Back to Journals » OncoTargets and Therapy » Volume 12
Fenofibrate potentiates chemosensitivity to human breast cancer cells by modulating apoptosis via AKT/NF-κB pathway
Authors Sun J, Zheng Z, Chen Q ![]() , Pan Y, Quan M, Dai Y
, Pan Y, Quan M, Dai Y
Received 17 October 2018
Accepted for publication 21 December 2018
Published 23 January 2019 Volume 2019:12 Pages 773—783
DOI https://doi.org/10.2147/OTT.S191239
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Jianguo Sun,1,2,* Zhibao Zheng,1,* Qi Chen,2 Yin Pan,1 Mingming Quan,1 Yuechu Dai1
1Department of Surgical Oncology, Taizhou Central Hospital (Taizhou University Hospital), Taizhou, Zhejiang, People’s Republic of China; 2Precision Medicine Center, Taizhou Central Hospital (Taizhou University Hospital), Taizhou, Zhejiang, People’s Republic of China
*These authors contributed equally to this work
Background: Cumulatively, evidences revealed that fenofibrate used in the therapy of hyperlipidemia and hypercholesterolemia has anti-cancer effect in multiple cancer types. However, its function and underlying mechanism of chemosensitization in breast cancer remain poorly understood.
Materials and methods: The cytotoxicity of fenofibrate and anti-cancer drugs in breast cancer cells was determined by MTT. Apoptosis and mitochondrial membrane potential were measured using flow cytometry. Caspases and PARP cleavage, the Bcl-2 family members’ protein expression, as well as the activation of AKT and NF-κB signaling pathways were evaluated using Western blot assay. Real-time PCR was used to determine the mRNA expression of Bcl-2 family members.
Results: Our data indicated that fenofibrate suppressed SKBR3 and MDA-MB-231 cell growth in a dose-dependent manner, in the same way as paclitaxel, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), ABT-737, and doxorubicin. Subtoxic levels of fenofibrate significantly augmented paclitaxel, TRAIL, ABT-737, and doxorubicin-induced apoptosis in both these two cell lines. Fenofibrate-promoted chemosensitivity is predominantly mediated by caspase-9 and caspase-3 activation and mitochondrial outer membrane permeabilization. Meanwhile, chemosensitivity promoted by fenofibrate also increased the expression of Bax and Bok and decreased the expression of Mcl-1 and Bcl-xl. Mechanistically, fenofibrate effectively reduced the phosphorylation levels of AKT and NF-κB. In addition, imiquimod, an NF-κB activator, could reverse fenofibrate-induced susceptibility to ABT-737-triggered apoptosis.
Conclusion: The present study provided the evidence of the underlying mechanisms on chemosensitization of fenofibrate by inducing the apoptosis of breast cancer in an AKT/NF-κB-dependent manner and implicated the potential application of fenofibrate in potentiating chemosensitivity in breast cancer therapy.
Keywords: human breast cancer, fenofibrate, chemosensitization, AKT, NF-κB
Introduction
Globally, human breast cancer is the second cause of cancer-correlated mortality in females.1 In China, breast cancer alone accounts for ~15% of all new cancers in women, mostly aged 30–59 years.2 Chemotherapy is effective in decreasing the measure of the primary cancer in neoadjuvant setting; however, it not only causes adverse effects in breast cancer patients, but also inevitably induces apoptosis resistance.3 Therefore, a better understanding of the theory and mechanism of chemoresistance is of great value to exploit novel therapeutic strategy to improve prognosis.
Resistance mechanism by which breast cancer escapes drug-induced cell death has been ascribed to variations in the apoptosis pathway.4 The key apoptotic regulators frequently inhibited by the AKT/NF-κB pathway in breast cancer, which blocks the activity of pro-apoptosis, are Bax, Bok, and Bim.5,6 Thus, it is necessary to investigate the molecular mechanisms of AKT/NF-κB pathway, which is responsible for apoptosis resistance, and to identify potential sensitizers that are competent of inhibiting apoptosis resistance.
Increasingly, evidences showed that fenofibrate, which was utilized in the therapy of hyperlipidemia and hypercholesterolemia, has anti-cancer effects on endometrial cancer, prostate cancer, triple-negative breast cancer, oral cancer, pancreatic cancer, and lung cancer, but only few studies have reported its effect on chemosensitivity of breast cancer.7–13 Researchers found that fenofibrate alone resulted in decreasing the semaphorin 6B gene expression in breast cancer,14 arresting G1 phase in human glioblastoma cells,15 but the detailed mechanisms by which fenofibrate sensitizes breast cancer cells to chemotherapy.
In the present study, we investigated the sensitization effects of fenofibrate on several anti-cancer agents including paclitaxel, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), ABT-737, and doxorubicin in breast cancer cells and provided insight into the molecular mechanisms of the action on fenofibrate-potentiated chemosensitivity.
Materials and methods
Cell culture
The human breast cancer cell lines MDA-MB-231 and SKBR3 were purchased from the American Type Culture Collection (Manassas, VA, USA) and cultured in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS (Hyclone; GE Healthcare Life Sciences, Uppsala, Sweden), 100 U/mL penicillin (Thermo Fisher Scientific), and 100 g/mL streptomycin (Thermo Fisher Scientific). All cell lines were cultured in a humidified incubator in an atmosphere of 5% (v/v) CO2 at 37°C.
Cell viability assay
Fenofibrate, paclitaxel, TRAIL, ABT-737, and doxorubicin alone or in combination were tested in vitro for cytotoxicity against breast cancer cell lines in 96-well plates using the MTT assay to determine the cell viability. In short, cells were seeded in 96-well plates and incubated with different concentrations of fenofibrate, paclitaxel, TRAIL, ABT-737, and doxorubicin alone or in combination for indicated times. First, the medium was discarded, and then 50 μL of MTT solution (1 mg/mL in PBS) was added to each well and the plate was incubated at 37°C for additional 4 hours. Then, 150 μL of dimethylsulfoxide was added to dissolve the formazan crystals. Finally, the absorbance of each sample was read at 570 nm using a microplate reader (Molecular Devices LLC, Sunnyvale, CA, USA).
Apoptotic assay by flow cytometry
Flow cytometry was used to assess externalization by fluorescein isothiocyanate (FITC)-labeled Annexin-V and propidium iodide (PI). Briefly, human breast cancer cells were collected after treatment and washed with ice-cold PBS before staining in 500 μL solution containing Annexin-V-FITC in dark at 4°C. After that, PI was added and incubated for 5 minutes at room temperature. Soon afterwards, the fluorescent signal in the cells was detected by flow cytometry (FACS Calibur; BD Biosciences, San Jose, CA, USA). Data analysis was performed by the software WinMDI 2.9 (The Scripps Research Institute, La Jolla, CA, USA).
Mitochondrial membrane potential change assay
5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylc-arbocyanine iodide (JC-1) staining was used to detect the mitochondrial membrane potential change. Human breast cancer cells were seeded in cell culture dish and incubated with various concentrations of fenofibrate, paclitaxel, TRAIL, ABT-737, and doxorubicin alone or in combination for 24 hours. Then, the cells were harvested, washed by PBS, and resuspended in JC-1 staining solution (10 μM) at room temperature for 10 minutes. After that, the cells were detected by a FACSCanto flow cytometer (BD, Franklin Lakes, NJ, USA).
RNA isolation and reverse transcription-quantitative PCR (RT-qPCR)
Total RNA was isolated from cell lines using the Trizol (Thermo Fisher Scientific) according to the manufacturer’s protocols. Complementary DNA synthesis of mRNA was performed using M-MLV reverse transcriptase (Promega Corporation, Fitchburg, WI, USA). The mRNA expression levels of Mcl-1, Bcl-2, Bim, Bcl-xl, Bok, Bnip3, and Bax were evaluated using PCR with an SYBR green PCR master mix (Thermo Fisher Scientific) and calculated using the 2−ΔΔCq method by normalizing to GAPDH. The thermocycling conditions were as follows: 95°C for 10 minutes, 45 cycles of 95°C for 15 seconds, and 60°C for 1 minute. All the reactions were performed in triplicate and the primer sequences are listed in Table 1.
 | Table 1 The sequences of primers used in real-time PCR |
Western blotting
Subsequent to the treatment indicated, the cells were lysed in lysis buffer (2.1 μg/mL aprotinin, 0.5 μg/mL leupeptin, 4.9 mM MgCl2, 1 mM orthovanadate, 1% Triton X 100, and 1 mM phenylmethylsulfonyl fluoride). The protein concentration was determined using a bicinchoninic acid assay. Subsequent to electrophoresis on a 12% or 15% SDS-PAGE gel, proteins were transferred onto polyvinylidene difluoride membranes. The membranes were blocked with 5% non-fat milk and incubated with primary antibodies at 4°C overnight. The corresponding horseradish peroxidase (HRP)-conjugated secondary antibody was added and incubated at room temperature for 2 hours. Signals were visualized using an enhanced chemiluminescence reaction with an HRP substrate. The primary antibodies against PARP, caspase-3, caspase-9, Mcl-1, Bcl-2, Bim, Bcl-xl, Bok, Bnip3, Bax, AKT, p-AKT, NF-κB, p-NF-κB, and histone 3 were purchased from Cell Signaling Technologies (Beverly, MA, USA). The antibody against β-actin was purchased from Sigma-Aldrich Co. (St Louis, MO, USA).
Statistical analysis
All data are expressed as mean ± SD from at least three separate experiments. All statistical analyses were performed using GraphPad Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA). Statistical significance was determined using a two-sided Student’s t-test for all data. For statistical analysis, P<0.05 was considered to indicate a statistically significant difference.
Results
Cytotoxicity of fenofibrate, paclitaxel, TRAIL, ABT-737, and doxorubicin on human breast cancer cells
To determine whether fenofibrate could suppress human breast cancer or not, two human breast cancer cell lines and paclitaxel, TRAIL, ABT-737, and doxorubicin were obtained, and the cytotoxicity was evaluated using MTT assay. The results revealed that fenofibrate slightly inhibited SKBR3 cell growth, but significantly suppressed MDA-MB-231 cell growth (Figure 1A). The IC50 of fenofibrate in MDA-MB-231 cells is >100 μM for 24 hours and 79.42±6.25 μM for 48 hours. The IC50 of fenofibrate in SKBR3 cells is >100 μM for both 24 and 48 hours. In addition, cell viability was measured in breast cancer cell lines treated with paclitaxel, TRAIL, ABT-737, and doxorubicin for 24 hours. As presented in Figure 1B–E, human breast cancer cell lines SKBR3 and MDA-MB-231 involved in the study are highly resistant to paclitaxel and TRAIL, while relatively sensitive to ABT-737 and doxorubicin. The IC50 values of paclitaxel, TRAIL, ABT-737, and doxorubicin in SKBR3 cells are >80 nM, 35±0.69 ng/mL, 11.56±0.93 μg/mL, and 0.71±0.08 μg/mL, respectively. The IC50 values of paclitaxel, TRAIL, ABT-737, and doxorubicin in MDA-MB-231 cells are >80 nM, >200 ng/mL, 4.25±0.21 μg/mL, and 32.41±1.12 μg/mL, respectively.
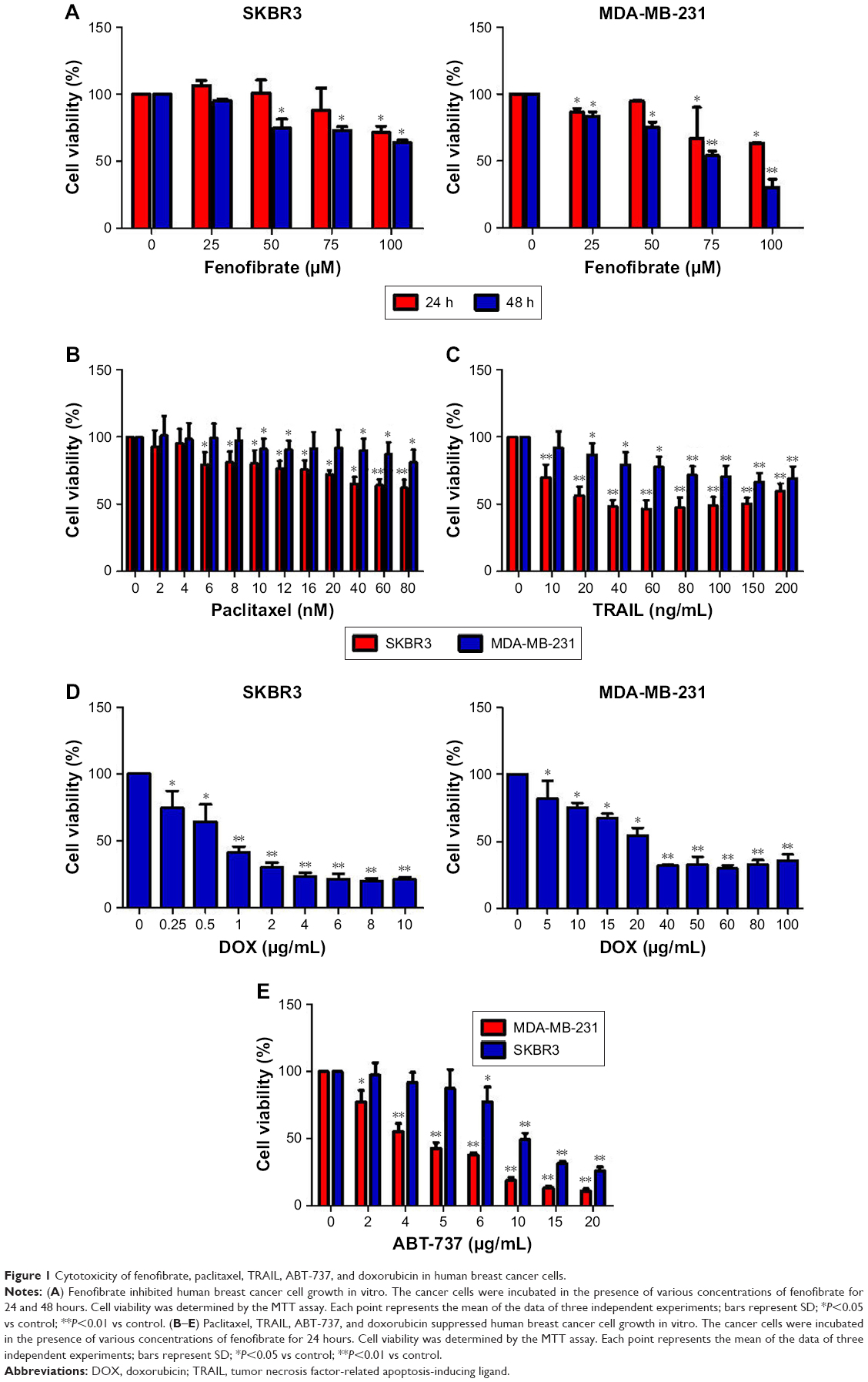 | Figure 1 Cytotoxicity of fenofibrate, paclitaxel, TRAIL, ABT-737, and doxorubicin in human breast cancer cells. |
Fenofibrate potentiates chemosensitivity of human breast cancer by modulating apoptosis
Next, we examined whether combined treatment of fenofibrate and paclitaxel, TRAIL, ABT-737, and doxorubicin exerts enhanced lethality in human breast cancer cell lines. After co-treatment with indicated concentrations of fenofibrate and paclitaxel, TRAIL, ABT-737, and doxorubicin for 24 hours, MTT assay was performed. Interestingly, combination of fenofibrate and paclitaxel, TRAIL, ABT-737, and doxorubicin dramatically inhibits the cell growth in both human breast cancer cell lines SKBR3 and MDA-MB-231 (Figure 2A and D).
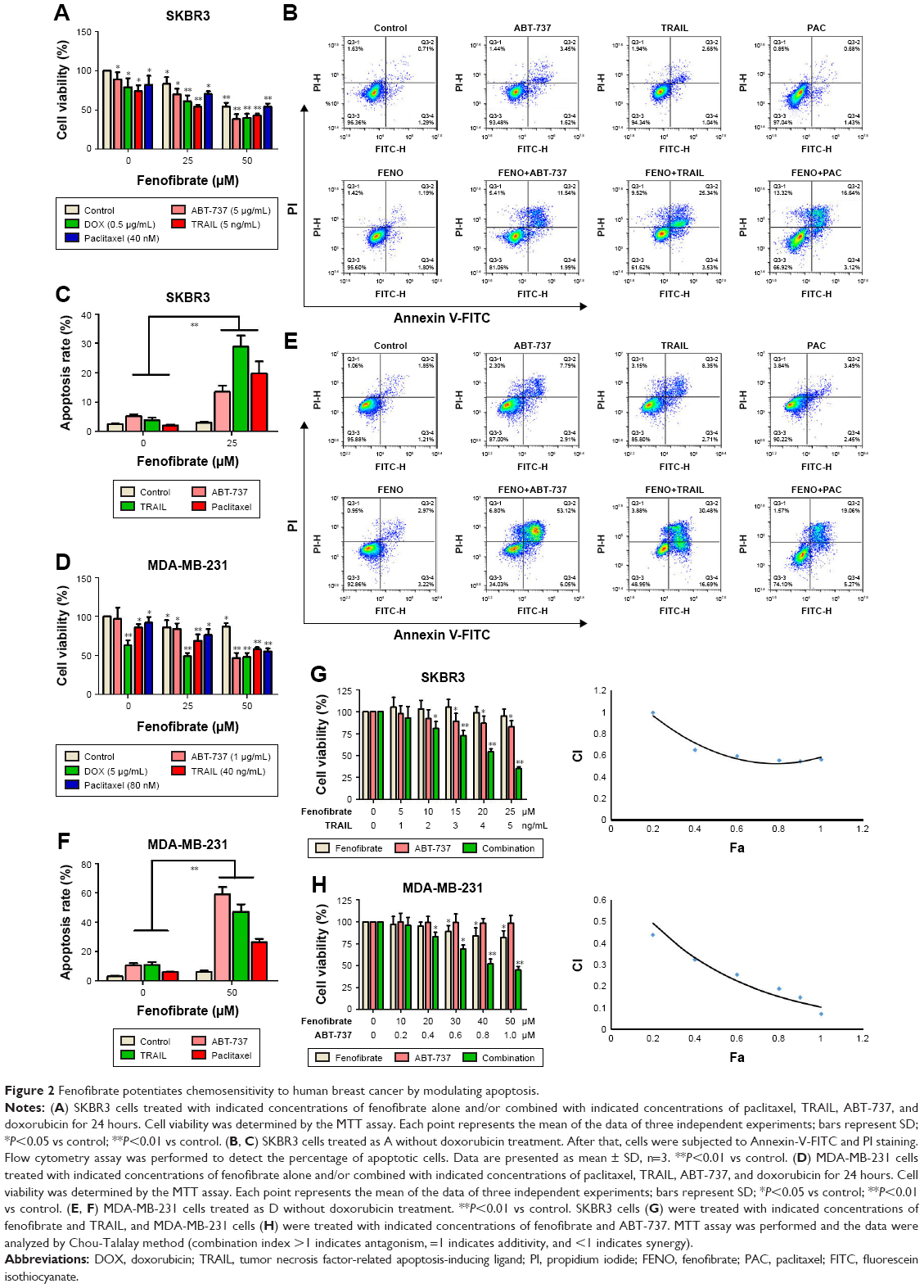 | Figure 2 Fenofibrate potentiates chemosensitivity to human breast cancer by modulating apoptosis. |
To confirm whether the sensitization effect of fenofibrate involves apoptosis induction, SKBR3 and MDA-MB-231 cells were subjected to flow cytometry analysis after cells were treated with fenofibrate, paclitaxel, TRAIL, and ABT-737, alone or in combination. As shown in Figure 2B, C, E, and F, fenofibrate dramatically enhanced paclitaxel-, TRAIL-, and ABT-737-induced apoptosis in SKBR3 and MDA-MB-231 cells (the percentage of Annexin-V-positive cells probably increased from 7.1% and 15.29% to 19.76% and 24.33% when combined with paclitaxel, from 8.71% and 20.31% to 28.87% and 47.07% when combined with TRAIL, from 10.06% and 19.95% to 13.53% and 59.17% when combined with ABT-737 in SKBR3 and MDA-MB-231, respectively). More importantly, combined treatment of fenofibrate and TRAIL (in SKBR3 cells) or ABT-737 (in MDA-MB-231 cells) presented well as synergism (combination index; CI <1, as shown in Figure 2G and H). These results suggest that fenofibrate potentiates chemosensitivity to human breast cancer cell via inducing apoptosis.
Initiation of intrinsic apoptotic pathway in the sensitization effect of fenofibrate on ABT-737, TRAIL, and paclitaxel in human breast cancer cells
Here, the underlying mechanisms of the sensitization effect of fenofibrate in breast cancer cells were further elucidated. Our observation indicated that PARP is cleaved in fenofibrate-and-TRAIL- and paclitaxel-treated SKBR3 cells and fenofibrate-and-TRAIL- and ABT-737-treated MDA-MB-231 cells (Figure 3A) which supported that the apoptosis is indeed involved in fenofibrate-and-TRAIL-, paclitaxel-, and ABT-737-induced cell death. We further examined the cleavage of caspases, and as shown in Figure 3A, co-treatment with fenofibrate and TRAIL, paclitaxel, and ABT-737 in SKBR3 and MDA-MB-231 cells significantly increased the cleavage of caspase-3 and caspase-9 (caspase-8 was not detected and the data not shown). Therefore, the effect of fenofibrate and ABT-737, TRAIL, and paclitaxel on mitochondrial outer membrane potential (MOMP) was determined by JC-1 staining and flow cytometry experiment. As shown in Figure 4, the disruption of MOMP induced by fenofibrate and ABT-737, TRAIL, and paclitaxel was significantly increased. After co-treatment with 25 μM fenofibrate and indicated concentration of ABT-737, TRAIL, and paclitaxel, the percentage of cells with depolarized MOMP increased to 19.17%, 36.46%, and 18.03% in SKBR3 cells and 62.5%, 53.2%, and 43.3% in MDA-MB-231 cells, respectively.
 | Figure 3 Treatment with fenofibrate resulted in downregulation of Mcl-1 and Bcl-xl and upregulation of Bok and Bax. |
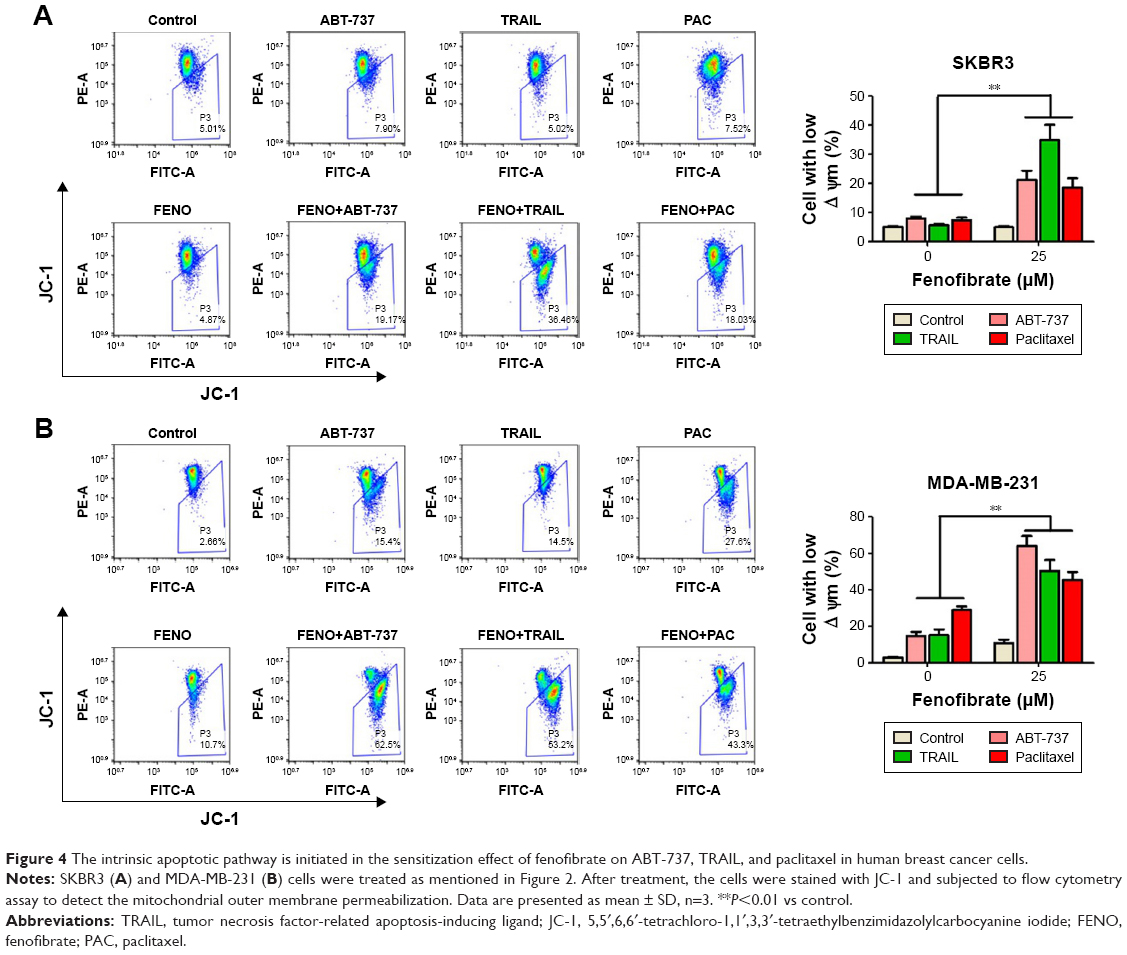 | Figure 4 The intrinsic apoptotic pathway is initiated in the sensitization effect of fenofibrate on ABT-737, TRAIL, and paclitaxel in human breast cancer cells. |
Treatment with fenofibrate resulted in downregulation of Mcl-1 and Bcl-xl and upregulation of Bok and Bax in human breast cancer cells
Previous studies have shown that fenofibrate inhibits mTOR-p70S6K signaling, regulates autophagy and endoplasmic reticulum stress in human prostate cancer cells, and induces apoptosis of triple-negative breast cancer cells via activation of NF-κB pathway.8–10 However, the role of fenofibrate in human breast cancer is still ambiguous. At first, we evaluated the effect of fenofibrate on Bcl-2 family proteins expression by Western blotting. Surprisingly, as shown in Figure 3B, treatment with fenofibrate dramatically inhibited the protein level of Mcl-1 and Bcl-xl and increased the protein level of Bok and Bax in SKBR3 and MDA-MB-231 cells.
To draw the conclusion whether fenofibrate-mediated decrease of Mcl-1 and Bcl-xl and increase of Bok and Bax in human breast cancer cells was at the transcriptional level, RT-PCR was used to identify the effects of fenofibrate on the mRNA level of Bcl-2 family proteins. It was found that fenofibrate repressed transcription level of Mcl-1 and Bcl-xl and activated transcription level of Bok and Bax in SKBR3 and MDA-MB-231 cells (Figure 3C), suggesting that fenofibrate-mediated Mcl-1 and Bcl-xl decrease and Bok and Bax increase was likely at the transcriptional level.
Fenofibrate potentiated the apoptosis induced by anti-cancer drugs in human breast cancer cells via inhibiting the activation of AKT/NF-κB pathway
AKT/NF-κB pathway is important for the balance between cell survival and apoptosis.16,17 To determine whether AKT/NF-κB signaling pathways are involved in fenofibrate-induced apoptosis, we checked phosphorylated AKT/NF-κB by Western blotting (Figure 5A). Fenofibrate treatment significantly decreased the level of phosphorylated AKT and NF-κB p65, but did not change the total levels of AKT and NF-κB p65 in SKBR3 and MDA-MB-231 cells. As shown in Figure 5B, after treatment of indicated concentration of fenofibrate, nuclear p65 decreased and cytosolic p65 increased both in a dose-dependent manner for 24 hours in MDA-MB-231 cells. These observations indicate that AKT/NF-κB pathway might be a potential target by which fenofibrate can potentiate human breast cancer cells to paclitaxel, TRAIL, ABT-737, and doxorubicin.
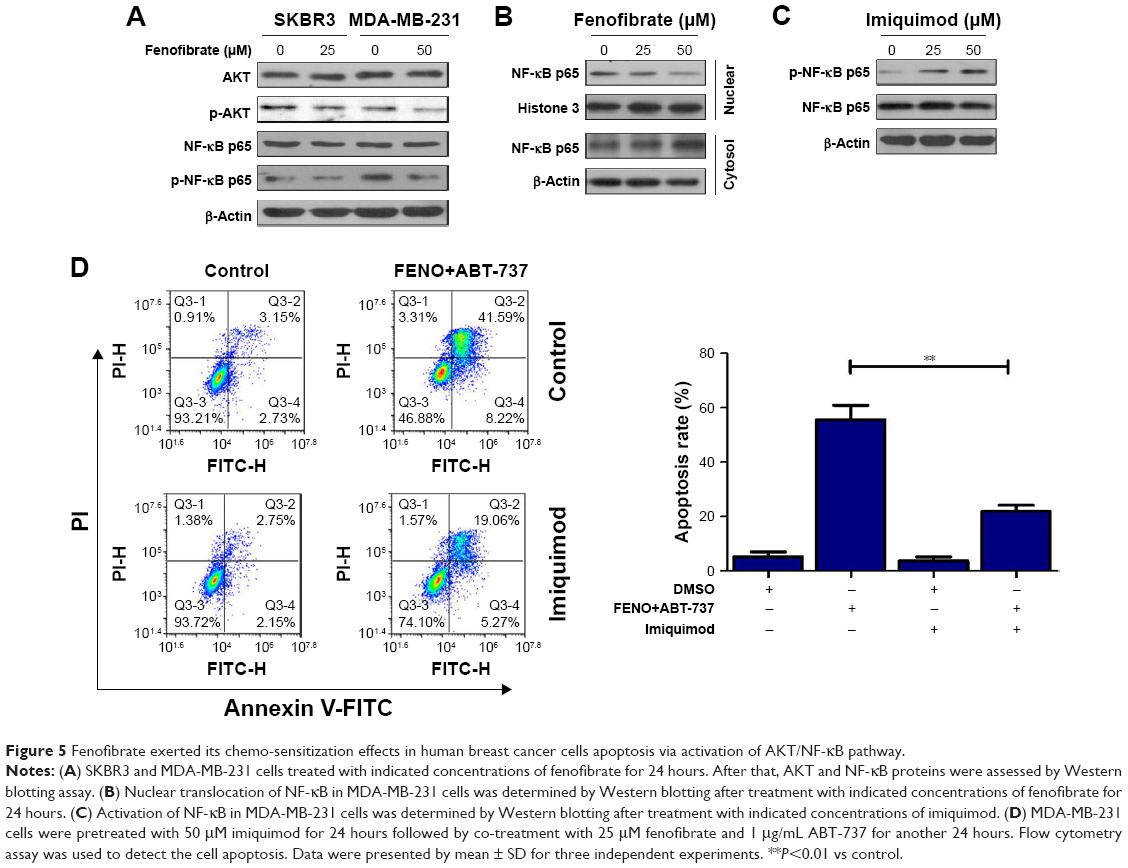 | Figure 5 Fenofibrate exerted its chemo-sensitization effects in human breast cancer cells apoptosis via activation of AKT/NF-κB pathway. |
To further confirm whether fenofibrate enhances the apoptosis-induced ability of paclitaxel, TRAIL, and ABT-737 via AKT/NF-κB pathway, MDA-MB-231 cells were pretreated with 50 μM imiquimod, an NF-κB agonist, to activate NF-κB pathway. After pretreatment, MDA-MB-231 cells were co-treated with 25 μM fenofibrate and 1 μg/mL ABT-737 for 24 hours. As shown in Figure 5C, phosphorylation of NF-κB p65 is highly activated in MDA-MB-231 cells treated with imiquimod compared with its control cells. More importantly, high level of phosphorylated NF-κB p65 significantly induced by imiquimod effectively attenuates the apoptosis mediated by the combination of fenofibrate and ABT-737 (Figure 5D). Taken together, fenofibrate sensitizes human breast cancer cells to paclitaxel, TRAIL, ABT-737, and doxorubicin at least partially through inhibiting the activation of AKT/NF-κB signaling pathway.
Discussion
Chemoresistance is one of the most important challenges in cancer treatment and believed to be a main reason for cancer therapy failure, and multiple mechanisms are reported to take part in the development of drug resistance in cancer cells. In addition to Bcl-2 family mentioned above, the abnormal regulation of cell cycle could also mediate the drug resistance in multiple cancer types.18 Furthermore, it was reported that mitochondrial reactive oxygen species play an important role in cancer drug resistance.19 As a key tumor suppressor, P53 exerts a pivotal role in protecting from malignancies. However, overexpression of mutated p53 is frequently associated with resistance to several anti-cancer drugs, including cisplatin, temozolomide, doxorubicin, gemcitabine, tamoxifen, and EGFR-inhibitors (such as cetuximab).20 Although tremendous progress has been made to help us to understand the underlying mechanism of drug resistance of cancer cells, it is still urgent to find novel and more effective sensitizer for chemotherapy.
As an important therapy of hyperlipidemia and hypercholesterolemia, fenofibrate is recently studied in anti-tumor effects in multiple cancer types.10,13,21–23 Its anti-tumor functions such as suppression of cancer cell growth,12 restraining cancer cell metastasis24 and inhibiting carcinogenesis,25 inducing cell-cycle arrest and apoptosis by suppression of Bcl-2, and AKT phosphorylation in prostate cancer26 were discovered during recent years. More recently, fenofibrate was reported to overcome the drug resistance of human prostate cancer cells in a peroxisome proliferator-activated receptor alpha/reactive oxygen species-independent manner.27 However, to date, the function and underlying mechanism of fenofibrate-enhanced chemosensitivity in other cancers have not been well documented, particularly in breast cancer.
The current study attempted to illustrate the role and the underlying mechanism of fenofibrate in the drug susceptibility in human breast cancer. First, the present study verified that fenofibrate inhibited human breast cancer cell growth in a dose- and time-dependent manner. In addition, MTT assay results were also validated by flow cytometry assay with Annexin-V/PI staining. We further illuminated the subcytotoxic level of fenofibrate-sensitized paclitaxel, TRAIL, and ABT-737 on human breast cancer cells by inducing apoptosis. These results suggested that fenofibrate modulates apoptotic pathway which results in an increase in cell sensitivity to paclitaxel-, TRAIL-, and ABT-737-mediated signaling.
Next, since cleavage of caspases and PARP are hallmarks of activation of the apoptosis pathway, we used Western blotting to check the result of cleaved caspase-3, caspase-9, and PARP, which were upregulated in the SKBR3 and MDA-MB-231 cells co-treated with fenofibrate and paclitaxel, TRAIL, as well as ABT-737. Moreover, the augment of pro-apoptosis Bcl-2 family members, such as Bok and Bax, and decrease of pro-survival Bcl-2 family members, such as Mcl-1 and Bcl-xl, resulted in apoptosis by the activation of caspase-9 and caspase-3.28–30 However, in the present study, we detected notable reduction of Mcl-1 and Bcl-xl and activation of Bok and Bax but no significant changes in Bim, Bnip3, and Bcl-2. Thus, we consider downregulation of Mcl-1 and Bcl-xl as well as upregulation of Bok and Bax is an important cause of the augmentation of paclitaxel-, TRAIL-, and ABT-737-induced apoptosis mediated by fenofibrate.
Finally, we investigated the activation of AKT/NF-κB pathway under fenofibrate treatment, since the serine-threonine kinase AKT is best known as a regulator of cell proliferation and survival.31 It is well known that NF-κB has two-way regulation effects on cell apoptosis.32 Our study demonstrated that the level of phosphorylated AKT and NF-κB p65 was reduced by fenofibrate in SKBR3 and MDA-MB-231 cells. Moreover, imiquimod induced phosphorylated NF-κB p65 expression significantly and reversed the apoptosis mediated by the combination of fenofibrate and ABT-737. Thus, these data obviously confirm that fenofibrate potentiates chemosensitivity of human breast cancer cell to paclitaxel, TRAIL, ABT-737, and doxorubicin by suppressing phosphorylated NF-κB p65 at least partially.
Conclusion
Although other downstream target genes of fenofibrate may also take part in modulating apoptosis, the present data demonstrated that fenofibrate could potentiate chemosensitivity to human breast cancer by suppressing the activation of AKT/NF-κB p65 signaling pathway at least partially, which served a prominent function as an activator in apoptosis by downregulating Mcl-1 and Bcl-xl and upregulating Bok and Bax. Therefore, the results of the present study prove that fenofibrate with paclitaxel, TRAIL, ABT-737, and doxorubicin therapies may turn out to be effective new strategies for the treatment of chemo-resistant human breast cancer.
Data availability
All data for this study are presented in this published article.
Acknowledgments
This work was supported in part by Zhejiang Medical Association (No 2013ZYC-A134), Technology Division of Taizhou (No 1501KY19), Department of Education of Zhejiang Province (No 201738081), and Department of Science and Technology of Zhejiang Province (No 2016C33230).
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. | ||
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Pan Y, Zhang F, Zhao Y, et al. Berberine enhances chemosensitivity and induces apoptosis through Dose-orchestrated AMPK signaling in breast cancer. J Cancer. 2017;8(9):1679–1689. | ||
Morin PJ. Drug resistance and the microenvironment: nature and nurture. Drug Resist Updat. 2003;6(4):169–172. | ||
Bentires-Alj M, Dejardin E, Viatour P, et al. Inhibition of the NF-kappa B transcription factor increases Bax expression in cancer cell lines. Oncogene. 2001;20(22):2805–2813. | ||
Inta I, Paxian S, Maegele I, et al. Bim and Noxa are candidates to mediate the deleterious effect of the NF-kappa B subunit RelA in cerebral ischemia. J Neurosci. 2006;26(50):12896–12903. | ||
Saidi SA, Holland CM, Charnock-Jones DS, Smith SK. In vitro and in vivo effects of the PPAR-alpha agonists fenofibrate and retinoic acid in endometrial cancer. Mol Cancer. 2006;5:13. | ||
Lian X, Gu J, Gao B, et al. Fenofibrate inhibits mTOR-p70S6K signaling and simultaneously induces cell death in human prostate cancer cells. Biochem Biophys Res Commun. 2018;496(1):70–75. | ||
Tao T, Zhao F, Xuan Q, Shen Z, Xiao J, Shen Q. Fenofibrate inhibits the growth of prostate cancer through regulating autophagy and endoplasmic reticulum stress. Biochem Biophys Res Commun. 2018;503(4):2685–2689. | ||
Li T, Zhang Q, Zhang J, et al. Fenofibrate induces apoptosis of triple-negative breast cancer cells via activation of NF-κB pathway. BMC Cancer. 2014;14:96. | ||
Tsai SC, Tsai MH, Chiu CF, et al. AMPK-dependent signaling modulates the suppression of invasion and migration by fenofibrate in CAL 27 oral cancer cells through NF-κB pathway. Environ Toxicol. 2016;31(7):866–876. | ||
Hu D, Su C, Jiang M, et al. Fenofibrate inhibited pancreatic cancer cells proliferation via activation of p53 mediated by upregulation of LncRNA MEG3. Biochem Biophys Res Commun. 2016;471(2):290–295. | ||
Goncalves MD, Hwang SK, Pauli C, et al. Fenofibrate prevents skeletal muscle loss in mice with lung cancer. Proc Natl Acad Sci U S A. 2018;115(4):E743–E752. | ||
Murad H, Collet P, Huin-Schohn C, et al. Effects of PPAR and RXR ligands in semaphorin 6B gene expression of human MCF-7 breast cancer cells. Int J Oncol. 2006;28(4):977–984. | ||
Han DF, Zhang JX, Wei WJ, et al. Fenofibrate induces G0/G1 phase arrest by modulating the PPARα/FoxO1/p27 kip pathway in human glioblastoma cells. Tumour Biol. 2015;36(5):3823–3829. | ||
Srinivas KP, Viji R, Dan VM, et al. DEPTOR promotes survival of cervical squamous cell carcinoma cells and its silencing induces apoptosis through downregulating PI3K/AKT and by up-regulating p38 MAP kinase. Oncotarget. 2016;7(17):24154–24171. | ||
Miyata S, Wang LY, Kitanaka S. 3EZ, 20Ac-ingenol induces cell-specific apoptosis in cyclin D1 over-expression through the activation of ATR and downregulation of p-Akt. Leuk Res. 2018;64:46–51. | ||
Shah MA, Schwartz GK. Cell cycle-mediated drug resistance: an emerging concept in cancer therapy. Clin Cancer Res. 2001;7(8):2168–2181. | ||
Okon IS, Zou MH. Mitochondrial ROS and cancer drug resistance: Implications for therapy. Pharmacol Res. 2015;100:170–174. | ||
Hientz K, Mohr A, Bhakta-Guha D, Efferth T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget. 2017;8(5):8921–8946. | ||
Kusunoki M, Sato D, Tsutsumi K, Tsutsui H, Nakamura T, Oshida Y. Black soybean extract improves lipid profiles in fenofibrate-treated type 2 diabetics with postprandial hyperlipidemia. J Med Food. 2015;18(6):615–618. | ||
Oikawa S, Yamashita S, Nakaya N, Sasaki J, Kono S, for the Effect of Fenofibrate and Ezetimibe Combination Treatment on Lipid (EFECTL) Study Investigators. Efficacy and safety of long-term coadministration of fenofibrate and ezetimibe in patients with combined hyperlipidemia: results of the EFECTL study. J Atheroscler Thromb. 2017;24(1):77–94. | ||
Flores-Castillo C, Zamora-Pérez JÁ, Carreón-Torres E, et al. Atorvastatin and fenofibrate combination induces the predominance of the large HDL subclasses and increased apo AI fractional catabolic rates in New Zealand White rabbits with exogenous hypercholesterolemia. Fundam Clin Pharmacol. 2015;29(4):362–370. | ||
Piwowarczyk K, Wybieralska E, Baran J, et al. Fenofibrate enhances barrier function of endothelial continuum within the metastatic niche of prostate cancer cells. Expert Opin Ther Targets. 2015;19(2):163–176. | ||
Gong Y, Shao Z, Fu Z, et al. Fenofibrate inhibits cytochrome P450 epoxygenase 2C activity to suppress pathological ocular angiogenesis. EBioMedicine. 2016;13:201–211. | ||
Lian X, Wang G, Zhou H, Zheng Z, Fu Y, Cai L. Anticancer properties of fenofibrate: a repurposing use. J Cancer. 2018;9(9):1527–1537. | ||
Luty M, Piwowarczyk K, Wróbel T, et al. PO-235 Fenofibrate overcomes the drug-resistance of human prostate cancer cells. ESMO Open. 2018;3(Suppl 2):A112. | ||
Hsu SY, Kaipia A, Mcgee E, Lomeli M, Hsueh AJ. Bok is a pro-apoptotic Bcl-2 protein with restricted expression in reproductive tissues and heterodimerizes with selective anti-apoptotic Bcl-2 family members. Proc Natl Acad Sci U S A. 1997;94(23):12401–12406. | ||
Ke FFS, Vanyai HK, Cowan AD, et al. Embryogenesis and adult life in the absence of intrinsic apoptosis effectors Bax, Bak, and Bok. Cell. 2018;173(5):1217–1230. | ||
Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–290. | ||
Benbrook DM, Masamha CP. The pro-survival function of Akt kinase can be overridden or altered to contribute to induction of apoptosis. Curr Cancer Drug Targets. 2011;11(5):586–599. | ||
Yang G, Xiao X, Rosen DG, et al. The biphasic role of NF-kappaB in progression and chemoresistance of ovarian cancer. Clin Cancer Res. 2011;17(8):2181–2194. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.