")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Fatostatin in Combination with Tamoxifen Induces Synergistic Inhibition in ER-Positive Breast Cancer
Authors Liu Y, Zhang N, Zhang H, Wang L, Duan Y, Wang X, Chen T, Liang Y , Li Y, Song X, Li C, Han D, Chen B, Zhao W, Yang Q
Received 29 March 2020
Accepted for publication 31 July 2020
Published 26 August 2020 Volume 2020:14 Pages 3535—3545
DOI https://doi.org/10.2147/DDDT.S253876
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Manfred Ogris
Ying Liu, 1,* Ning Zhang, 1,* Hanwen Zhang, 1 Lijuan Wang, 2 Yi Duan, 1 Xiaolong Wang, 1 Tong Chen, 1 Yiran Liang, 1 Yaming Li, 1 Xiaojin Song, 1 Chen Li, 1 Dianwen Han, 1 Bing Chen, 2 Wenjing Zhao, 2 Qifeng Yang 1, 2
1Department of Breast Surgery, Qilu Hospital of Shandong University, Ji’nan, Shandong, People’s Republic of China; 2Pathology Tissue Bank, Qilu Hospital of Shandong University, Ji’nan, Shandong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qifeng Yang Email [email protected]
Background: Tamoxifen is the cornerstone of adjuvant therapy for hormone receptor-positive breast cancer. Despite its efficacy, limited drug sensitivity and endocrine resistance remain the important clinical challenges. The main objective of this study was to investigate fatostatin, which was found to sensitize breast cancer to the antitumour effect of tamoxifen both in vitro and in vivo.
Methods: Fatostatin-induced ER degradation was detected by immunoprecipitation assay. The antitumour effect of fatostatin and tamoxifen on MCF-7 and T47D cells was assessed by MTT and colony forming assays. Cell cycle arrest was detected by flow cytometric analysis. Apoptosis was detected by annexin V/propidium iodide double staining and TUNEL assay. Autophagy was detected by MDC assay and acridine orange staining. Migration and invasion assays were performed using a Transwell system, and the efficacy of the synergistic use of fatostatin and tamoxifen in vivo was evaluated using an MCF-7 xenograft model in BALB/c nu/nu female mice.
Results: The synergistic use of fatostatin and tamoxifen significantly suppressed cell viability and invasion, induced cell cycle arrest, and regulated apoptosis and autophagy in MCF-7 and T47D cell lines via PI3K-AKT-mTOR signalling. Additionally, the expression levels of Atg7/12/13, beclin and LC3B increased while p-mTOR and P62 expression levels decreased after treatment with fatostatin and tamoxifen. Tumor growth in the xenograft model was suppressed significantly with the synergistic treatment of fatostatin and tamoxifen.
Conclusion: Fatostatin could induce ER degradation by K48-linked polyubiquitination, which was the key mechanism contributing to tamoxifen inhibition of PI3K-AKT-mTOR signalling in breast cancer. Fatostatin may have a promising clinical use for ER-positive breast cancer patients.
Keywords: tamoxifen, fatostatin, oestrogen-positive breast cancer, polyubiquitination
Corrigendum for this paper has been published
Introduction
Breast cancer alone accounted for 30% of all new cancer diagnoses in women in the US in 2019. An estimated 42,260 breast cancer deaths (41,760 women, 500 men) occurred in the US in 2019.1 Among all breast cancer patients, approximately two-thirds are ER (oestrogen receptor) positive. ER is believed to control the growth and death of breast cancer cells through oestrogen-regulated signal transduction, thus playing an important role in regulating the progression of ER-positive breast cancers.2–4 Treatment of premenopausal and postmenopausal patients with ER-positive breast cancer usually involves tamoxifen, which directly targets ER-mediated signalling, and ER antagonists.5 Endocrine therapy is the most important treatment modality in this subgroup of patients, and tamoxifen remains the standard front-line endocrine therapy complementary to surgery.6 However, the development of drug resistance is a major cause of treatment failure for those patients who initially respond to tamoxifen. Approximately half of the patients who receive tamoxifen as first-line therapy for recurrent disease do not respond to the treatment due to low sensitivity.7
To overcome resistance to tamoxifen in clinical practice, new strategies are being developed to improve the response and maintain its antitumour function. Drug combinations may be a practical strategy to prolong the survival rate and improve quality of life. Previous studies have reported that combined therapy exhibited higher efficiency than monotherapy in various tumours.8,9 Some clinically used drugs that were not initially developed for the treatment of cancer may have antitumour effects and can be applied to enhance the sensitivity of classic antitumour drugs. For example, it was reported that the antidiabetic drug metformin could enhance tamoxifen-mediated tumour growth inhibition in ER-positive breast cancer.10 It has also been reported that rapamycin, an inhibitor of mammalian target of rapamycin (mTOR) signalling, enhanced the sensitivity of ER-positive breast cancer cells to tamoxifen both in vitro and in vivo.11 Furthermore, carnosic acid and tamoxifen synergistically induced apoptosis in a caspase-3-dependent manner in breast cancer.12 According to a recent study, palbociclib, a CDK4/6 inhibitor, showed an additive inhibitory effect on breast cancer cells when combined with tamoxifen.13
Fatostatin is a chemical inhibitor of the sterol regulatory element-binding protein (SREBP) pathway that directly binds (SREBP cleavage activating protein) SCAP and blocks its intracellular transport.14,15 Previously, we found that SREBP1 played an important role in breast cancer metastasis,16 which indicated that fatostatin might be a promising drug in breast cancer treatment. In the present study, we aimed to determine whether fatostatin could exert an additive effect on tamoxifen sensitivity. We found that fatostatin could enhance the sensitivity of ER-positive breast cancer cells to tamoxifen both in vitro and in vivo. Furthermore, our study demonstrated for the first time that fatostatin could degrade the ER protein by polyubiquitination pathways. Overall, combined therapy with fatostatin and tamoxifen may provide new insight into the development of therapeutic strategies for the treatment of ER-positive breast cancer.
Methods
Cell Lines and Materials
The breast cancer cell lines MCF-7, T47D, MDA-MB-231, and MDA-MB-468 were obtained from American Type Culture Collection (ATCC). All cells were cultured at 37°C and 5% CO2. The cell lines were characterized by Genetic Testing Biotechnology Corporation using short tandem repeat (STR) markers. Tamoxifen (Sigma-Aldrich, USA) was dissolved in ethanol and ethanol was used as the vehicle control. Fatostatin (Selleck, USA) was dissolved in DMSO and DMSO was used as the vehicle control.
Plasmid Transfection
MCF‑7 and T47D cells were transfected with plasmids using Lipofectamine 2000 according to the manufacturer’s instructions. All the plasmids including pCMV-Flag-ER, pCMV-HA, pCMV-HA-UB, pCMV-HA-K48 and pCMV-HA-K63 were constructed by researchers in our lab. The cells were cultured in a 6-well plate for 24 to 48 h, and the expression level was detected by Western blot analysis.
Western Blot and Immunoprecipitation Assays
Proteins were collected from different cell groups lysed in lysis buffer in the presence of protease inhibitors, and Western blotting was performed using the standard method. For the co-immunoprecipitation assay, cells were harvested at 48 h posttransfection and lysed in NP40 lysis buffer for 30 minutes on ice. The indicated primary antibody and control IgG were added to the lysate separately and incubated on a rotator at 4°C for 1 h. Afterwards, 20 µL protein A/G agarose beads were added and incubated at 4°C overnight on a rotator. The immunoprecipitation assay was examined using the indicated primary antibodies in the same way as the immunoblotting assay.
Cell Viability Assay and Colony Formation Assay
MCF-7 and T47D cells were cultured in 96-well plates with different concentrations of fatostatin and tamoxifen. Afterwards, MTT was added to each well and incubated for another 4 h at 37°C. The absorbance values were obtained using a microplate reader (Perkin Elmer) at 570 nm after DMSO incubation. For the colony formation assay, the cells mentioned above were seeded in six-well plates. After 24 h, the cells were treated with fatostatin and/or tamoxifen for 48 h. Then, the medium was replaced, and the cells were allowed to grow for 15 days. Finally, cells were fixed and stained with 1% crystal violet solution.
TUNEL Assay
TUNEL staining was performed according to the manufacturer’s instructions. After treatment, the cells were fixed with 4% paraformaldehyde in phosphate-buffered saline, rinsed with PBS, and then permeabilized with 0.1% Triton X-100 for 2 minutes on ice followed by the application of the TUNEL reagents for 1 h at 37°C. The TUNEL-positive cells were imaged using fluorescence microscopy.
MDC and Acridine Orange Staining
MCF-7 and T47D cells were treated with fatostatin and tamoxifen as described. First, the culture medium was discarded, 1× PBS was added for washing twice, and MDC or acridine orange staining solution was added to each well. After 15 minutes of staining in warm and dark conditions, the staining solution was discarded, and the cells were washed with 1×PBS three times, after which the cell slides were placed in glass slides. The slides were observed under a fluorescence microscope.
Migration and Invasion Assays
The Transwell system (Corning Costar) was used in these assays. In the migration assay, cells were starved in serum-free medium for 12 h at 37°C. Then, medium with 20% FBS was added to the lower well of each chamber, and cells suspended in serum-free medium were added to the upper Transwell inserts (BD Biosciences). After incubation for 48 h, the total number of cells adhering to the lower surface of the membrane was quantified in six representative fields. The invasion assay was performed in the same way as the migration assay except that the membrane was coated with Matrigel (BD Biosciences).
Xenograft Tumorigenicity Assay and Immunohistochemical Analysis
Cells were injected subcutaneously into 4-week-old BALB/c nu/nu female mice. For the xenograft tumorigenicity assay of MCF-7 cells, a 17β-oestradiol pellet was implanted into each mouse 5 days before injection. After 2 days, the mice were randomly assigned to four groups. The fatostatin group was given 15mg/kg intraperitoneal (i.p.) injections for 30 days, while the tamoxifen dosage was 5 mg/kg administered by gavage once every three days. Tumour growth was measured every 3 days, and tumour volume was calculated using the following equation: volume=(width2×length)/2. After 30 days, the mice were sacrificed, and the xenografts were removed for immunohistochemical staining and Western blotting, which were performed using standard methods.
Statistical Analysis
SPSS software (version 18.0) was used for statistical analysis. One-way ANOVA were performed to determine significance. Statistical differences are indicated as *P < 0.05, **P < 0.01 and ***P < 0.001. All quantitative data are presented as the mean ± SD from three independent experiments.
Results
The Degradation of ER Induced by Fatostatin is Ubiquitination-Dependent
To demonstrate whether the sensitivity to fatostatin was restricted to ER-positive breast cancer cells, we used ER-positive (MCF-7 and T47D) and ER-negative (MDA-MB-231 and MDA-MB-468) cell lines in MTT assays (Figure 1A). Fatostatin inhibited the growth of ER-positive breast cancer cells more effectively than that of ER-negative breast cell lines. These results showed that the inhibitory effect of fatostatin was more potent in ER-positive breast cancer cell lines. To further explore whether fatostatin functioned by affecting ER expression, we performed Western blotting and found that fatostatin could reduce the expression of ER at the protein level in a time-dependent and concentration-dependent manner (Figure 1B). Then, we wondered whether deregulated ER expression resulted from the deregulation of protein synthesis or degradation. Thus, we set different time periods of treatments with protein synthesis inhibitors (actinomycin, CHX), protease inhibitors (MG132) and lysosome inhibitors (chloroquine) in combination with fatostatin. The results showed that fatostatin could accelerate the degradation of ER through the proteasome-mediated pathway (Figure 1C and D). To verify whether fatostatin could induce ER degradation via the ubiquitination pathway, co-immunoprecipitation (co-IP) and ubiquitination assays were performed. We chose K48 and K63 sites because these two sites occurred most frequently in the polyubiquitination pathway. The results demonstrated that fatostatin accelerated ER degradation through ubiquitination at the K48 site (Figure 1E and F), indicating that fatostatin might be a new strategy for ER degradation.
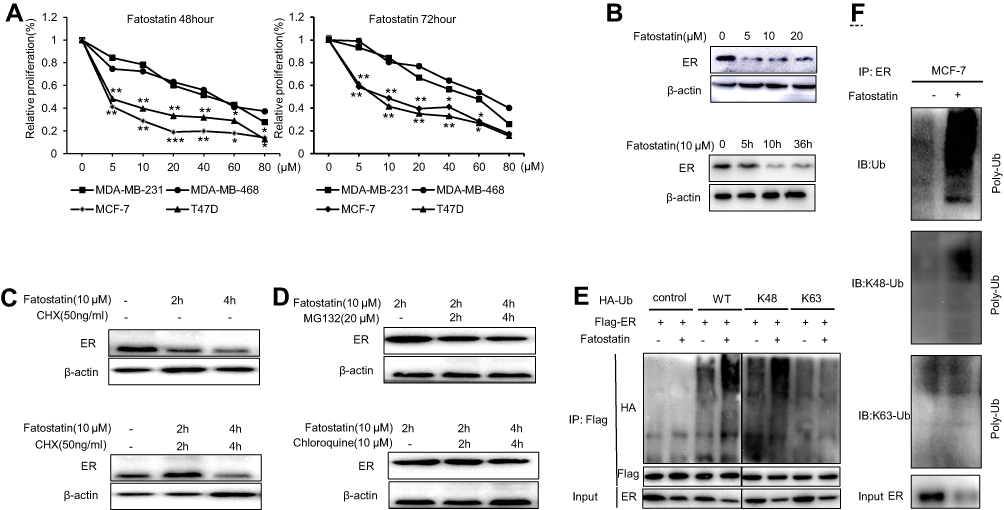 |
Figure 1 The mechanism of degradation of ER induced by fatostatin. (A) T47D, MCF-7, MDA-MB-231 and MDA-MB-468 cells were treated with fatostatin at various concentrations (0, 5, 10, 20, 40, 60, 80 µM). After 48 h and 72 h, cell viability was measured by MTT assays. (B) MCF-7 cells were treated with fatostatin in a time- and concentration-dependent manner and then measured by Western blot. MCF-7 cells were treated with CHX (C), MG132 and chloroquine (D) for 0, 2, and 4 h and co-treated with fatostatin at the same time point. Protein lysates were collected and subjected to Western blot assay for ER protein level. (E) Total protein was collected from MCF-7 cells transfected with pCMV-Flag-ER, pCMV-HA, pCMV-HA-UB, pCMV-HA-K48 and pCMV-HA-K63 and then treated with fatostatin for 48h. The cells were immunoprecipitated with Flag antibody and immunoblotted for HA. (F) Total protein was collected from MCF-7 cells and then treated with fatostatin for 48h. The cells were immunoprecipitated with ER antibody and immunoblotted for Ub antibody, K48-specific antibody and K63-specific antibody. All of the experiments were performed in triplicate, and the data are presented as the mean ± SD of three separate experiments. The data are representative of independent experiments (means ± SD) using one-way analysis of variance (ANOVA) to analyze the differences among groups. *p < 0.05; **p < 0.01; ***p < 0.001 vs. the control group. Abbreviations: Tam, tamoxifen; Fato, fatostatin. |
The Combination of Fatostatin and Tamoxifen Reduced the Viability and Growth of ER-Positive Breast Cancer Cells
To further investigate whether fatostatin can enhance the tamoxifen sensitivity of ER-positive breast cancer cell lines, we exposed the cells to fatostatin and tamoxifen in a serial concentration and time gradient and examined the effects of the combination of tamoxifen and fatostatin on the viability of MCF-7 and T47D cells with the MTT assay. As shown in Figure 2A, after 48 h, the inhibitory rate observed with the combined treatment of fatostatin (60 μM) plus tamoxifen (25 μM) reached 78% compared to that observed with treatment with tamoxifen alone (24%) in T47D cells. Likewise, treatment with fatostatin (20 μM) in combination with tamoxifen (15 μM) suppressed cell growth by approximately 55% compared to treatment with tamoxifen alone in MCF-7 cells. In order to evaluate whether fatostatin and tamoxifen worked in a synergistic manner, a combination index (CI) was determined by using the computer software CompuSyn. As shown in Figure S1, CI of fatostatin and tamoxifen in 10 μM is less than 1 and could play an synergistic role in combination treatment (CI<1) with the minimal toxicity, which illustrated these two drugs have combination effect and demonstrated that fatostatin could enhance the sensitivity of ER-positive breast cancer cells to tamoxifen. We further verified that the combined treatment significantly inhibited T47D and MCF-7 cells, as demonstrated by the clone formation assay (Figure 2B and C). These results demonstrated that fatostatin was capable of enhancing the inhibitory effect of tamoxifen on the growth of T47D and MCF-7 cells.
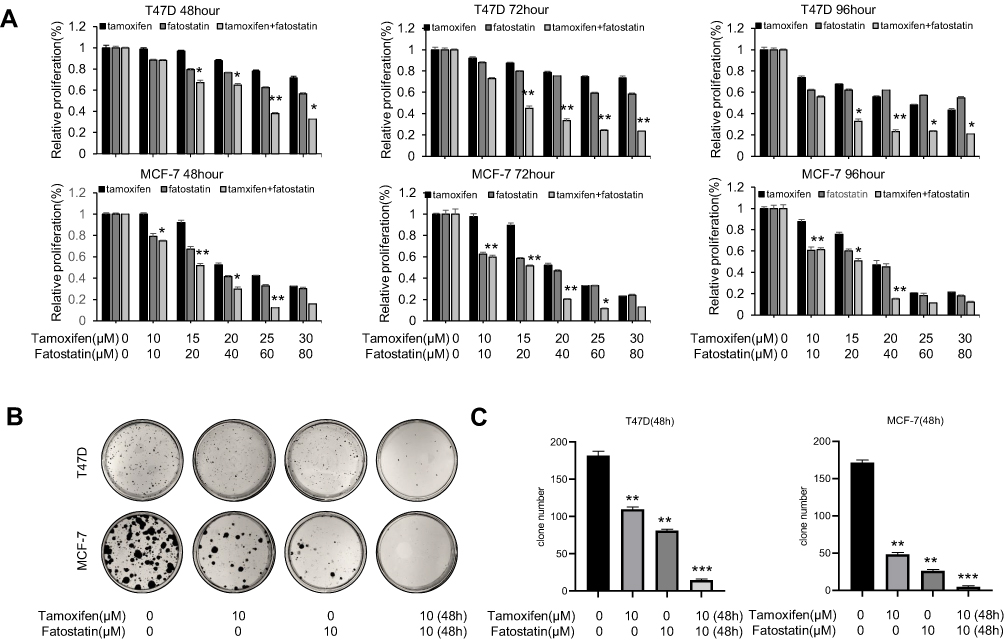 |
Figure 2 The combination of fatostatin and tamoxifen reduced the viability and growth of ER-positive breast cancer cells. (A) T47D and MCF-7 cells were treated with tamoxifen at various concentrations (0, 10, 15, 20, 25, 30 µM), and fatostatin (0, 10, 20, 40, 60, 80 µM). After 48 h, 72 h and 96 h, cell viability was measured by the MTT assay. (B) Colony formation assay showed growth inhibition of each treated group. Representative images of colonies stained with crystal violet solution. (C) T47D and MCF-7 cell colonies were counted after treatment with tamoxifen and/or fatostatin. All experiments were performed in triplicate, and the data are presented as the mean ± SD of three separate experiments. The data are representative of independent experiments (means ± SD) using one-way analysis of variance (ANOVA) to analyze the differences among groups. *p < 0.05; **p < 0.01; ***p < 0.001 vs. the control group. |
Fatostatin and Tamoxifen Induced Cell Cycle Arrest in T47D and MCF-7 Cells
To further understand the mechanisms of cell growth inhibition induced by fatostatin and tamoxifen, flow cytometry was used to determine the cell cycle distribution. ER-positive breast cancer cells were treated with tamoxifen (10 μM) and fatostatin (10 μM) for 48 h and then harvested for cell cycle analysis. Remarkably, tamoxifen arrested the cell cycle at G0/G1 phase, while fatostatin arrested the cell cycle at G2/M phase (Figure 3A and B). The combination of fatostatin and tamoxifen significantly decreased the proportion of both T47D and MCF-7 cells in S phase. Western blot analyses confirmed that fatostatin and tamoxifen could downregulate the expression of Cyclin D1 but increase the expression of Cyclin B1 (Figure 3C). The above results indicated that fatostatin and tamoxifen inhibited S phase entry in T47D and MCF-7 cells by downregulating Cyclin D1 and upregulating Cyclin B1 expression.
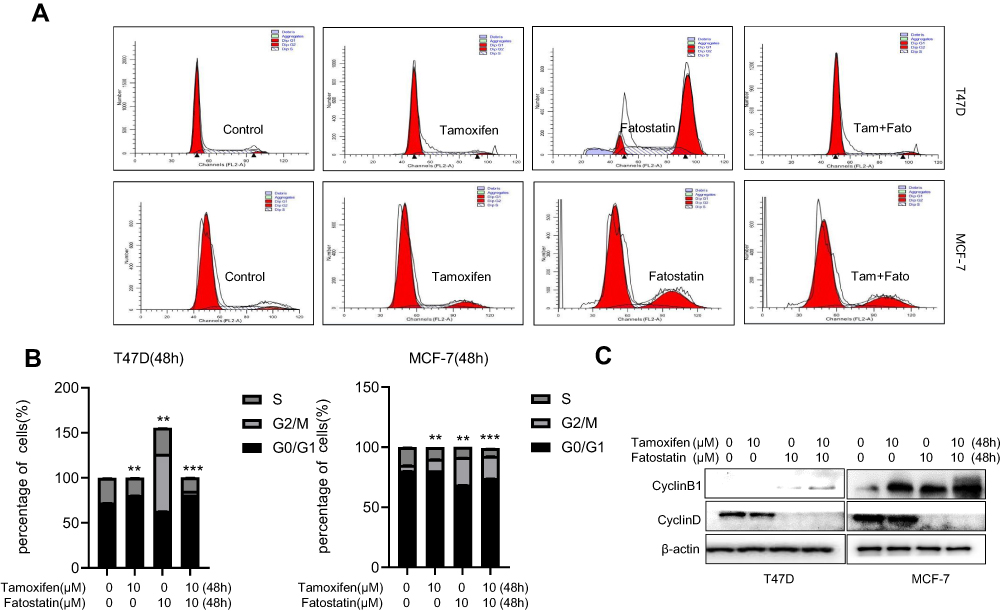 |
Figure 3 Fatostatin and tamoxifen suppressed cell cycle progression in T47D and MCF-7 cells. (A) Tamoxifen (0, 10 µM) and/or fatostatin (0, 10 µM) suppressed the cell cycle progression in MCF-7 cells and T47D cells. Cell cycle distribution in T47D and MCF-7 cells was assessed by flow cytometry after staining with propidium iodide (PI). (B) Cell cycle results were quantitatively analysed. (C) Western blot results of cyclin D1 and cyclin B1 protein expression levels in cells treated with tamoxifen (0, 10 µM) and/or fatostatin (0, 10 µM). All of the experiments were performed in triplicate, and the data are presented as the mean ± SD of three separate experiments. The data are representative of independent experiments (means ± SD) using one-way analysis of variance (ANOVA) to analyze the differences among groups. **p < 0.01; ***p < 0.001 vs. the control group. |
Fatostatin Enhanced Tamoxifen-Induced Apoptosis and Autophagy in T47D and MCF-7 Cells
Flow cytometry was used to test the apoptosis rate after different treatments in both cell lines. As a result, in T47D cells, treatment with fatostatin plus tamoxifen led to an 12.2% of early apoptosis compared to treatment with tamoxifen alone (5.67%) (Figure 4A and B). In MCF-7 cells, the apoptotic rate observed with combined treatment with tamoxifen and fatostatin was 9.14% compared to treatment with tamoxifen alone (8.0%) (Figure 4A and B). We also used the TUNEL assay to detect cell apoptosis induced by fatostatin and tamoxifen (Figure 4C). Western blot analyses confirmed that fatostatin and tamoxifen could downregulate the protein levels of p-AKT and Bcl-2 but increased the expression of cleaved PARP, cleaved caspase 3/8/9 and Bax (Figure 4D). These results demonstrated that fatostatin was capable of working synergistically with tamoxifen to enhance tamoxifen-induced apoptosis in T47D and MCF-7 cells.
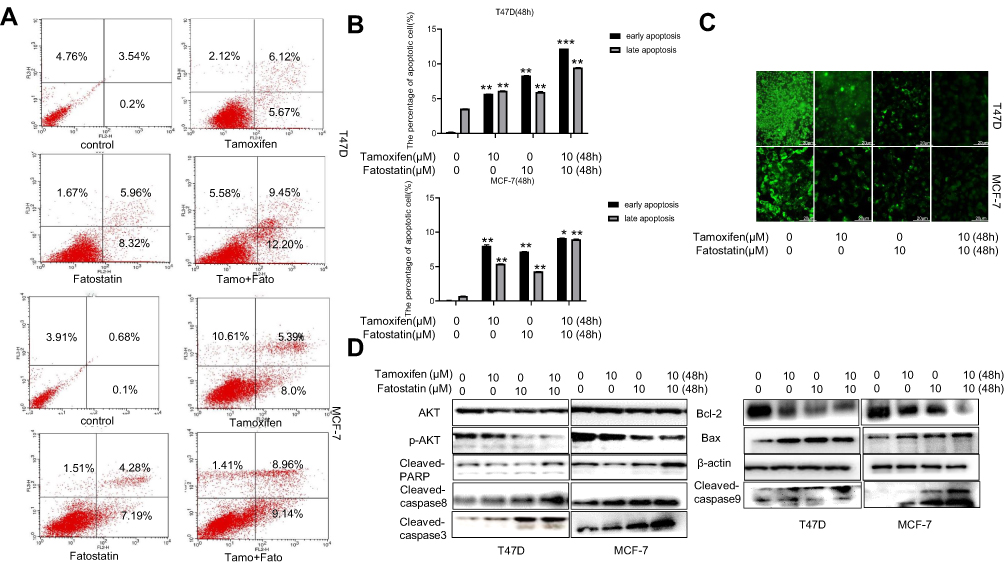 |
Figure 4 Fatostatin enhanced tamoxifen-induced apoptosis in T47D and MCF-7 cells. (A) Flow cytometric analysis of PI-annexin-V to quantify apoptosis in T47D and MCF-7 cells after treatment with tamoxifen (0, 10 µM) and/or fatostatin (0, 10 µM). (B) The results were quantitatively analysed. (C) Representative TUNEL staining by fluorescence microscopy of T47D and MCF-7 cells treated with tamoxifen (0, 10 µM) and/or fatostatin (0, 10 µM). Scale bars: 20 µm. (D) Western blot indicating the protein expression levels of AKT, p-AKT, cleaved PARP, cleaved caspase3/8/9, Bcl-2 and Bax. Columns are the average of three independent experiments. The data are representative of independent experiments (means ± SD) using one-way analysis of variance (ANOVA) to analyze the differences among groups. *p < 0.05; **p < 0.01 vs. the control group. |
Fatostatin Enhanced Tamoxifen-Induced Autophagy in T47D and MCF-7 Cells
Fatostatin also synergized with tamoxifen to induce autophagy in ER-positive breast cancer cells. MDC staining and A/O staining assays were used to observe autophagy in ER-positive breast cancer cells treated with fatostatin and tamoxifen, and the results showed that the combination of fatostatin and tamoxifen could promote autophagy in ER-positive breast cancer cells (Figure 5A and B). We also used Western blotting to demonstrate that the combination of the two drugs could induce autophagy by activating the mTOR pathway. More specifically, the expression levels of Atg7/12/13, Beclin and LC3B increased while p-mTOR and P62 expression levels decreased after treatment with fatostatin and tamoxifen (Figure 5C). These results indicated that fatostatin was capable of functioning synergistically with tamoxifen to enhance tamoxifen-induced autophagy in T47D and MCF-7 cells.
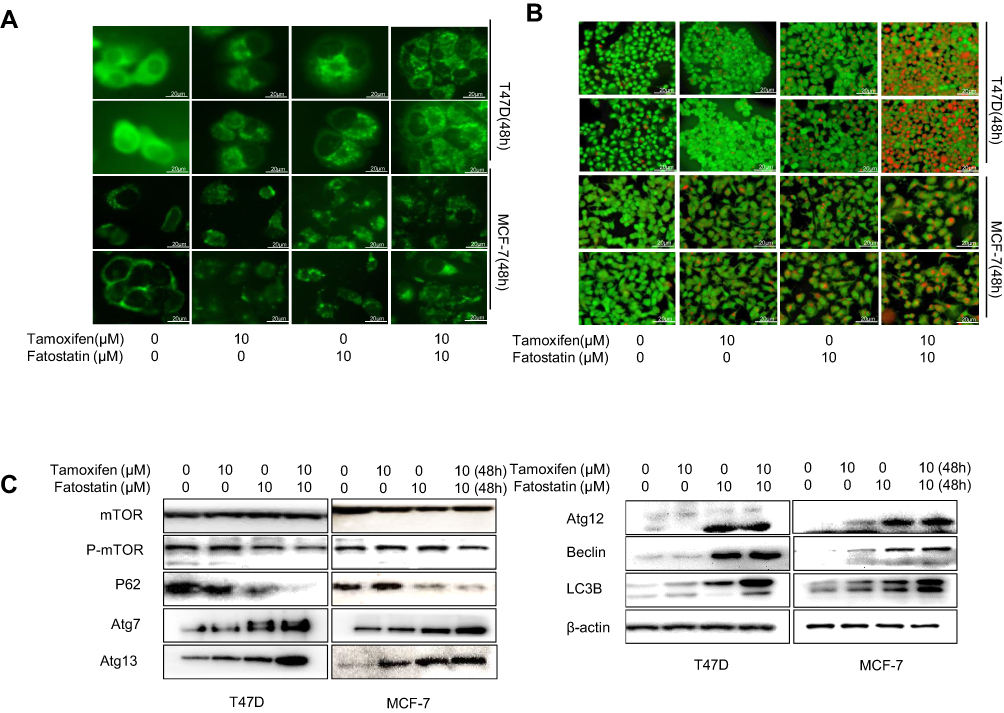 |
Figure 5 Fatostatin enhanced tamoxifen-induced autophagy in T47D and MCF-7 cells. (A) T47D and MCF-7 cells stained with MDC after treatment with tamoxifen (0, 10 µM) and/or fatostatin (0, 10 µM) and then measured by fluorescence microscopy. Scale bars: 20 µm. (B) T47D and MCF-7 cells stained with O/A after treatment with tamoxifen (0, 10µM) and/or fatostatin (0, 10µM) measured by fluorescence microscopy. Scale bars: 20 µm. (C) Western blot results of autophagy-associated protein expression levels treated with tamoxifen (0, 10 µM) and/or fatostatin (0, 10 µM). |
The Combination of Tamoxifen and Fatostatin Suppressed the Migration and Invasion of ER-Positive Breast Cancer Cells
The impact of fatostatin, tamoxifen and the combination of the two drugs on the migration and invasion ability of ER-positive breast cancer cells was investigated by the wound healing assay (Figure 6A and C) and Transwell assay (Figure 6 and D). These results showed that the combination of the two drugs significantly reduced the migration and invasion ability of both T47D and MCF-7 cells.
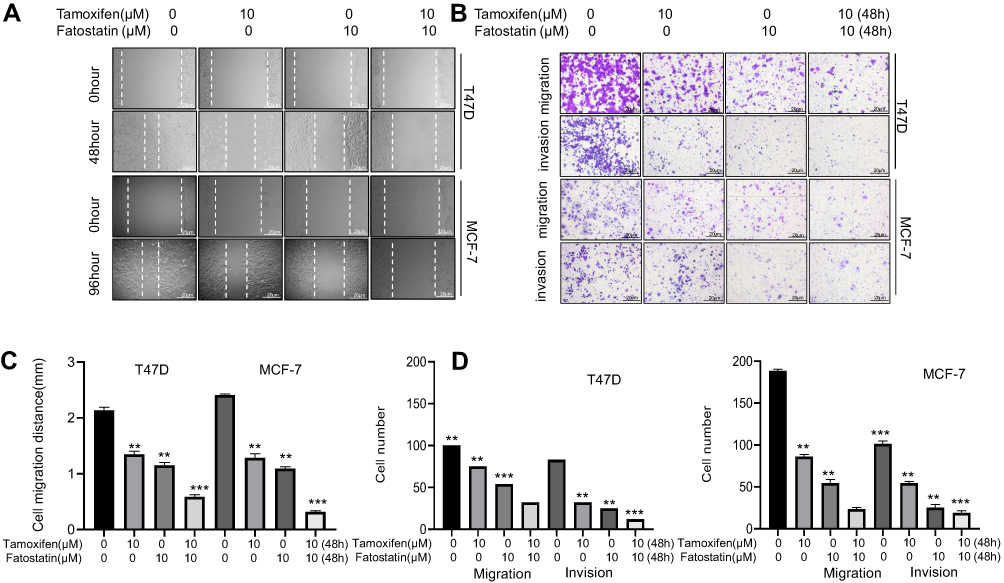 |
Figure 6 The combination of tamoxifen and fatostatin suppressed metastasis in ER-positive breast cancer. (A) Wound healing assays showed that cell mobility was strongly inhibited by synergistic treatment with tamoxifen and fatostatin. Scale bars: 20 µm. (B) Transwell assays showed that fatostatin sensitized cells to tamoxifen to suppress migration and invasion in both T47D and MCF-7 cells. Scale bars: 20 µm. Migration (C) and invasion (D) of the above cells were quantitatively analysed. Crystal violet solution was used as a staining agent, and cell numbers were counted in six representative fields. All experiments were performed in triplicate, and the data are presented as the mean ± SD of three separate experiments. The data are representative of independent experiments (means ± SD) using one-way analysis of variance (ANOVA) to analyze the differences among groups. **p < 0.01; ***p < 0.001 vs. the control group. |
Effects of Fatostatin and Tamoxifen on the Growth of ER-Positive Breast Cancer Cells in vivo
To determine whether combined therapy was superior to monotherapy in vivo, MCF-7 breast cancer cells were subcutaneously injected into the right sides of BALB/c nu/nu mice. As shown in Figure 7A, the volume of transplanted tumours in the combined treatment group was significantly smaller than that in the control group and the single drug treatment group. The combination of fatostatin and tamoxifen significantly inhibited the tumour growth rate (Figure 7B). Moreover, to explore the mechanism of the inhibitory effect of the drug combination, we measured LC3B, Ki67 and ER protein levels by immunohistochemical staining and found that the expression levels of Ki67 and ER were significantly decreased, while LC3B levels were obviously increased, in the combined treatment group (Figure 7D). In addition, according to the Western blot assay (Figure 7C), we discovered that compared with the single drug group, the combined treatment increased the level of apoptosis-associated proteins, such as cleaved PARP, and decreased the anti-apoptotic Bcl-2 protein level. It also regulated the levels of cell cycle-related proteins, such as decreasing Cyclin D1 and increasing Cyclin B1 levels. At the same time, we found that combined treatment could impact autophagy because it decreased p-AKT, p-mTOR and P62 protein levels and increased Atg7 and LC3B expression levels. Notably, ER decreased significantly at the protein level. These results suggested that the combination of fatostatin and tamoxifen may induce apoptosis, cell cycle arrest and autophagy in MCF-7 cells.
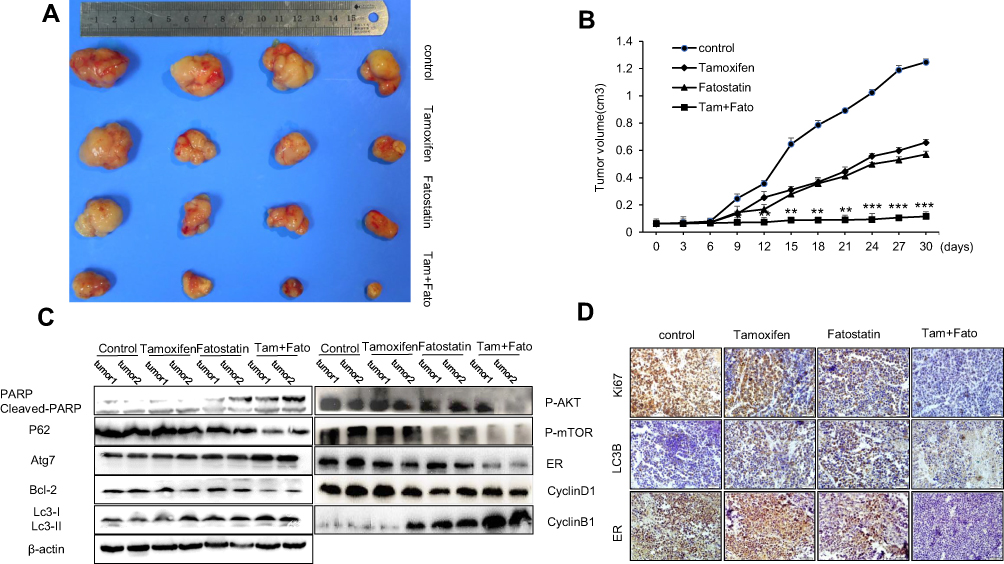 |
Figure 7 Effects of fatostatin and tamoxifen on the growth of ER-positive breast cancer in vivo. (A) Images of the xenograft tumours in nude mice implanted with MCF-7 cells (1×107) and treated with PBS, tamoxifen, fatostatin or a combination of tamoxifen and fatostatin (n=4 in each group). (B) Tumour growth curves of each treated group. The tumour volumes were assessed on the indicated days. (C) Western blot assay showing the protein expression levels of cleaved PARP, P62, Atg7, Bcl-2, LC3B, p-AKT, p-mTOR, ER, Cyclin D1 and Cyclin B1 among each group. (D) IHC analysis of Ki67, LC3B and ER expression in breast cancer at 20x magnification. Scale bars: 20 µm. The data are representative of independent experiments (means ± SD) using one-way analysis of variance (ANOVA) to analyze the differences among groups. *p < 0.05; **p < 0.01; ***p < 0.001 vs. the control group. |
Discussion
ER plays a pivotal role in breast cancer initiation and progression and is used not only as a prognostic marker but also as a predictor of the response to endocrine therapies.17 Recent studies have indicated that ER activation by oestrogen promotes the proliferation and occurrence of breast cancer cells by triggering downstream signalling pathways such as MAPK and PI3K,17,18 so endocrine therapy is the main treatment method for patients with ER-positive breast cancer.19 In recent years, endocrine therapies for breast cancer have been designed to interrupt oestrogen signalling by either decreasing the amount of oestrogen available for binding or by blocking ERs, which is still the most successful systemic therapy in the management of ER-positive breast cancer.20 Tamoxifen, a selective ER modulator (SERM) that antagonizes oestrogens for ER binding, is the first-line treatment for both early and advanced breast cancer patients.21 However, some patients gradually develop chemoresistance, which limits the efficacy of the treatment, especially for metastatic breast cancer patients who are treated with tamoxifen.22 Therefore, drug synergistic therapies have become an important strategy for tumour progression.23–25
Previously, our study discovered that depletion of SREBP1 in breast cancer cells was sufficient to decrease the metastatic ability of cancer cells by interfering with the EMT programme.16 SREBP1, the main regulator in lipid metabolism,26 also plays an important role in driving endocrine resistance in invasive lobular breast cancer.27 Recently, fatostatin was discovered as a chemical inhibitor of the SREBP pathway and was shown to inhibit the maturation and nuclear translocation of SREBPs.28 One study revealed that fatostatin could suppress prostate cancer development by blocking SREBP-regulated metabolic pathways and androgen receptor (AR) signalling in vitro and in vivo,29 and it was also reported that fatostatin could decrease cell viability and proliferation in pancreatic cancer.30 Based on our previous research, we decided to explore whether fatostatin in combination with tamoxifen could induce synergistic inhibition in breast cancer cancers. Our study reports for the first time that the combination of fatostatin and tamoxifen could downregulate the PI3K-AKT-mTOR pathway and significantly influence the phosphorylation of AKT and mTOR at the protein level. The PI3K/AKT/mTOR pathway is an important signalling pathway that is involved in many cell activities,31 such as survival, proliferation, growth, metastasis, energy metabolism and autophagy.32–34 We found that synergetic drugs of fatostatin and tamoxifen could induce autophagy by downregulating P62 while upregulating Atg7 and LC3B-II levels.35–37 Moreover, we also demonstrated that the combined drugs induced apoptosis by increasing cleaved PARP levels while decreasing the level of Bcl-2 and caused cell cycle arrest by deregulating the protein levels of Cyclin D1 and Cyclin B1. In conclusion, synergetic use of fatostatin and tamoxifen could suppress cell growth, induce cell cycle arrest, apoptosis and autophagy, and inhibit cell mobility in breast cancer cells.
In this study, we found that fatostatin could exert a more obvious inhibitory effect on ER-positive breast cancer cells than ER-negative breast cancer cells, which was consistent with the results by Brovkovych et al.38 In addition, both Western blotting and immunohistochemistry staining demonstrated that fatostatin could significantly downregulate ER, which is worth exploring further. We wondered whether the deregulated ER resulted from the deregulation of protein synthesis or protein degradation. Therefore, we set different time periods of treatments with protein synthesis inhibitors (actinomycin, CHX), protease inhibitors (MG132) and lysosome inhibitors (chloroquine) in combination with fatostatin. The results showed that fatostatin could accelerate the degradation of ER through the proteasome-mediated pathway, suggesting that fatostatin may induce ER degradation in a ubiquitin-mediated manner.39 Accordingly, many studies have indicated that proteasome-mediated ER degradation is an important step in maintaining ER expression and transactivation function.40 It has been reported that monoubiquitination regulates receptor internalization, lysosomal degradation and protein circulation.41 In addition to monoubiquitination, other modification types have been identified, such as K48-linked and K63-linked polyubiquitination.42 By co-immunoprecipitation assay, we verified that fatostatin could degrade ER by K48-linked rather than K63-linked polyubiquitination, which might be an important finding. In our study, we also found that fatostatin could downregulate the protein level of ER comparable to fulvestrant (a selective ER degrader used in the clinic), indicating that fatostatin may have a promising application in ER-positive breast cancer in the future.
In conclusion, our study found that fatostatin could increase the sensitivity to tamoxifen in ER-positive breast cancer cells in vivo and in vitro. The combination of fatostatin and tamoxifen could inhibit proliferation and invasiveness as well as induce apoptosis and autophagy in ER-positive breast cancer cells. Fatostatin in combination with tamoxifen might be a new therapeutic treatment for ER-positive breast cancer. Importantly, our study clarified that fatostatin degraded ER protein by K48-linked ubiquitination, which may shed light on the underlying mechanism. Overall, fatostatin may have a promising clinical use for ER-positive breast cancer patients.
Study Approval
All animal experiments were performed with the approval of the Ethics Committee on Scientific Research of Qilu Hospital, Shandong University. All experimental procedures and protocols were reviewed and approved by the Animal Care and Use Committee of Qilu Hospital of Shandong University and were in accordance with the Guide for the Care and Use of Laboratory Animals.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81502285, No. 81972475, No. 81874119, No. 81672613), National Key R&D Program of China (No. 2018YFC0114705), China Postdoctoral Science Foundation (No. 2018M630787, No. 2019T120593), Key Research and Development Program of Shandong Province (No. 2019GSF108140) and Special Support Plan for National High Level Talents (Ten Thousand Talents Program).
Disclosure
The authors declare no conflicts of interest for this work.
References
1. DeSantis CE, Miller KD, Goding Sauer A, Jemal A, Siegel RL. Breast cancer statistics, 2019. CA Cancer J Clin. 2019;69(6):438–451. doi:10.3322/caac.21583
2. Morgan MM, Johnson BP, Livingston MK, et al. Personalized in vitro cancer models to predict therapeutic response: challenges and a framework for improvement. Pharmacol Ther. 2016;165:79–92. doi:10.1016/j.pharmthera.2016.05.007
3. Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9(9):631–643. doi:10.1038/nrc2713
4. Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi:10.1038/nature11412
5. Demir K, Unuvar T, Eren S, Abaci A, Bober E. Tamoxifen as first-line treatment in a premenarchal girl with juvenile breast hypertrophy. J Pediatr Adolesc Gynecol. 2010;23(5):e133–e136. doi:10.1016/j.jpag.2009.11.003
6. Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev. 2001;81(1):153–208. doi:10.1152/physrev.2001.81.1.153
7. Early Breast Cancer Trialists’ Collaborative, G. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365(9472):1687–1717. doi:10.1016/S0140-6736(05)66544-0
8. Early Breast Cancer Trialists’ Collaborative, G. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet.2011;378(9793):771–784. doi:10.1016/S0140-6736(11)60993-8
9. Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008;7(10):3129–3140. doi:10.1158/1535-7163.MCT-08-0013
10. Kumar BN, Rajput S, Dey KK, et al. Celecoxib alleviates tamoxifen-instigated angiogenic effects by ROS-dependent VEGF/VEGFR2 autocrine signaling. BMC Cancer. 2013;13(1):273. doi:10.1186/1471-2407-13-273
11. Hare SH, Harvey AJ. mTOR function and therapeutic targeting in breast cancer. Am J Cancer Res. 2017;7(3):383–404.
12. Han NN, Zhou Q, Huang Q, Liu KJ. Carnosic acid cooperates with tamoxifen to induce apoptosis associated with caspase-3 activation in breast cancer cells in vitro and in vivo. Biomed Pharmacother. 2017;89:827–837. doi:10.1016/j.biopha.2017.01.084
13. Fanning SW, Jeselsohn R, Dharmarajan V, et al. The SERM/SERD bazedoxifene disrupts ESR1 helix 12 to overcome acquired hormone resistance in breast cancer cells. Elife. 2018;7. doi:10.7554/eLife.37161
14. Choi Y, Kawazoe Y, Murakami K, Misawa H, Uesugi M. Identification of bioactive molecules by adipogenesis profiling of organic compounds. J Biol Chem. 2003;278(9):7320–7324. doi:10.1074/jbc.M210283200
15. Kamisuki S, Mao Q, Abu-Elheiga L, et al. A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem Biol. 2009;16(8):882–892. doi:10.1016/j.chembiol.2009.07.007
16. Zhang N, Zhang H, Liu Y, et al. SREBP1, targeted by miR-18a-5p, modulates epithelial-mesenchymal transition in breast cancer via forming a co-repressor complex with Snail and HDAC1/2. Cell Death Differ. 2019;26(5):843–859. doi:10.1038/s41418-018-0158-8
17. Su X, Xu X, Li G, Lin B, Cao J, Teng L. ER-alpha36: a novel biomarker and potential therapeutic target in breast cancer. Onco Targets Ther. 2014;7:1525–1533.
18. Hall JM, Couse JF, Korach KS. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem. 2001;276(40):36869–36872. doi:10.1074/jbc.R100029200
19. Renoir JM, Marsaud V, Lazennec G. Estrogen receptor signaling as a target for novel breast cancer therapeutics. Biochem Pharmacol. 2013;85(4):449–465. doi:10.1016/j.bcp.2012.10.018
20. Lumachi F, Luisetto G, MM Basso S, Basso U, Brunello A, Camozzi V. Endocrine therapy of breast cancer. Curr Med Chem. 2011;18(4):513–522. doi:10.2174/092986711794480177
21. Gradishar WJ. Tamoxifen–what next? Oncologist. 2004;9(4):378–384. doi:10.1634/theoncologist.9-4-378
22. Smith RA, Cokkinides V, Brooks D, Saslow D, Brawley OW. Cancer screening in the United States, 2011: a review of current American Cancer Society guidelines and issues in cancer screening. CA Cancer J Clin. 2011;61(1):8–30. doi:10.3322/caac.20096
23. Cho SK, Pedram A, Levin ER, Kwon YJ. Acid-degradable core-shell nanoparticles for reversed tamoxifen-resistance in breast cancer by silencing manganese superoxide dismutase (MnSOD). Biomaterials. 2013;34(38):10228–10237. doi:10.1016/j.biomaterials.2013.09.003
24. Motawi TK, Abdelazim SA, Darwish HA, Elbaz EM, Shouman SA. Modulation of tamoxifen cytotoxicity by caffeic acid phenethyl ester in MCF-7 breast cancer cells. Oxid Med Cell Longev. 2016;2016:3017108. doi:10.1155/2016/3017108
25. Darakhshan S, Ghanbari A, Gholami FR, Bidmeshki AP. Tamoxifen and tranilast show a synergistic effect against breast cancer in vitro. Bratisl Lek Listy. 2015;116(1):69–73.
26. Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol. 2017;13(12):710–730. doi:10.1038/nrendo.2017.91
27. Du T, Sikora MJ, Levine KM, et al. Key regulators of lipid metabolism drive endocrine resistance in invasive lobular breast cancer. Breast Cancer Res. 2018;20(1):106. doi:10.1186/s13058-018-1041-8
28. Shao W, Machamer CE, Espenshade PJ. Fatostatin blocks ER exit of SCAP but inhibits cell growth in a SCAP-independent manner. J Lipid Res. 2016;57(8):1564–1573. doi:10.1194/jlr.M069583
29. Li X, Chen YT, Hu P, Huang WC. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol Cancer Ther. 2014;13(4):855–866. doi:10.1158/1535-7163.MCT-13-0797
30. Sekar S, Gopalakrishnan V, Taghibiglou C. Sterol regulatory element-binding protein 1 inhibitors decrease pancreatic cancer cell viability and proliferation. Biochem Biophys Res Commun. 2017;488(1):136–140. doi:10.1016/j.bbrc.2017.05.023
31. de Melo AC, Paulino E, Garces AH. A review of mTOR pathway inhibitors in gynecologic cancer. Oxid Med Cell Longev. 2017;2017:4809751. doi:10.1155/2017/4809751
32. Zhu H, Liu Q, Tang J, et al. Alpha1-ACT functions as a tumour suppressor in hepatocellular carcinoma by Inhibiting the PI3K/AKT/mTOR signalling pathway via activation of PTEN. Cell Physiol Biochem. 2017;41(6):2289–2306. doi:10.1159/000475648
33. Zhang D, Wang S, Chen J, et al. Fibulin-4 promotes osteosarcoma invasion and metastasis by inducing epithelial to mesenchymal transition via the PI3K/Akt/mTOR pathway. Int J Oncol. 2017;50(5):1513–1530. doi:10.3892/ijo.2017.3921
34. Woo SU, Sangai T, Akcakanat A, Chen H, Wei C, Meric-Bernstam F. Vertical inhibition of the PI3K/Akt/mTOR pathway is synergistic in breast cancer. Oncogenesis. 2017;6(10):e385. doi:10.1038/oncsis.2017.86
35. Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov. 2017;16(7):487–511.
36. Shoji-Kawata S, Sumpter R, Leveno M, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494(7436):201–206. doi:10.1038/nature11866
37. White E. The role for autophagy in cancer. J Clin Invest. 2015;125(1):42–46. doi:10.1172/JCI73941
38. Brovkovych V, Izhar Y, Danes JM, et al. Fatostatin induces pro- and anti-apoptotic lipid accumulation in breast cancer. Oncogenesis. 2018;7(8):66. doi:10.1038/s41389-018-0076-0
39. Alferez DG, Simões BM, Howell SJ, Clarke RB. The role of steroid hormones in breast and effects on cancer stem cells. Curr Stem Cell Rep. 2018;4(1):81–94. doi:10.1007/s40778-018-0114-z
40. Dwane L, Gallagher WM, Chonghaile TN, O’Connor DP. The emerging role of non-traditional ubiquitination in oncogenic pathways. J Biol Chem. 2017;292(9):3543–3551. doi:10.1074/jbc.R116.755694
41. Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I. Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat Cell Biol. 2003;5(5):461–466. doi:10.1038/ncb983
42. Bist P, Cheong WS, Ng A, et al. E3 ubiquitin ligase ZNRF4 negatively regulates NOD2 signalling and induces tolerance to MDP. Nat Commun. 2017;8:15865. doi:10.1038/ncomms15865
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.