Back to Journals » Stem Cells and Cloning: Advances and Applications » Volume 8
Fetal exposure to a diabetic intrauterine environment resulted in a failure of cord blood endothelial progenitor cell adaptation against chronic hypoxia
Authors Dincer UD
Received 2 September 2014
Accepted for publication 25 September 2014
Published 18 December 2014 Volume 2015:8 Pages 1—14
DOI https://doi.org/10.2147/SCCAA.S73658
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Bernard Binetruy
U Deniz Dincer
Department of Basic and Clinical Pharmacology, School of Medicine, Bezmialem Vakif University (BAVU), Fatih/Istanbul, Turkey
Abstract: Gestational diabetes mellitus (GDM) has long-term health consequences, and fetal exposure to a diabetic intrauterine environment increases cardiovascular risk for her adult offspring. Some part of this could be related to their endothelial progenitor cells (EPCs). Understanding the vessel-forming ability of human umbilical cord blood (HUCB)-derived endothelial colony-forming cells (ECFCs) against pathological stress such as GDM response to hypoxia could generate new therapeutic strategies. This study aims to investigate the role of chronic hypoxia in EPCs functional and vessel-forming ability in GDM subjects. Each ECFC was expressed in endothelial and pro-angiogenic specific markers, namely endothelial nitric oxide synthase (eNOS), platelet (PECAM-1) endothelial cell adhesion molecule 1, vascular endothelial-cadherin CdH5 (Ca-dependent cell adhesion molecule), vascular endothelial growth factor A, (VEGFA) and insulin-like growth factor 1 (IGF1). Chronic hypoxia did not affect CdH5, but PECAM1 MRNA expressions were increased in control and GDM subjects. Control hypoxic and GDM normoxic VEGFA MRNA expressions and hypoxia-inducible factor 1-alpha (HIF1α) protein expressions were significantly increased in HUCB ECFCs. GDM resulted in most failure of HUCB ECFC adaptation and eNOS protein expressions against chronic hypoxia. Chronic hypoxia resulted in an overall decline in HUCB ECFCs' proliferative ability due to reduction of clonogenic capacity and diminished vessel formation. Furthermore, GDM also resulted in most failure of cord blood ECFC adaptation against chronic hypoxic environment.
Keywords: endothelial progenitor cells, gestational diabetes mellitus, chronic hypoxia, human cord blood
Introduction
Gestational diabetes mellitus (GDM) has long-term health consequences for her adult offspring.1,2 GDM demonstrates higher prevalence of obesity, insulin resistance, type 2 diabetes mellitus (DM) (during adolescence), development of metabolic syndrome (in early childhood), cardiovascular disease, and dyslipidemia in her offspring.1,2 Some of them could be related to decreased endothelial cells (ECs) and their progenitors.
Until 1997, it was believed that the differentiation of mesodermal cells into angioblasts and endothelial differentitation occur during embryonic development.3,4 When Asahara et al isolated endothelial progenitor cells (EPCs) from human peripheral blood, they started a predominant paradigm about new vessel formation in adult subjects.4 After they isolated hematopoietic progenitor cells from adults, they demonstrated that those progenitor cells can actually differentiate into ECs.5,6
The role of the endothelium is to receive, integrate, and respond against systemic and local environments.7 ECs and their progenitors, which form the vascular endothelium, play a key role in sensing and responding to different physiological and pathological stresses such as hypoxia.4,6 This can be described as a compensatory mechanism. During this period, a larger amount of EPCs are released and mobilized into the peripheral blood and cause vasodilatation and increased vessel formation. On the other hand, chronic hypoxia mostly exhibits apoptosis, with the cells unable to survive.
Adult peripheral blood and cord blood contain EPCs, which represent a minor subpopulation of blood mononuclear cells (MNCs). EPCs also contain different subpopulations such as colony-forming unit ECs (CFU-ECs) and endothelial colony-forming cells (ECFCs). They exhibit different functional properties.6,8 Decrease in CFU-EC colonies is oppositely correlated with hypercholesterolemia, hypertension, and type 2 DM.6,9 On the other hand, ECFCs are associated with angiogenesis and apoptosis.6,8 The number of ECFCs and their functional capacity determine de novo vessel formation and coordinate the physiological and pathological vessel-forming ability, including cell survival, proliferation, migration, and capillary-like structure formation in ECs. ECFCs directly contribute to the re-endothelization process, which plays an important role in the maintenance of the endothelium.10–12 Importantly, they have the capacity to repair the injured endothelium and the ability to promote vessel formation.11–13 Especially during acute hypoxia, a large number of EPCs mobilize into peripheral blood and they directly contribute to the revascularization process by a series of reactions.14–16
This project mainly designed to investigate the adaptation ability and/or deficiency of EPCs against long-term hypoxia and a fetal diabetic environment. It clarifies whether fetal exposure to a diabetic intrauterine environment would result in EPCs’ adaptation deficiency against a hypoxic environment. Collectively, it illuminates the role of hypoxia on the development of apoptosis, and gives beneficial information related to future therapeutic modalities.
Research design and methods
Clinical data, cord blood collection, and human blood samples
This work was undertaken according to the Declaration of Helsinki based on the primary ethical obligations of subjects, especially to collect human cord blood samples. Pregnant subjects from uncomplicated pregnancies were identified during outpatient clinics and/or delivery (previously approved by the Institutional Review Board (IRB), Department of Pediatrics, Indiana University School of Medicine).
Maternal data
GDM subjects demonstrated symptoms of diabetes during their pregnancy plus high blood glucose (BG) concentration (BG ≥11.1 mM [200 mg/dL], or fasting plasma glucose ≥7.0 mM [126 mg/dL], or 2-hour plasma glucose ≥11.1 mM [200 mg/dL] during an oral glucose tolerance test). HgbA1Cs 39 mmol/mol 117 mg/dL estimated average glucose [eAG]).
ECFC isolation
Around 50 mL blood was collected from the cord blood (from IRB-approved patients) of freshly delivered placentas by a research nurse or trained research personnel. Mononuclear cells (MNCs) were isolated as described previously.8,17 MNCs were washed three times with EBM-2 medium (Cambrex, Walkersville, MD, USA) supplemented with 10% fetal bovine serum (FBS; Hyclone, Logan, UT, USA), 2% penicillin/streptomycin, 5 mM dextrose, and 0.25 μg/mL amphotericin (Thermo Fisher Scientific, Waltham, MA, USA) (complete EBM-2 medium) and seeded onto six-well tissue culture plates precoated with collagen. After 24 hours, the adherent cells were washed and the medium changed daily until first ECFC colonies appeared between 5 days and 11 days, as previously described.8,17 The number of colonies was counted for each cord blood sample. The colonies were trypsinized and replated in a flask precoated with collagen. The cells were passaged upon reaching 80%–90% confluence. Early passage (<5) ECFC cell lines were used for the present study. For that purpose, five controls and five GDM human umbilical cord blood (HUCB) ECFCs were selected.
Long-term hypoxia
Early passage (<5) HUCB ECFCs obtained from GDM (n=5) and control (n=5) subjects were cultured with plates precoated with collagen under hypoxic condition (5% CO2/94% N2 and 1% O2 in a triple-gas incubator)7,18 as well as under normoxic condition17 for 7 days using EGM2 Medium with 10% FBS.
Characterization of HUCB ECFCs and real-time polymerase chain reaction
HUCB ECFCs were identified with previously described classical cell culture methods.8,17 Endothelial and pro-angiogenic-specific primers were designed to identify their endothelial origin and angiogenic capacity for real-time polymerase chain reaction (PCR):endothelial specific markers: platelet endothelial cell adhesion molecule (PECAM, CD31), vascular endothelial-cadherin (CdH5, Ca-dependent cell adhesion molecule) [CdH5], endothelial nitric oxide synthase (eNOS), pro-angiogenic specific markers: vascular endothelial growth factor A (VEGFA) and insulin-like growth factor 1 (IGF-1).
Total ribonucleic acid (RNA) was extracted according to manufacturer’s instructions (SV total RNA isolation system [Promega Corporation, Fitchburg, WI, USA]). At the end of the isolation, RNA samples were dissolved in nuclease-free water (pH 7.5). RNA samples were used as templates for the synthesis of first-strand cDNAs, as described previously.19,20 The total RNA quantity and quality were determined using the Experion™ semiautomated electrophoresis system (Bio-Rad Laboratories Inc., Hercules, CA, USA). In the Experion priming station, the microfluidic-based LabChip sample wells (RNA StdSens Chips, Experion, Bio-Rad Laboratories Inc.) were filled with a polymer-sieving matrix containing a fluorescent dye and 1 μL of denatured total RNA.21 The concentration of total RNA was determined by using the ratio of the sample RNA area to the Experion RNA ladder area. RNA samples with distinct 18S and 28S ribosomal RNA fragments in the electropherogram were used to assess the RNA quality.21 Selected good-quality RNA samples were used as templates for the synthesis of first-strand cDNAs.21
Real-time PCR reactions were performed for PECAM1, VE-cadherin, eNOS, VEGFA, IGF-1, and β-actin duplicated with a custom-designed SYBR green mix [2.4 μL of 25 mM MgCl2, 5 μL of 1:10,000 dilution SYBR Green I (Molecular Probes), and 5 μL of 1 nM fluorescein calibration dye (1 mM/L in DMSO, Bio-Rad Laboratories Inc.)] in 50 μL of total reaction using Taq DNA polymerase (Promega).19,20 Primers were designed using the Genomatix and the National Center for Biotechnology Information database. The primer sequences used for transcripts were as follows: PECAM1 (CD31) sense GAGATGAGGTATGGGCTGGA and antisense TGCTGCATCAAGAGTGGTTC (accession number AH002931); VE-cadherin (CDH5) sense CCTACCAGCCCAAAGTGTGT and antisense GAGATGACCACGGGTAGGAA (accession number NM_001795); and eNOS sense ACCCTCACCGCTACAACATC and antisense GCTCATTCTCCAGGTGCTTC (accession number NM_000603). VEGFA primers (sense CCAACTTCTGGGCTGTTCTC and antisense CCCCTCTCCTCTTCCTTCTC) were designed with the GENOMATIX program based on previously published accession numbers (BC011177, NM_001025366, NM_003376, NM_001025367, NM_001025368, NM_001025369, NM_001025370, NM_001033756). IGF-1 primers (sense CATGTCCTCCTCGCATCTCT and antisense ATACCCTGTGGGCTTGTTGA) were designed with the Genomatix program based on previously published accession numbers (NM_000618, NM_001111283, NM_001111284, NM_001111285). β-Actin primers (sense GGACTTCGAGCAAGAGATGG and antisense AGCACTGTGTTGGCGTACAG) were designed for integral control of the experiment (accession number NM_001101).
Amplification was carried out with the iCycler iQ multicolor real-time PCR detection system (Bio-Rad Laboratories Inc.) as follows: 30-seconds denaturation (95°C) followed by 45-seconds annealing and 1-minute extension (72°C), repeated for a total of 33 cycles. β-Actin was amplified in each set of PCR reactions and served as an internal reference during quantitation to correct for operator and/or experimental variations. The data were analyzed in duplicate using the modified 2−ΔΔCT equation:19–22

The mean threshold cycle (CT) values for target and internal control (β-actin) genes were determined in each sample. On the other hand, the formula was further modified for the control and GDM samples under hypoxic and normoxic conditions using the specific gene primers as well as internal control β-actin primers in each duplicated set (Table 1).
 | Table 1 The primer sequences used for real-time PCRr |
Western blot analysis eNOS, P-eNOS, and hypoxia-inducible factor 1-alpha (HIF1α)
For 7 days, hypoxic and normoxic HUCB ECFCs were obtained from GDM (n=4) and control (n=4). Proteins were isolated and homogenized with lysis buffer containing protease inhibitors (50 mmol/L Tris-HCl, 1% NP-40, 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L phenylmethylsulfonyl fluoride, 1 mg/mL aprotinin, 1 mg/mL leupeptin, and 1 mg/mL pepstatin). The supernatants were collected and used for analysis. Protein quantity and quality were determined using the Experion semiautomated electrophoresis system (Bio-Rad Laboratories Inc.). Basically, 4 μL of denatured protein for each sample was loaded into the Pro260 chip (Experion, Bio-Rad Laboratories Inc.). The Pro260 ladder used in the chip was based on protein gel electrophoresis, and contains nine proteins ranging from 10 kDa to 260 kDa as well as 1.2 kDa lower markers. Electropherograms were obtained for each group. Equivalent amounts of protein were separated by gel electrophoresis (10% Tris-HCl Criterion Precast Gel, Bio-Rad Laboratories Inc.).
Proteins were transferred onto nitrocellulose membrane (0.2 μm, Schleicher and Schuell, Germany) by semidry electroblotting (Trans-Blot SD; Bio-Rad Laboratories Inc.) at 15 V for 1 hour. The nitrocellulose membrane was soaked in 10 mM Tris-HCl containing 5% nonfat dry milk (Bio-Rad Laboratories Inc.) and 0.7% polyoxyethylene-sorbitan monolaurate (Tween 20) at pH 7.2 overnight at 4°C to block nonspecific sites. The membranes were incubated with P-eNOS (1:200 dilution; Santa Cruz Biotechnology, Inc, Santa Cruz, CA, USA), eNOS (1:1,000 dilution; Cell Signaling Technology, Inc., Beverly, MA, USA), and hypoxia-inducible factor 1-alpha (HIF 1α) (1:100 dilution; EMD Millipore, Billerica, MA, USA) antibodies. Western blot experiment was completed using same protocol as previously described.17
Senescence assay
Acidic β-galactosidase (β-gal) assay was conducted to investigate the functional capacity of EPCs against long-term hypoxia. Two thousand cells were seeded and triplicated into a well of six-well plates for each experimental group (human umbilical cord blood ECFCs obtained from control pregnancies and plated under normoxic condition [C-HUCB ECFC-N], human umbilical cord blood ECFCs obtained from control pregnancies and plated under hypoxic condition [C-HUCB ECFC-H], human umbilical cord blood ECFCs obtained from GDM subjects and plated under normoxic condition [GDM-HUCB ECFC-N], and human umbilical cord blood ECFCs obtained from GDM subjects and plated under hypoxic condition [GDM-HUCB ECFC-H]) and exposed to hypoxia (5% CO2/94% N2 and 1% O2) in a triple gas incubator. After 7 days, the triple gas hypoxia cells were fixed with 2% formaldehyde and 0.2% gluteraldehyde combination. Then cells were stained with β-galactosidase solution (X-gal + β-gal; 1 mg/mL final concentration) and incubated with normoxic cell culture overnight. The next day, the blue stained cells were counted under a microscope.
Increased stained cells with β-gal contributed to the reduction of clonogenic capacity of the hypoxic samples.
Matrigel assay
After exposure to chronic hypoxia (5% CO2/94% N2 and 1% O2 in a triple gas incubator for 7 days), ECFCs from each experimental group (C-HUCB ECFC-N, C-HUCB ECFC-H, GDM-HUCB ECFC-N, and GDM-HUCB ECFC-H) were seeded and triplicated at 6,000 cells/well in 96-well tissue culture plates coated with 35 μL of matrigel (BD Biosciences, San Jose, CA, USA). After 6–10 hours, the vessel-forming capacity (number of closed network units) was quantitated with visual microscopy (Zeiss Axiovert 2 inverted microscope with a 5× CP-Achromat/0.12 NA objective). The images were collected for each experimental group with a SPOT RT camera.
Data and statistical analysis
Differences among group values were evaluated by one-way analysis of variance followed by a Newman–Keuls test. Data are presented as means ± SE (Standard Error).
Results were considered significantly different at P<0.05.
Results
Characterization of HUCB ECFCs using cell culture and real-time PCR
The HUCB MNCs were plated onto collagen-coated plates in endothelial-specific media, as previously described.6,23 When nonadherent cells were discarded, cells appeared in cobblestone formation. They proved to be phenotypically indistinguishable from cultured ECs and possessed de novo vessel-forming ability.8,9,24 Flow cytometry demonstrated the endothelial origin of human hematopoietic progenitor cells.17,25 Cells were obtained from IRB-approved (under Declaration of Helsinki) ready HUCB ECFCs from the EPC Bank, The Herman B Wells Pediatric Research Center, Department of Pediatrics, Riley Pediatric Foundation and University Pediatric Association, Indiana University School of Medicine, Indianapolis, IN, USA, previously isolated and declared as ECFC (early passage <5).
Real-time PCR results demonstrated that each HUCB ECFC expressed eNOS, PECAM1, VE-cadherin, VEGFA, and IGF-1 messenger RNA (mRNA) (Figures 1–3). All control and GDM HUCB ECFCs expressed eNOS, PECAM1 (CD31), VE-cadherin, VEGFA, and IGF-1 mRNAs under normoxic as well as hypoxic conditions.
 | Figure 1 Real-time PCR reactions performed to characterize and compare HUCB ECFCs. |
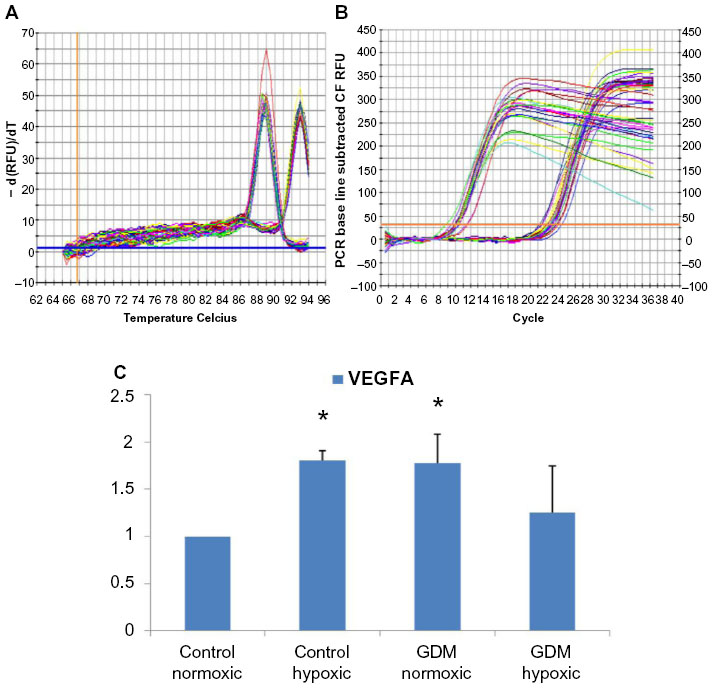 | Figure 2 Real-time PCR reactions were performed with endothelial growth factor A (VEGFA) mRNA for HUCB ECFCs obtained from gestational and control mothers under normoxic as well as chronic hypoxic condition. (A) The real time PCR melt curve, (B) The real time PCR AMP/Cycle, (C) Quantitative assessment of real time PCR. |
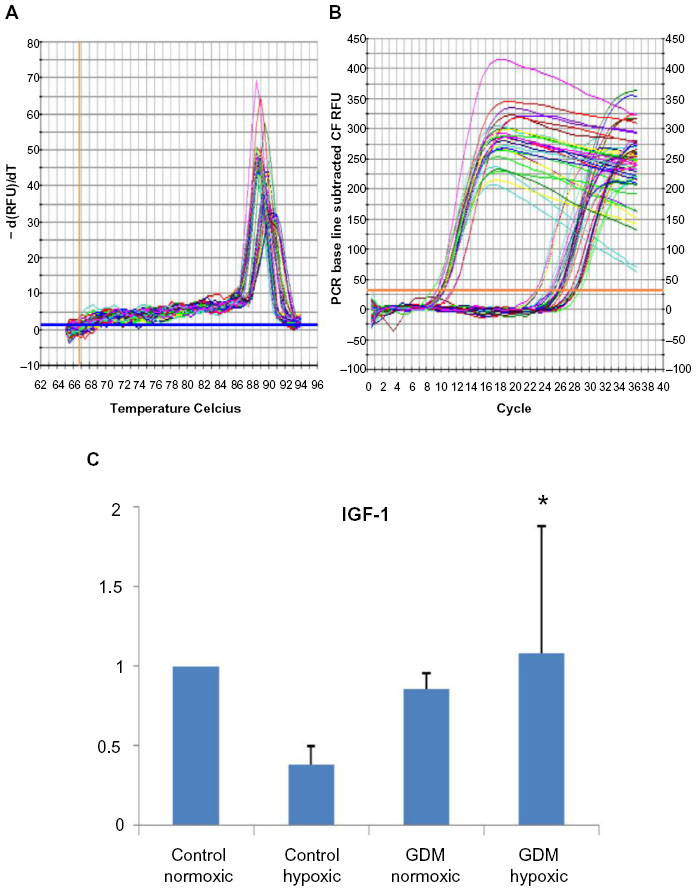 | Figure 3 Real-time PCR reactions were performed to identify the angiogenesis capability of HUCB ECFCs. (A) The real time PCR melt curve, (B) The real time PCR AMP/Cycle, (C) Quantitative assessment of real time PCR. |
HUCB ECFCs obtained from GDM and C subjects demonstrated increased PECAM1 (CD31) mRNA expressions with chronic hypoxic condition (Figure 1). PECAM1 (CD31) was only expressed in ECs. This indicates, first, that all samples have endothelial origin, and, second, type 1 transmembrane protein PECAM1 mRNA expressions were stimulated with chronic hypoxia. Using real-time PCR, significant differences were not detected in eNOS mRNA expression in HUCB ECFCs; on the other hand, it tended to increase in Control and GDM hypoxic samples. Significant differences were also not detected in VE-cadherin (CdH5) mRNA expressions in HUCB ECFCs with hypoxia. VE-cadherin is a Ca++-dependent cell adhesion molecule, and it indicated that chronic hypoxia did not affect Ca++-induced cell adhesion.
According to real-time PCR, C hypoxic and GDM normoxic HUCB ECFCs, VEGFA mRNA expressions were significantly increased compared to control HUCB ECFCs, (Figure 2). Chronic hypoxia triggered VEGFA mRNA expressions in HUCB ECFCs obtained from control pregnancies but did not affect the GDM samples. On the other hand, GDM itself resulted in increased VEGFA mRNA expressions. According to real-time PCR, significant differences were also detected in IGF-1 mRNA expression in HUCB ECFCs in GDM hypoxic samples when compared with control hypoxic HUCB ECFC mRNAs (Figure 3). Hypoxia alone did not cause any changes, but if long-term hypoxia was applied after diabetic intrauterine environment, IGF-1 mRNA expressions significantly increased.
Western blot analysis
eNOS, P-eNOS, and HIF 1α protein expressions were obtained from HUCB ECFCs with normoxic as well as hypoxic condition (Figure 4IA–C). eNOS protein expressions were significantly decreased in HUCB ECFCs in control as well as GDM subjects plated under hypoxic condition if compared with those plated under normoxic condition. Their quantitative assessments demonstrated that exposure to a gestational diabetic environment resulted in the most significant increase in eNOS protein expressions when plated under normoxic condition (Figure 4I and IIA). On the other hand, chronic hypoxia was significantly increased in P-eNOS protein expressions obtained from control and GDM HUCB-ECFCs when compared with their normoxic samples (Figure 4I and IIB). When combined with mRNA expression data, this means that the reduction of eNOS protein expressions against chronic hypoxic environment results from posttranslational modifications of those proteins.
 | Figure 4 (I) Representative gel pictures of (I) (A) eNOS, (B) p-eNOS, and (C) HIF 1α protein expressions; (II) represents quantitative assessments of eNOS (A), p-eNOS (B) and HIF1-alpha (C). Signal intensities were normalized with concomitant β-actin (n=4 C and n=4 GDM). HUCB ECFCs were exposed to 7 days of hypoxic condition. (IIA) eNOS. *GDM HUCB ECFC-N significantly different from C-HUCB ECFC-N, C-HUCB ECFC-H GDM HUCB ECFC-H; +C-HUCB ECFC-H and GDM-HUCB ECFC-H are significantly different from C-HUCB ECFC-N and GDM-HUCB ECFC-N (P<0.01); (IIB) p-eNOS; *C-HUCB ECFCH and GDM-HUCB ECFC-H are different from C-HUCB ECFC-N and GDM-HUCB ECFC-N (P<0.001); (IIC)-HIF 1α. *C-HUCB ECFC-H, GDM-HUCB ECFC-N, and GDM-HUCB ECFC-H are different from C-HUCB ECFC-N (P<0.001). +C-HUCB ECFC-H is different from CHUCB ECFC-N, GDM-HUCB ECFC-N, and GDM-HUCB ECFC-H (P<0.001). #GDM-HUCB ECFC-N is different from C-HUCB ECFC-N and GDM-HUCB ECFC-H (P<0.05). |
Chronic hypoxia resulted in increased HIF 1α protein expression in HUCB ECFCs obtained from control subjects, but decreased expression of GDM samples.
Gestational diabetic environment resulted in increased HIF 1α protein expression with regular condition when compared with control subjects. On the other hand, when chronic hypoxia was applied to GDM samples, the increase was not as high as in control subjects. The most significant increase was seen in control subjects (C-HUCB ECFC-N) that were plated under chronic hypoxia (Figure 4I and 4IIC).
Senescence assay
The photomicrographs in Figure 5 represent senescence assays, and when positively stained with senescence assay (SA) β-gal of HUCB ECFCs indicates the clonogenic capacity of those cells. HUCB ECFCs obtained from GDM and control subjects exhibited significant increase in senescence in vitro exposure to chronic hypoxia (5% CO2/94% N2 and 1% O2 in a triple gas incubator).7,18 It shows the reduction of clonogenic capacity against a chronic hypoxic environment (Figures 5 and 7A). The most significant enhanced senescence was observed in ECFCs obtained from GDM exposed to a chronic hypoxic condition (Figure 7A).
 | Figure 5 Photomicrographs representing senescence assay against chronic hypoxia. |
Matrigel assay
The photomicrographs represent matrigel assay, which represents the vessel-forming ability (Figure 6). A significant reduction in tube formation was observed in control and GDM HUCB ECFCs when cultured under chronic hypoxic conditions (Figure 7B). Their quantitative assessments demonstrated that exposure to long-term hypoxia exhibited impairment in HUCB ECFCs’ tube-forming ability. The most significant decrease in capillary tube formation was observed in ECFCs obtained from GDM mothers exposed to a chronic hypoxic condition.
 | Figure 6 Photomicrographs representing matrigel assay against chronic hypoxia, which represents the vessel-forming ability. |
 | Figure 7 (A) Senesence assay. Enhanced senescence of HUCB ECFCs (n=4 C and n=4 GDM) exposed to 7 days of hypoxic condition. SA β-gal assays demonstrated that C- and GDM-HUCB ECFCs exhibited significantly increase in senescence during in vitro exposure to chronic hypoxia which contributed to the reduction in clonogenic capacity and correleted with reduced proliferative ability. *C-HUCB ECFC-H and GDM-HUCB ECFCH are significantly different from C-HUCB ECFC-N and GDM-HUCB ECFC-H; ■C-HUCB ECFC-H significant difference from GDM-HUCB ECFC-H; ▲shows GDM-HUCB ECFCN significantly different from GDM-HUCB ECFC-H. (B) Matrigel assay. Quantitative assessment of tube formation was conducted by scoring the number of closed capillary tube networks per well. Y-axis represents the percentage of C-normoxic. Decreased number of closed capillary tube networks of HUCB ECFCs exposed to a chronic hypoxic condition correlates with reduced vessel-forming ability. *C-HUCB ECFC-N significant difference from C-HUCB ECFC-H, GDM-HUCB ECFC-N, and GDM-HUCB ECFC-H; ▲GDM-HUCB ECFC-N significant difference from GDM-HUCB ECFC-H; *C-HUCB ECFC-H significant difference from GDM-HUCB ECFC-H. |
Discussion
GDM is defined as any degree of glucose intolerance with onset or first recognition during pregnancy, and complicates around 2%–7% of all pregnancies in the US (more than 200,000 pregnancies annually).26 Major risk factors for developing GDM are family history of diabetes, increased pre-gravid body mass index, and a history of prior GDM.27–29 Women with multiple gestations, obesity, and bigger placental mass are at higher risk for GDM.26 On the other hand, GDM itself generates insulin resistance, and reduced sensitivity to insulin action. Furthermore, referer to GDM increases the release of human placental lactogen, growth, and corticotropin-releasing hormones from the placenta.28,30 The destiny of GDM is related to uncompensated insulin sensitivity or type II DM, whether only appearing during the gestational period or the condition persisting after pregnancy. The definition applies regardless of whether insulin or only diet modification is used during pregnancy for their treatment if the subject demonstrates symptoms of diabetes or high blood glucose (BG) concentration.27,29 GDM has serious long-term health consequences for her adult offspring, by inducing the development of metabolic syndrome, hypertension, type II DM, and premature cardiovascular disease.26,27,29 Development of metabolic syndrome in children with increasing age is correlated with maternal GDM and maternal obesity.1,2
EPCs are believed to differentiate into ECs, apart from angiogenesis.5,6 This belief has introduced new therapeutic strategies, and may be a potential therapeutic approach for a variety of vascular diseases.6 Previous studies have demonstrated that the number and migratory activity of EPCs are inversely correlated with the risk factors for cardiovascular disease and hypertension.31–34 The number and function of EPCs sometimes is accepted as a better predictor than the Framingham risk factor score to understand cardiovascular function.35 In adults, circulating EPCs directly participate in the formation of new blood vessels and angiogenesis.5,6 They contribute to the repair of injured endothelium and tissue repair.6,36,37 Identifying their role, whether development of angiogenesis or apoptosis against hypoxia, depends on their vessel-forming ability and clonogenic capacity. Furthermore, whether exposure of EPCs to the intrauterine diabetic environment results in apoptosis was investigated.
EPCs represent a minor subpopulation of blood MNCs. In vitro cell culture methods have been developed to identify and expand the EPC population from blood MNCs.6,10,38 ECFCs have proven to be phenotypically indistinguishable from cultured ECs and possess de novo vessel-forming ability.8,9,24 For that reason, ECFCs were selected in all experimental procedures. Gene-specific primers were also specifically designed to reveal their endothelial origin. All control and GDM HUCB ECFCs expressed eNOS, PECAM1 (CD31), VE-cadherin, VEGFA, and IGF-1 mRNAs. The “A type 1 trans-membrane protein” PECAM1 is expressed only in ECs. PECAM1 expressions were stimulated with chronic hypoxia in both control and GDM subjects.
Significant differences were not detected in eNOS mRNA expressions in hypoxic C and GDM HUCB ECFCs but they tended to increase. Chronic hypoxia resulted in significantly decreased eNOS and increased P-eNOS protein expressions obtained from C and GDM HUCB ECFCs. Their quantitative assessments demonstrated that exposure to a gestational diabetic environment resulted in the most significant decrease in eNOS and increase in P-eNOS protein expressions. If we combine these data, it can be concluded that the reduction of eNOS protein expression against chronic hypoxic environment results from posttranslational modification and phosphorylation of that protein. Diabetic intrauterine environment also negatively affects eNOS protein expression in HUCB ECFCs.
The Ca++-dependent cell adhesion molecule VE-cadherin (CdH5) is expressed in all cells, but chronic hypoxia did not affect Ca++-dependent cell adhesion molecule. This indicates that all that cells were able to express Ca++-regulated adhesion molecule in the hypoxic cell culture environment. Chronic hypoxia also demonstrated increased VEGFA mRNA expressions but not IGF-1. Even GDM-generated insulin resistance and the reduced sensitivity to insulin action of IGF-1 were not affected by hypoxia, though significant differences were detected in the GDM-hypoxic samples when compared to C-hypoxic samples. VEGFA expressions were also significantly increased in GDM-normoxic and C-hypoxic subjects but did not change in GDM-hypoxic samples. This indicates that GDM leads to the failure of ECFCs’ adaptation ability against a chronic hypoxic environment.
Vascular ECs exhibit adaptation to a hypoxic environment using complex compensatory mechanisms, and their progenitors play a key role in sensing and responding to insufficient O2 perfusion. Under hypoxia, ECs produce different metabolites such as nitric oxide, reactive oxygen species, and lactate.7 Hypoxia is described as a deficiency in oxygen and distinguishes three major cases according to the quantity of oxygen: moderate hypoxia (5% O2 triple gas vs normoxia 20% O2); severe hypoxia (1% O2 triple gas); and anoxia.7 The difference between physiological and pathological vessel formation under different hypoxic conditions for a certain amount of time determines the fate of the cells. The knowledge of how endothelial progenitors act under an existing hypoxic environment could lead to new therapeutic strategies. Furthermore, deficiency in EPCs’ adaptation ability against hypoxia obtained from GDM subjects demonstrates increased cardiovascular risk during adulthood. If we include their HbA1c level in GDM subjects, which were all normal and basically reflect a short duration of diabetes, we get another perspective. The question, “how did GDM affect their EPCs in a short period” also needs further investigation. The real difference between a clinical and an experimental setting is that we continuously treat the patients and their blood glucose (BG) is always under control except for some fluctuations. Behind that clinical reality, HUCB ECFCs demonstrated that GDM caused reduced proliferation capacity and vessel-forming ability under normoxic as well as hypoxic conditions.
HIF 1 activates the transcription of a large group of genes that encode many proteins in angiogenesis against response to hypoxia.39 It mainly regulates some genes that are involved in vasodilatation, increased vascular permeability, extracellular matrix remodeling, and EC proliferation. It plays an important role in developmental and pathological vessel formation under hypoxic conditions via O2-dependent and/or O2-independent pathways.7,16,40 It coordinates the proliferation and migration of ECs via increases in eNOS, VEGF, fibroblast growth factor (FGF), platelet-derived growth factor (PDGF), C-X-C chemokine receptor type 4 (CXCR-4), and stromal cell-derived factor 1 (SDF-1) expressions under hypoxic conditions.7,16,40 These physiological responses provide either increased O2 delivery or cell survival with hypoxia, which activates alternative metabolic pathways or reduces reactive oxygen species. On the other hand, HIF-1α (O2-regulated subunit) is a heterodimeric DNA-binding complex that continuously synthesizes and degrades during that period.15,16,40 Similarly, chronic hypoxia resulted in the highest increase in HIF 1α protein expression in HUCB ECFCs obtained from control subjects.
However, when GDM exists as a pathological condition, it demonstrated decreased expression of HIF 1α transcription factor compared to control subjects. It demonstrates that GDM results in lower vessel-forming ability compared to controls in response to hypoxia.
HUCB ECFCs obtained from GDM and control subjects demonstrated reduced proliferation capacity and vessel-forming ability against chronic hypoxia. They exhibited significantly increased senescence in vitro when exposed to a chronic hypoxia, which contributes to the reduction of their clonogenic capacity. The most significant decrease in capillary tube formation was observed in ECFCs obtained from GDM subjects when exposed to a chronic hypoxic condition. These data suggest that chronic hypoxia results in an overall decline in the proliferative capacity of ECFCs due to premature senescence and diminished vessel-forming ability, as seen from control and GDM subjects. If we combine these data with increased HIF 1α protein expressions, it mainly indicates that further degradation of that protein or chronic hypoxia-induced HIF 1α pathway does not affect the vessel-forming ability. The underlying mechanisms need further investigations.
Acknowledgments
The author wishes to thank Bezmialem Vakif University, Istanbul, Turkey; The Trustees of Indiana University; Herman B Wells Center for Pediatric research, Department of Pediatrics, Indiana University School of Medicine; the Riley Pediatric Foundation; and the University Pediatric Association, Indiana University School of Medicine, for financial support. Special thanks to Ethel Derr-Yellin and Dr Laura Haneline. A preliminary report of this manuscript was presented at the 4th International Meeting of Angiogenesis (Page 89/Poster 25), VU University Medical Center, March 2–4, 2011, Amsterdam, the Netherlands.
Disclosure
The author reports no conflicts of interest in this work.
References
Vohr BR, Boney CM. Gestational diabetes: the forerunner for the development of maternal and childhood obesity and metabolic syndrome? J Matern Fetal Neonatal Med. 2008;21(3):149–157. | |
Yogev Y, Visser GH. Obesity, gestational diabetes and pregnancy outcome. Semin Fetal Neonatal Med. 2009;14(2):77–84. | |
Heil M, Eitenmuller I, Schmitz-Rixen T, Schaper W. Arteriogenesis versus angiogenesis: similarities and differences. J Cell Mol Med. 2006;10(1):45–55. | |
Asahara T, Murohara T, Sullivan A, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275(5302):964–967. | |
Risau W. Mechanisms of angiogenesis. Nature. 1997;386(6626):671–674. | |
Prater DN, Case J, Ingram DA, Yoder MC. Working hypothesis to redefine endothelial progenitor cells. Leukemia. 2007;21(6):1141–1149. | |
Paternotte E, Gaucher C, Labrude P, Stoltz JF, Menu P. Review: behaviour of endothelial cells faced with hypoxia. Biomed Mater Eng. 2008;18(4–5):295–299. | |
Ingram DA, Mead LE, Tanaka H, et al. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood. 2004;104(9):2752–2760. | |
Ingram DA, Mead LE, Moore DB, Woodard W, Fenoglio A, Yoder MC. Vessel wall-derived endothelial cells rapidly proliferate because they contain a complete hierarchy of endothelial progenitor cells. Blood. 2005;105(7):2783–2786. | |
Hill JM, Zalos G, Halcox JP, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348(7):593–600. | |
Masuda H, Kalka C, Asahara T. Endothelial progenitor cells for regeneration. Hum Cell. 2000;13(4):153–160. | |
Masuda H, Asahara T. Post-natal endothelial progenitor cells for neovascularization in tissue regeneration. Cardiovasc Res. 2003;58(2):390–398. | |
Asahara T, Masuda H, Takahashi T, et al. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 1999;85(3):221–228. | |
Larsen M, Hog A, Lund EL, Kristjansen PE. Interactions between HIF-1 and Jab1: balancing apoptosis and adaptation. Outline of a working hypothesis. Adv Exp Med Biol. 2005;566:20311. | |
Semenza GL. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE. 2007;2007(407):cm8. | |
Weidemann A, Johnson RS. Biology of HIF-1alpha. Cell Death Differ. 2008;15(4):621–627. | |
Ingram DA, Lien IZ, Mead LE, et al. In vitro hyperglycemia or a diabetic intrauterine environment reduces neonatal endothelial colony-forming cell numbers and function. Diabetes. 2008;57(3):724–731. | |
Hwang HS, Maeng YS, Kim YH, Kwon YG, Park YW, Kim IK. Nestin expression during differentiation of fetal endothelial progenitor cells and hypoxic culture of human umbilical vein endothelial cells. Acta Obstet Gynecol Scand. 2008;87(6):643–651. | |
Dincer UD, Araiza AG, Knudson JD, Molina PE, Tune JD. Sensitization of coronary alpha-adrenoceptor vasoconstriction in the prediabetic metabolic syndrome. Microcirculation. 2006;13(7):587–595. | |
Dincer UD, Araiza A, Knudson JD, Shao CH, Bidasee KR, Tune JD. Dysfunction of cardiac ryanodine receptors in the metabolic syndrome. J Mol Cell Cardiol. 2006;41(1):108–114. | |
Dincer UD. Cardiac beta-adrenoceptor expression is markedly depressed in Ossabaw swine model of cardiometabolic risk. Int J Gen Med. 2011;4: 493–499. | |
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. | |
Lin Y, Weisdorf DJ, Solovey A, Hebbel RP. Origins of circulating endothelial cells and endothelial outgrowth from blood. J Clin Invest. 2000;105(1):71–77. | |
Yoder MC, Mead LE, Prater D, et al. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007;109(5):1801–1809. | |
Duda DG, Cohen KS, Scadden DT, Jain RK. A protocol for phenotypic detection and enumeration of circulating endothelial cells and circulating progenitor cells in human blood. Nat Protoc. 2007;2(4):805–810. | |
Cheng YW, Caughey AB. Gestational diabetes: diagnosis and management. J Perinatol. 2008;28(10):657–664. | |
Buchanan TA, Unterman TG, Metzger BE. The medical management of diabetes in pregnancy. Clin Perinatol. 1985;12(3):625–650. | |
Buchanan TA, Metzger BE, Freinkel N, Bergman RN. Insulin sensitivity and B-cell responsiveness to glucose during late pregnancy in lean and moderately obese women with normal glucose tolerance or mild gestational diabetes. Am J Obstet Gynecol. 1990;162(4):1008–1014. | |
Metzger BE, Buchanan TA, Coustan DR, et al. Summary and recommendations of the Fifth International Workshop-Conference on Gestational Diabetes Mellitus. Diabetes Care. 2007;30(Suppl 2):S251–S260. | |
Ryan EA. Hormones and insulin resistance during pregnancy. Lancet. 2003;362(9398):1777–1778. | |
Liew A, Barry F, O’Brien T. Endothelial progenitor cells: diagnostic and therapeutic considerations. Bioessays. 2006;28(3):261–270. | |
Gulati R, Jevremovic D, Peterson TE, et al. Diverse origin and function of cells with endothelial phenotype obtained from adult human blood. Circ Res. 2003;93(11):1023–1025. | |
Hill JM, Zalos G, Halcox JP, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348(7):593–600. | |
Rehman J, Li J, Parvathaneni L, et al. Exercise acutely increases circulating endothelial progenitor cells and monocyte-/macrophage-derived angiogenic cells. J Am Coll Cardiol. 2004;43(12):2314–2318. | |
Hill JM, Zalos G, Halcox JP, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348(7):593–600. | |
Iwami Y, Masuda H, Asahara T. Endothelial progenitor cells: past, state of the art, and future. J Cell Mol Med. 2004;8(4):488–497. | |
Timmermans F, Plum J, Yoder MC, Ingram DA, Vandekerckhove B, Case J. Endothelial progenitor cells: identity defined? J Cell Mol Med. 2009;13(1):87–102. | |
Ito H, Rovira II, Bloom ML, et al. Endothelial progenitor cells as putative targets for angiostatin. Cancer Res. 1999;59(23):5875–5877. | |
Pereira T, Zheng X, Poellinger L. Degradation of the hypoxia-inducible factor 1alpha: where does it happen? Cell Cycle. 2006;5(23):2720–2722. | |
Nagaya N, Mori H, Murakami S, Kangawa K, Kitamura S. Adrenomedullin: angiogenesis and gene therapy. Am J Physiol Regul Integr Comp Physiol. 2005;288(6):R1432–R1437. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.