial fat links obesity to cardiovascular diseases. Prog Cardiovasc Dis. 2023;78:27–33. doi:10.1016/j.pcad.2023.04.006
43. Quek J, Chan KE, Wong ZY, et al. Global prevalence of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in the overweight and obese population: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol. 2023;8(1):20–30. doi:10.1016/S2468-1253(22)00317-X
44. Robinson PD. Obesity and its impact on the respiratory system. Paediatr Respir Rev. 2014;15(3):219–226. doi:10.1016/j.prrv.2014.06.003
45. Lambert AA, Putcha N, Drummond MB, et al. Obesity is associated with increased morbidity in moderate to severe COPD. Chest. 2017;151(1):68–77. doi:10.1016/j.chest.2016.08.1432
46. De leeuw AJM, Oude Luttikhuis MAM, Wellen AC, et al. Obesity and its impact on COVID-19. J Mol Med. 2021;99(7):899–915. doi:10.1007/s00109-021-02072-4
47. Forno E, Han -Y-Y, Mullen J, et al. Overweight, obesity, and lung function in children and adults: a meta-analysis. J Allergy Clin Immunol Pract. 2018;6(2):570–581.e10. doi:10.1016/j.jaip.2017.07.010
48. Park Y, Kim J, Kim YS, et al. Longitudinal association between adiposity changes and lung function deterioration. Respir Res. 2023;24(1):44. doi:10.1186/s12931-023-02322-8
49. Vrieling F, Stienstra R. Obesity and dysregulated innate immune responses: impact of micronutrient deficiencies. Trends Immunol. 2023;44(3):217–230. doi:10.1016/j.it.2023.01.003
50. Almond M, Farne HA, Jackson MM, et al. Obesity dysregulates the pulmonary antiviral immune response. Nat Commun. 2023;14(1):6607. doi:10.1038/s41467-023-42432-x
51. Shaikh SR, MacIver NJ, Beck MA. Obesity dysregulates the immune response to influenza infection and vaccination through metabolic and inflammatory mechanisms. Annu Rev Nutr. 2022;42(1):67–89. doi:10.1146/annurev-nutr-062320-115937
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.