Back to Journals » Journal of Inflammation Research » Volume 14
Ferroptosis is Involved in Hyperoxic Lung Injury in Neonatal Rats
Authors Jia D ![]() , Zheng J, Zhou Y
, Zheng J, Zhou Y ![]() , Jia J, Ye X
, Jia J, Ye X ![]() , Zhou B, Chen X
, Zhou B, Chen X ![]() , Mo Y, Wang J
, Mo Y, Wang J ![]()
Received 19 August 2021
Accepted for publication 11 October 2021
Published 18 October 2021 Volume 2021:14 Pages 5393—5401
DOI https://doi.org/10.2147/JIR.S335061
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Danyun Jia,1 Jinyu Zheng,2 Yiyang Zhou,2 Jinqiu Jia,3 Xiaoxiao Ye,4 Bingbing Zhou,4 Xingxing Chen,4 Yunchang Mo,1 Junlu Wang1
1Department of Anesthesiology, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, 325000, Zhejiang, People’s Republic of China; 2Department of Clinical Medicine, Wenzhou Medical University, Wenzhou, 325000, Zhejiang, People’s Republic of China; 3Department of Pediatric, Taizhou Women and Children’s Hospital of Wenzhou Medical University, Taizhou, 317599, Zhejiang, People’s Republic of China; 4Department of Nursing, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, 325000, Zhejiang, People’s Republic of China
Correspondence: Yunchang Mo; Junlu Wang Email [email protected]; [email protected]
Purpose: To evaluate whether ferroptosis is involved in hyperoxic acute lung injury (HALI) and its mechanisms through the HALI model.
Methods: HE staining was used to assess lung injury pathology after the establishment of neonatal rat HALI model. ELISA was used to detect ROS, GPX4, and GSH expression. Prussian blue staining and Western Blot were used to detect iron deposition and the expression of ferroptosis-related proteins, respectively.
Results: The HALI group showed pathological changes with larger and fewer alveoli and thicker alveolar septa after HE staining. Prussian blue staining detected significant iron deposition in the lung tissue of the HALI group. GPX4, GSH, GSS, and SLC7A11 expressions were significantly decreased in the HALI group than in the normal control group. In contrast, ROS, TFRC, FHC, and FLC expressions showed opposite results (p< 0.05).
Conclusion: Ferroptosis may be involved in the pathological process of hyperoxic lung injury in neonatal rats.
Keywords: ferroptosis, hyperoxic lung injury, neonatal rats
Introduction
Oxygen therapy can rescue and treat neonates with hypoxemia or acute respiratory failure by preventing hypoxia and ensuring normal oxygen supply to tissues and organs. However, prolonged exposure to high concentrations of oxygen can lead to different degrees of damage to the newborn,1,2 with the lungs being the most common organ damaged by hyperoxia,3 causing bronchopulmonary dysplasia (BPD) in severe cases.4 Besides, there is no exact and effective prevention and treatment for lung injury caused by hyperoxia. Therefore, the pathogenesis of hyperoxia-induced lung injury should be assessed to improve the prognosis and find effective and practical treatment methods.
An imbalance in the oxidative-antioxidant system causes hyperoxic lung injury. The excessive release of reactive oxygen species (ROS) and their derivatives can lead to the inactivation of various cellular enzymes, DNA damage and lipid membrane peroxidation. These impair the structural function of alveolar epithelial cells causing acute pulmonary edema and acute pulmonary atelectasis.5,6 However, the exact mechanism of damage has not been fully elucidated. Ferroptosis is a programmed cell death caused by excess accumulation of iron-dependent lipid peroxidation (LPO). Ferroptosis inducing factors can directly or indirectly affect glutathione peroxidase through different pathways, decreasing intracellular antioxidant capacity and lipid ROS accumulation, thus leading to oxidative cell death. Among them, dysregulation of iron metabolism, accumulation of lipid peroxides and failure of Glutathione peroxidase 4 (GPX4) antioxidant system are the main triggering mechanisms of ferroptosis.7 Abnormal expression of a series of iron regulatory proteins such as transferrin and ferritin will lead to an imbalance of intracellular iron homeostasis, and the accumulation of redox-active Fe2+ generates large amounts of highly toxic ROS through the redox cycle of Fenton reaction and Haber-Weiss reaction,8 ultimately triggering ferroptosis and lipid oxidative damage. GPX4, glutathione (GSH) and SLC7A11 and other proteins, on the other hand, play an important role in the antioxidant process of the body and are key proteins in the occurrence of ferroptosis.7
Existing studies suggest that ferroptosis is associated with lung cancer, pulmonary fibrosis, chronic obstructive pulmonary disease, pulmonary ischemia-reperfusion injury, and acute lung injury (ALI). Besides, increased lipid ROS and abnormal levels of ferroptosis-associated proteins have been found in the above diseases. However, ferroptosis inhibitors can improve symptoms.9–13 However, there are very few studies on ferroptosis and hyperoxic lung injury. Robbins et al found that defects in the GSH system would exacerbate hyperoxic lung injury.14 Mouse models of lung injury with deletion of the ROS repressor gene exhibited increased inflammation with tissue iron accumulation.15 However, these studies still cannot show a direct link between ferroptosis and hyperoxic lung injury. Therefore, the present study was conducted to preliminarily investigate whether ferroptosis is involved in the pathological process of HALI and its related mechanisms through a neonatal rat HALI model.
Materials and Methods
HALI Model Construction and Grouping
All animal care and experiments were performed following the guidelines for the care and use of laboratory animals. The Laboratory Animal Ethics Committee of Wenzhou Medical University approved the experiments. All Sprague-Dawley (SD) rats (200–250 g) were obtained from Charles River (Beijing, China) and housed in the Laboratory Animal Center under pathogen-free conditions. Adult rats were freely mated to produce offspring for subsequent studies. The experiment was divided into a hyperoxia model group and an air control group. Neonatal rats (along the dam) in the hyperoxia model group were put in an airtight rearing tank immediately after birth, and oxygen was rapidly introduced into the tank. The oxygen flow rate was changed to 1 L/min when the oxygen concentration in the tank reached 85%, and the exposure was continued for seven days to simulate hyperoxic lung injury.16,17 The incubator was left with ventilation holes to keep the pressure inside the incubator consistent with the outside world, so it can be assumed that this is an atmospheric pressure hyperoxia-induced lung injury. CO2 was adsorbed using calcium hydroxide, while ammonia and other toxic gases were adsorbed using activated carbon. The temperature was controlled via a ventilation system, and the humidity was controlled through a dehumidification box (50–70%) (12 h/12 h light/dark cycle). The box was opened for 30 min every day during cleaning, bedding change, feeding, and when the rats needed to be operated on. The air control group was placed in a normal feeding box and fed normally. The mother rats of the hyperoxia model group and the air control group were exchanged every 24 h to avoid oxygen toxicity that affects the feeding of the neonatal rats. The body weight and survival of the infant rats were observed and recorded daily. No rat developed seizures during hyperbaric oxygen exposure.
HE Staining
The rats were sacrificed. After exposure of the lungs and heart, the left atrium was dissected to allow adequate evacuation of the perfusate, and phosphate buffered saline was immediately injected into the right ventricle to remove blood from the pulmonary circulation. The lungs were then perfused with 4% paraformaldehyde through the right ventricle at a constant pressure of 20 cm H2O for 20 min.18 Then the left lung was removed, fixed in 4% PFA at 4 °C overnight, dehydrated, paraffin-embedded, and patched after cutting into 4 μm thick coronal sections. The sections were stained using hematoxylin, differentiation solution, and eosin reagent (Solarbio, China). The sections were later permeabilized with xylene and sealed with neutral resin. Light microscopy (Olympus, Tokyo, Japan) was then used to assess histopathological changes. According to previous studies,19–22 we selected images with scale bars = 100 μm to measure radial alveolar count (RAC), mean linear intercept (MLI) and mean alveolar diameter (MAD) to quantify inter-alveolar distance and size. MAD indicates mean alveolar diameter. MLI indicates the ratio of alveolar volume to surface area, and was determined by drawing 5 lines in each field of view and dividing the length of each line by the number of alveoli passing through that line. The RAC was obtained by drawing a line from the center of the terminal fine bronchus to the nearest connective tissue septa and counting the number of alveoli on this line. All images were evaluated by researchers unaware of the experimental group and repeated 6 times.
Prussian Blue Staining
The deposition of iron-containing heme in lung tissue was detected by using Prussian blue staining kit (Solarbio, China). Paraffin sections with a thickness of 4 µm were prepared, routinely dehydrated and transparent, and then stained with Perls staining solution for 30 min. The sections were washed three times with distilled water; the background was lightly stained with Eosin staining solution for 15–30 s, and rinsed with tap water for 2–3 s. The sections were dehydrated with anhydrous ethanol and transparent with xylene, sealed with neutral gum, and placed under a light microscope for observation. The mean percentage positive area was used to quantify the difference between the two groups of Prussian blue results.23 This is a commonly used method of immunohistochemical analysis that counts the percentage of positive staining area over the measured area and finally takes the mean value as the percentage value of the mean positive staining area for this case section, thus being less affected by some human-induced interference. Color threshold quantification was performed on six non-overlapping areas/groups by image J 1.53 software (NIH, USA). All images were evaluated by researchers who were unaware of the experimental group.
ELISA
The right and left lung tissues were quickly removed after sacrificing the pups. The specimens were added to an amount of PBS (pH 7.4) and homogenized using a homogenizer. The supernatant was collected by centrifugation at 12,000 rpm for 10 min at 4°C. Protein concentration was assessed using a BCA protein assay kit (Proteintech, Wuhan, China). Protein was diluted a certain number of times for ELISA assay. The ELISA kit (Affandi, China) was operated according to its instructions. Antibodies are used to coat the ELISA plate. The antigen in the sample or standard will bind to the coating antibody during the experiment and the free components are washed away. Biotinylated antibodies and horseradish peroxidase-labeled affinities are added sequentially. The antibody binds to the antigen bound to the encapsulated antibody and the biotin specifically binds to the affinity to form an immune complex, and the free components are washed away. A chromogenic substrate (TMB) is added, and TMB appears blue catalyzed by horseradish peroxidase and turns yellow after the addition of termination solution. The OD value was measured at 450 nm with an enzyme marker, and there was a positive ratio between the antigen concentration and OD450 value, and the contents of ROS, GPX4 and GSH in neonatal rat lung tissue were calculated by drawing a standard curve.
Western Blot
Rats in both groups were deeply anesthetized with isoflurane after seven days of modeling and sacrificed. Lung tissues were quickly removed, and RIPA lysis buffer (Beyotime, China) and protease inhibitors were added. The supernatant was collected after centrifugation at 12,000 g, 4°C for 10 min. An enhanced BCA protein assay kit (Proteintech, Wuhan, China) was used to determine protein concentration. Protein samples were separated on 8–12% SDS-PAGE gels and transferred to polyvinylidene fluoride PVDF membranes (Millipore, USA). The membranes were blocked with 5% skim milk at room temperature for 2 h, then incubated with primary antibodies at 4°C overnight. The primary antibodies used were: GPX4 (1:1000, A11243, ABclonal), Glutathione Synthetase (GSS, 1:1000, A14535, ABclonal), SLC7A11 (1:1000, A2413, ABclonal), Transferrin receptor (TFRC, 1:1000, A5865, ABclonal), ferritin heavy chain (FHC, 1: 1000, A19544, ABclonal), ferritin light chain (FLC, 1:1000, A11241, ABclonal). The membranes were washed thrice with Tris-buffered saline and Tween 20 (TBST), then incubated with appropriate secondary antibodies (goat anti-mouse, 1:10,000, BS12478, Bioworld; goat anti-rabbit, 1:30,000, ab6721) at room temperature for 1 h. The ChemiDoc-XRS+ imaging system (Bio-Rad, USA) was used to detect these strips. The grayscale values of each band were analyzed using Image Lab 6.1 software (Bio-Rad, USA) via image J 1.53 software (NIH, USA). All experiments were repeated at least thrice.
Statistical Analysis
All data were expressed as mean ± SD. Comparisons between the two groups were made using the independent samples t-test. Data were statistically analyzed using SPSS 26.0 software (IBM Inc., Chicago, IL, USA) and GraphPad Prism 9.1.1 software (GraphPad Software Inc., San Diego, CA, USA). P<0.05 was considered a statistically significant difference.
Results
Results of Modeling Treatment
The effect of hyperoxia on the degree of lung injury in neonatal rats was observed by HE staining (Figure 1). After 7 days of hyperoxia, the volume of alveolar cavity increased, while the number of alveoli decreased and the alveolar septa thickened. The MLI and MAD per unit area increased, while the RAC decreased. Lung histopathological changes were significant, indicating that sustained hyperoxia successfully induced acute lung injury.
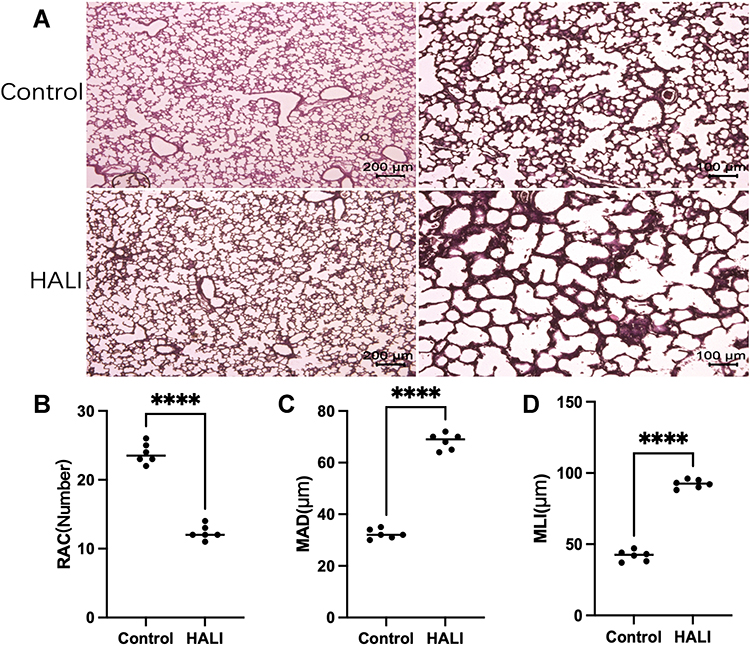 |
Figure 1 (A) In HE staining, the lung tissue structure in the HALI group showed a decrease in the number of alveoli, an increase in the volume of the alveolar cavity, and a thickening of the alveolar septa compared with the control. Scale bars = 200 μm, 100 μm. (B) Semi-quantitative pathology score of RAC in lung tissues. (C) Semi-quantitative pathology score of MAD in lung tissues. (D) Semi-quantitative pathology score of MLI in lung tissues. The values are the mean ± SD; n = 6 nonoverlapping fields/group; ****P < 0.0001. |
Altered Lung Tissue Cells
The nuclei and other tissues were red after Prussian blue staining, while the sites of iron-containing heme deposition were blue. The iron deposition was significantly more in the lung tissue of the HALI group than that of the control group (Figure 2). Moreover, there was significant blue iron deposition in the lung tissue of the HALI group (arrow). The above results suggest that iron metabolism disorders and iron deposition occur in HALI lung tissues.
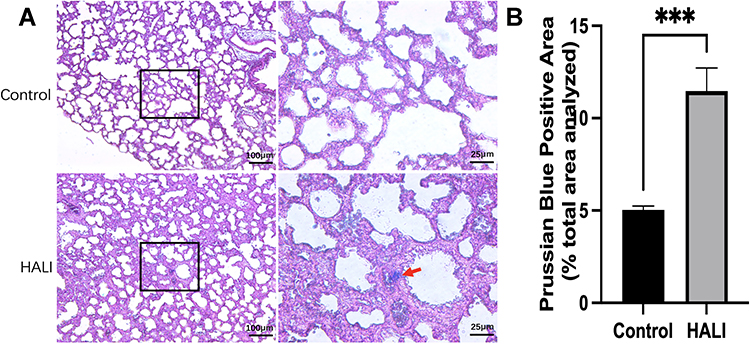 |
Figure 2 (A) Prussian blue staining in alveolar tissue. The columns on the right show magnified images of the black boxes in the left column. Scale bars = 25 μm, 100 μm. (B) Prussian Blue Positive Area (% total area analyzed). Iron deposition in lung tissue was significantly higher in the HALI group than in the control group (***P < 0.001). The values are the mean ± SD; n = 6 nonoverlapping fields/group. |
Cellular Oxidative Stress
Ferroptosis is closely related to oxidative stress. Herein, ELISA was used to detect the key protein GPX4 and ROS in lung tissue (Figure 3). GPX4 protein significantly decreased after seven days of hyperoxia compared with the normal group (P<0.01). WB results (Figure 4) also demonstrated that GPX4 expression decreased after hyperoxia (P<0.01). The hyperoxia also significantly decreased GSH content in lung tissue (P<0.0001), while it significantly increased ROS (P<0.0001), indicating that oxidative stress occurred in lung tissue. GSS is a key enzyme for GSH synthesis. WB (Figure 4) showed that GSS expression was significantly decreased after hyperoxia (P<0.01). System Xc- is also a key protein in the GSH synthesis pathway and an upstream ferroptosis regulator. It consists of SLC7A11 and SLC3A2 subunits.24 WB results (Figure 4) showed that SLC7A11 was significantly down-regulated after hyperoxia (P<0.01). These results suggest that the cellular capacity for antioxidants is significantly diminished due to the blocked synthesis and decreased GSH expression.
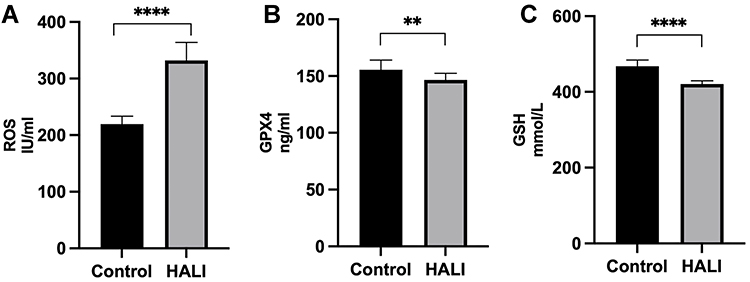 |
Figure 3 ELISA detection of ROS (A), GPX4 (B) and GSH (C) expressions in rats. ****P < 0.0001, **P < 0.01 and ****P < 0.0001 vs the control group. The values represent the mean ± SD, n = 10. |
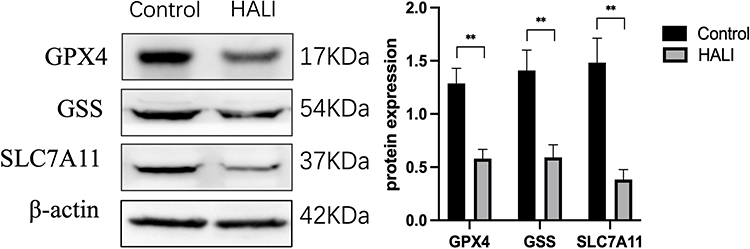 |
Figure 4 Western blotting analyzed the expression of GPX4, GSS, SLC7A11, which is ferroptosis key protein. **P < 0.01 vs the control group. Data were shown as mean ± SD, n = 3. |
Iron Metabolism-Related Proteins
The regulation of ferritin and iron metabolism is essential in the ferroptosis mechanism.25 TFRC is required for the uptake of the transferrin-iron complex into cells. WB showed (Figure 5) that TFRC expression was significantly increased in the HALI group (P<0.05). Ferritin is an essential protein used to store iron ions in cells. It comprises two subunits, FHC and FLC. This study (Figure 5) showed that FHC and FLC expressions were significantly increased after hyperoxia treatment (P<0.05), indicating a substantial increase in intracellular free ferric ions.
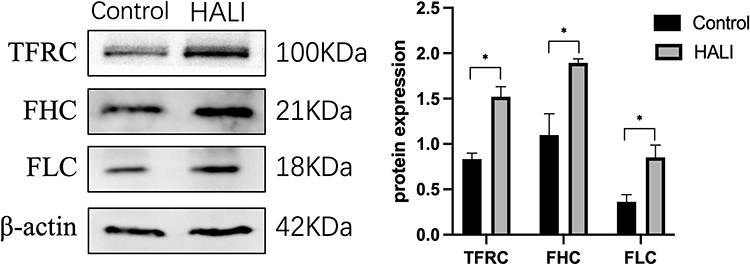 |
Figure 5 Western blotting analyzed the expression of TFRC, FHC, FLC, which is involved in iron transport storage. *P < 0.05 vs the control group. Data were shown as mean ± SD, n = 3. |
Discussion
Existing studies have confirmed that continuous exposure to high levels of oxygen will cause damage to the lungs.26 In this study, neonatal rats were exposed to 85% oxygen concentration for seven days to simulate the pathological process of HALI.27–29 HE staining showed a decrease in the number of alveoli, an increase in the volume of the alveolar cavity, and a thickening of the alveolar septa, suggesting damage to lung tissue from continuous high oxygen exposure.
Some studies have confirmed the presence of ferroptosis in various adult murine models of ALI. A study showed that ferroptosis-like pathological changes occur in lung cells through a mouse model of acute lung injury induced via tail vein injection with oleic acid. Besides, the study identified significant iron overload, reduced GSH content, and significantly lower ferritin expression levels in lung tissue.10 Liu et al also showed that ferroptosis promotes LPS-induced acute lung injury. They also indicated that ferroptosis inhibitor ferrostatin-1 (Fer-1) could effectively alleviate LPS-induced acute lung injury and inflammatory response.30 Li et al and Dong et al have identified the involvement of ferroptosis in an ischemia-reperfusion-induced ALI mouse model and attempted to alleviate ALI by inhibiting ferroptosis.11,31 In mice, ferroptosis also promotes acute radiation-induced lung injury (RILI), probably via radiation-induced ROS.32 In the present study, we found ferroptosis-like changes in the lung tissues of rats in the HALI group, including iron deposition, elevated ROS expression, and decreased GSH expression.
Ferroptosis is characterized by impaired GSH-GPX4 antioxidant system, iron accumulation, and lipid ROS generation. Lipid metabolism is closely related to the sensitivity of cells to ferroptosis. Lipid peroxides induce ferroptosis. Furthermore, excessive accumulation of lipid peroxides causes plasma membrane damage, leading to the onset of cellular ferroptosis.33 In the HALI model group, GPX4, a key regulator of ferroptosis,34,35 expression was significantly reduced. GPX4 is crucial in ferroptosis, and its inhibition can lead to ferroptosis from a genetic or pharmacological perspective.36,37 GPX4 inhibition can also cause the production of several polyunsaturated fatty acid (PUFAs) radicals, causing ferroptosis.38 Therefore, GPX4 downregulation can trigger ferroptosis.37 Moreover, GPX4 and GSH form an important antioxidant system in cells. Two GSH molecules act as electron donors to reduce free hydrogen peroxide (H2O2) or organic peroxides (ROOH), especially phospholipid peroxides (PL-OOH), in water or corresponding alcohols via GPX4. Herein, ROS were significantly increased and GSH was substantially depleted in the HALI group compared with the normal group, indicating that the antioxidant capacity of lung tissue is reduced after hyperoxia injury.
Amino acid metabolism also plays an important role in ferroptosis. System Xc- transports intracellular glutamate from the cell while transporting cystine into the cytoplasm. It is also an essential amino acid transporter protein. Previous studies have shown that inhibition of system Xc- decreases cystine uptake and reduces its intracellular conversion to cysteine, impairing glutathione peroxidase 4 (GPX4) function.39 Inhibition of system Xc- through salazosulfapyridine induces ferroptosis, whereas β-mercaptoethanol increases intracellular cystine uptake via HT1080, thereby inhibiting Erastin-induced ferroptosis.40 SLC7A11 is the transport catalytic subunit in System Xc- and an important marker in ferroptosis.41 Inhibition of SLC7A11 function with olaparib inhibits cystine uptake, significantly reducing GSH levels and promoting lipid peroxidation. However, SLC7A11 overexpression significantly restores GSH levels and attenuates lipid peroxidation, restoring cell viability.42 Similarly, SLC7A11 inhibition inhibits system Xc- activity.43 Herein, SLC7A11 protein content decreased in lung tissue after hyperoxia injury, suggesting that system Xc- was also inhibited. System Xc- is a key pathway for the synthesis of the cellular antioxidant GSH. Therefore, System Xc- can impair the metabolism of related amino acids and reduce GSH synthesis, decreasing cellular antioxidant capacity and causing loss of cellular protection.
Abnormal iron metabolism and iron accumulation contribute to the development of ferroptosis. Previous studies have shown that reducing iron overload in cells through iron chelators, such as desferrioxamine, desferrioxamine mesylate, and cyclopiroxamine, can inhibit suppression of erastin-mediated ferroptosis. In contrast, providing exogenous iron sources (ferric ammonium citrate, ferric citrate, and ferric chloride hexahydrate) enhances erastin-induced death.40 Prussian blue staining indicated that iron accumulation was significantly increased in the HALI group than in the normal group. Elevated iron levels can cause cellular damage by generating a large number of ROS through the Fenton reaction.24 Therefore, abnormal distribution and increased levels of iron can affect normal physiological processes. Besides, the fragile iron homeostasis may be disrupted during ferroptosis, leading to a sustained increase in intracellular free iron content.44 This study investigated the cause of iron accumulation by examining proteins related to iron transport and found increased levels of TFRC, a key protein receptor responsible for extracellular Fe3+ input. TFRC upregulation can lead to ferroptosis by increasing intracellular iron levels.45 However, TFRC silencing can inhibit ferroptosis induced by Erastin.46 Ferritin is an iron storage protein complex consisting of FLC and FHC. It plays a central role in iron metabolism by storing excess cellular iron. It also prevents the iron-mediated catalysis of ROS, thus preventing tissue damage.47 Ferroptosis-sensitive cells with RAS mutations have significantly elevated TFRC and reduced expression of FHC and FLC compared with ferroptosis-tolerant cells.48 Therefore, increased iron uptake and reduced iron storage may lead to iron overload during ferroptosis. FHC and FLC translation is regulated via the iron regulatory protein (IRP) mechanism. IRP binds to the iron-responsive element (IRE) in the 5ʹ UTR of the ferritin transcript under low iron conditions to block translation.49 However, when sufficient iron is available, IRP expresses aconitase activity and no longer binds to IRE, ultimately leading to enhanced ferritin expression.50 Herein, TFRC, FHC, and FLC expressions were significantly increased in hyperoxic lung injury. This could be due to the massive influx of iron ions into the cytosol during the initial phase of injury. This allows cellular stress to increase the expression of iron storage proteins to maintain iron homeostasis by increasing iron storage.
In summary, hyperoxia-induced lung tissue undergoes several oxidative stresses with impaired function of multiple antioxidant pathways, making it sensitive to ferroptosis. Also, abnormal iron accumulation occurs in lung tissue with dysregulation of intracellular iron transport, storage, and regulatory proteins. Excess iron leads to increased levels of lipid peroxidation. Therefore, ferroptosis-related mechanisms, such as abnormalities in iron metabolism, amino acid metabolism, and lipid peroxidation regulation, play an important role in HALI. This study suggests that ferroptosis may be involved in the pathological process of hyperoxic lung injury in neonatal rats. Therefore, the inhibition of ferroptosis in the lung is a promising strategy for the treatment of HALI. This study provides insights into the mechanisms of other oxidative stress-related diseases.
However, this study has some limitations. First, iron chelators, ferrostatin-1 and liproxstatin-1, and other ferroptosis inhibitory drugs were not used to explore the protective and therapeutic effects of ferroptosis inhibition on hyperoxic lung injury. Second, the role of ferroptosis in hyperoxic damage to alveolar cells was not verified at the cellular level. Third, it is uncertain whether ferroptosis occurs in both alveolar tissue and pulmonary vasculature. Therefore the above factors deserve further exploration.
Disclosure
Danyun Jia, Jinyu Zheng and Yiyang Zhou contributed equally to this work and should be considered co-first authors. The authors declare no conflict of interest.
References
1. Vogel ER, Britt RD, Trinidad MC, et al. Perinatal oxygen in the developing lung. Can J Physiol Pharmacol. 2015;93(2):119–127. doi:10.1139/cjpp-2014-0387
2. Baraldi E, Filippone M. Chronic lung disease after premature birth. N Engl J Med. 2007;357(19):1946–1955. doi:10.1056/NEJMra067279
3. Carnesecchi S, Deffert C, Pagano A, et al. NADPH oxidase-1 plays a crucial role in hyperoxia-induced acute lung injury in mice. Am J Respir Crit Care Med. 2009;180(10):972–981. doi:10.1164/rccm.200902-0296OC
4. Mosca F, Colnaghi M, Fumagalli M. BPD: old and new problems. J Matern Fetal Neonatal Med. 2011;24(Suppl 1):80–82. doi:10.3109/14767058.2011.607675
5. Slutsky AS. Lung injury caused by mechanical ventilation. Chest. 1999;116(1 Suppl):9S–15S. doi:10.1378/chest.116.suppl_1.9S-a
6. Tauseef M, Knezevic N, Chava KR, et al. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J Exp Med. 2012;209(11):1953–1968. doi:10.1084/jem.20111355
7. Tang D, Chen X, Kang R, et al. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107–125. doi:10.1038/s41422-020-00441-1
8. Ayala A, Muñoz MF, Argüelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014:360438. doi:10.1155/2014/360438
9. Chabot F, Mitchell JA, Gutteridge JM, et al. Reactive oxygen species in acute lung injury. Eur Respir J. 1998;11(3):745–757.
10. Zhou H, Li F, Niu JY, et al. Ferroptosis was involved in the oleic acid-induced acute lung injury in mice. Sheng li Xue Bao: [Acta Physiologica Sinica]. 2019;71(5):689–697.
11. Dong H, Qiang Z, Chai D, et al. Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging. 2020;12(13):12943–12959. doi:10.18632/aging.103378
12. Xu Y, Li X, Cheng Y, et al. Inhibition of ACSL4 attenuates ferroptotic damage after pulmonary ischemia-reperfusion. FASEB J. 2020;34(12):16262–16275. doi:10.1096/fj.202001758R
13. Huang Y, Dai Z, Barbacioru C, et al. Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res. 2005;65(16):7446–7454. doi:10.1158/0008-5472.CAN-04-4267
14. Robbins ME, Cho HY, Hansen JM, et al. Glutathione reductase deficiency alters lung development and hyperoxic responses in neonatal mice. Redox Biol. 2021;38:101797. doi:10.1016/j.redox.2020.101797
15. Ryter SW, Choi AMK. Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Transl Res. 2016;167(1):7–34.
16. Ali M, Heyob K, Tipple TE, et al. Alterations in VASP phosphorylation and profilin1 and cofilin1 expression in hyperoxic lung injury and BPD. Respir Res. 2018;19(1):229. doi:10.1186/s12931-018-0938-1
17. Ruan Y, Dong W, Kang L, et al. The changes of twist1 pathway in pulmonary microvascular permeability in a newborn rat model of hyperoxia-induced acute lung injury. Front Pediatr. 2020;8:190. doi:10.3389/fped.2020.00190
18. Burkhardt W, Kraft S, Ochs M, et al. Persurf, a new method to improve surfactant delivery: a study in surfactant depleted rats. PLoS One. 2012;7(10):e47923. doi:10.1371/journal.pone.0047923
19. Teng RJ, Jing X, Michalkiewicz T, et al. Attenuation of endoplasmic reticulum stress by caffeine ameliorates hyperoxia-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2017;312(5):L586–L598. doi:10.1152/ajplung.00405.2016
20. Bhaskaran M, Xi D, Wang Y, et al. Identification of microRNAs changed in the neonatal lungs in response to hyperoxia exposure. Physiol Genomics. 2012;44(20):970–980. doi:10.1152/physiolgenomics.00145.2011
21. Wu Q, Chong L, Shao Y, et al. Lipoxin A4 reduces hyperoxia-induced lung injury in neonatal rats through PINK1 signaling pathway. Int Immunopharmacol. 2019;73:414–423. doi:10.1016/j.intimp.2019.05.046
22. Chen S, Wu Q, Zhong D, et al. Caffeine prevents hyperoxia-induced lung injury in neonatal mice through NLRP3 inflammasome and NF-κB pathway. Respir Res. 2020;21(1):140. doi:10.1186/s12931-020-01403-2
23. Liu S, Grigoryan MM, Vasilevko V, et al. Comparative analysis of H&E and Prussian blue staining in a mouse model of cerebral microbleeds. J Histochem Cytochem. 2014;62(11):767–773. doi:10.1369/0022155414546692
24. Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10(1):9–17. doi:10.1038/nchembio.1416
25. Manz DH, Blanchette NL, Paul BT, et al. Iron and cancer: recent insights. Ann N Y Acad Sci. 2016;1368(1):149–161. doi:10.1111/nyas.13008
26. Xuefei Y, Xinyi Z, Qing C, et al. Effects of hyperoxia on mitochondrial homeostasis: are mitochondria the Hub for bronchopulmonary dysplasia? Front Cell Dev Biol. 2021;9:642717. doi:10.3389/fcell.2021.642717
27. Yang H, Fu J, Xue X, et al. Epithelial-mesenchymal transitions in bronchopulmonary dysplasia of newborn rats. Pediatr Pulmonol. 2014;49(11):1112–1123. doi:10.1002/ppul.22969
28. Sun H, Choo-Wing R, Fan J, et al. Small molecular modulation of macrophage migration inhibitory factor in the hyperoxia-induced mouse model of bronchopulmonary dysplasia. Respir Res. 2013;14:27. doi:10.1186/1465-9921-14-27
29. Warner BB, Stuart LA, Papes RA, et al. Functional and pathological effects of prolonged hyperoxia in neonatal mice. Am J Physiol. 1998;275(1):L110–L117.
30. Liu P, Feng Y, Li H, et al. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell Mol Biol Lett. 2020;25:10. doi:10.1186/s11658-020-00205-0
31. Li Y, Cao Y, Xiao J, et al. Inhibitor of apoptosis-stimulating protein of p53 inhibits ferroptosis and alleviates intestinal ischemia/reperfusion-induced acute lung injury. Cell Death Differ. 2020;27(9):2635–2650. doi:10.1038/s41418-020-0528-x
32. Li X, Zhuang X, Qiao T. Role of ferroptosis in the process of acute radiation-induced lung injury in mice. Biochem Biophys Res Commun. 2019;519(2):240–245. doi:10.1016/j.bbrc.2019.08.165
33. Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26(3):165–176. doi:10.1016/j.tcb.2015.10.014
34. Imai H, Matsuoka M, Kumagai T, et al. Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr Top Microbiol Immunol. 2017;403:143–170.
35. Chen D, Fan Z, Rauh M, et al. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene. 2017;36(40):5593–5608. doi:10.1038/onc.2017.146
36. Friedmann Angeli JP, Schneider M, Proneth B, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16(12):1180–1191. doi:10.1038/ncb3064
37. Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–331. doi:10.1016/j.cell.2013.12.010
38. Yang WS, Kim KJ, Gaschler MM, et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113(34):E4966–E4975. doi:10.1073/pnas.1603244113
39. Du K, Oh SH, Dutta RK, et al. Inhibiting xCT/SLC7A11 induces ferroptosis of myofibroblastic hepatic stellate cells but exacerbates chronic liver injury. Liver Int. 2021;41(9):2214–2227. doi:10.1111/liv.14945
40. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
41. Wang Y, Yang L, Zhang X, et al. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep. 2019;20(7):e47563. doi:10.15252/embr.201847563
42. Hong T, Lei G, Chen X, et al. PARP inhibition promotes ferroptosis via repressing SLC7A11 and synergizes with ferroptosis inducers in BRCA-proficient ovarian cancer. Redox Biol. 2021;42:101928. doi:10.1016/j.redox.2021.101928
43. Dixon SJ, Patel DN, Welsch M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523. doi:10.7554/eLife.02523
44. Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425–1428. doi:10.1080/15548627.2016.1187366
45. Ooko E, Saeed ME, Kadioglu O, et al. Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine. 2015;22(11):1045–1054. doi:10.1016/j.phymed.2015.08.002
46. Gao M, Monian P, Quadri N, et al. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59(2):298–308. doi:10.1016/j.molcel.2015.06.011
47. Pham CG, Bubici C, Zazzeroni F, et al. Ferritin heavy chain upregulation by NF-κB inhibits TNFα-induced apoptosis by suppressing reactive oxygen species. Cell. 2004;119(4):529–542. doi:10.1016/j.cell.2004.10.017
48. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15(3):234–245. doi:10.1016/j.chembiol.2008.02.010
49. Huang BW, Miyazawa M, Tsuji Y. Distinct regulatory mechanisms of the human ferritin gene by hypoxia and hypoxia mimetic cobalt chloride at the transcriptional and post-transcriptional levels. Cell Signal. 2014;26(12):2702–2709. doi:10.1016/j.cellsig.2014.08.018
50. Toth I, Bridges KR. Ascorbic acid enhances ferritin mRNA translation by an IRP/aconitase switch. J Biol Chem. 1995;270(33):19540–19544. doi:10.1074/jbc.270.33.19540
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.