")
Back to Journals » Drug Design, Development and Therapy » Volume 15
Fasudil Dichloroacetate Alleviates SU5416/Hypoxia-Induced Pulmonary Arterial Hypertension by Ameliorating Dysfunction of Pulmonary Arterial Smooth Muscle Cells
Authors Liu P , Huang W, Ding Y , Wu J, Liang Z, Huang Z, Xie W, Kong H
Received 17 December 2020
Accepted for publication 18 March 2021
Published 22 April 2021 Volume 2021:15 Pages 1653—1666
DOI https://doi.org/10.2147/DDDT.S297500
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Ping Liu,1,* Wen Huang,1,* Yirui Ding,1 Jianbing Wu,2 Zhuangzhuang Liang,2 Zhangjian Huang,2 Weiping Xie,1 Hui Kong1
1Department of Respiratory and Critical Care Medicine, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, 210029, People’s Republic of China; 2State Key Laboratory of Natural Medicines, China Pharmaceutical University, Nanjing, 210009, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Weiping Xie; Hui Kong
Department of Respiratory and Critical Care Medicine, The First Affiliated Hospital of Nanjing Medical University, 300 Guangzhou Road, Nanjing, Jiangsu, 210029, People’s Republic of China
Tel +86-25-68136426
Fax +86-25-68136269
Email [email protected]; [email protected]
Background: Pulmonary arterial hypertension (PAH) is an incurable disease that urgently needs therapeutic approaches. Based on the therapeutic effects of fasudil and dichloroacetate (DCA) on PAH, we aimed to explore the effects and potential mechanism of a new salt, fasudil dichloroacetate (FDCA), in a SU5416 plus hypoxia (SuHx)-induced rat model of PAH.
Methods: The rat model of PAH was established by a single subcutaneous injection of SU5416 (20 mg/kg) followed by hypoxia (10% O2) exposure for 3 weeks. FDCA (15, 45, or 135 mg/kg i.g. daily) or the positive control, bosentan (100 mg/kg i.g. daily), were administered from the first day after SU5416 injection. After 3-week hypoxia, hemodynamic parameters, and histological changes of the pulmonary arterial vessels and right ventricle (RV) were assessed. Additionally, in vitro, the effects of FDCA (50 μM), compared with equimolar doses of fasudil, DCA, or fasudil+DCA, on the proliferation, migration, and contraction of human pulmonary arterial smooth muscle cell (PASMC) under hypoxia (1% O2) were evaluated.
Results: FDCA dose-dependently attenuated SuHx-induced PAH, with significant reductions in RV systolic pressure, pulmonary artery wall thickness, pulmonary vessel muscularization, perivascular fibrosis, as well as RV hypertrophy and fibrosis. In vitro, FDCA inhibited hypoxia-induced PASMC proliferation, migration, and contraction to a greater degree than fasudil or DCA alone by restoring mitochondrial function, reducing intracellular Ca2+, and inhibiting calcium/calmodulin-dependent kinase (Ca2+/CaMK) activity as well as Rho-kinase activity.
Conclusion: FDCA ameliorates hypoxia-induced PASMC dysfunction by inhibiting both Ca2+/CaMK and Rho-kinase signaling pathways, as well as maintaining mitochondrial homeostasis, thus alleviating SuHx-induced PAH.
Keywords: pulmonary arterial hypertension, fasudil dichloroacetate, pulmonary arterial smooth muscle cells, hypoxia, calcium
Introduction
Pulmonary arterial hypertension (PAH), a progressive lung vascular disease with a poor prognosis and limited therapeutic options, is characterized by vasoconstriction and vascular remodeling of the distal pulmonary arteries (PAs), which eventually result in right ventricular (RV) hypertrophy and heart failure.1,2 Despite the development of new therapies targeting the endothelin, nitric oxide, and prostacyclin pathways in the past two decades, there are still no curative treatments for PAH, making it a life-shortening disease.3 Strategies to improve outcomes need to be focused on innovative approaches for treating PAH.
Irreversible pulmonary vascular remodeling and reversible pulmonary vasoconstriction are the two main pathophysiological processes of PAH. Pulmonary arterial smooth muscle cell (PASMC) dysfunction is thought to be a dominant mechanism involved both pulmonary vascular remodeling and vasoconstriction. Biological characteristics of PASMCs from patients with PAH are similar to those of tumor cells, such as continuous proliferation, apoptosis resistance, excessive migration, and deregulation of cellular energetics,4 leading to thickening and remodeling of distal PAs. Moreover, PASMC directly drive the contraction of the vascular wall, thus regulating the size of the vascular lumen.5 The dysfunction of PASMCs in PAH are largely dependent on the intracellular calcium concentration ([Ca2+]i),6,7 which is controlled by complex regulatory mechanisms. Among these mechanisms, both mitochondrial function and Rho-kinase-mediated Ca2+ sensitization perform indispensable roles.6,8,9
In view of the complexity of the pathogenesis of PAH, combination therapy simultaneously targeting multiple targets has become an important recommendation for treating PAH.10 The Rho-kinase inhibitor fasudil is used to treat cerebral vasospasm after subarachnoid hemorrhage, coronary artery disease, and PAH. It inhibits vasoconstriction and spasm by antagonizing intracellular Ca2+ and blocking myosin light chain (MLC) phosphorylation. Emerging evidence suggests that fasudil alleviates pulmonary vascular remodeling in various experimental animal models of PAH,11,12 and it has exhibited therapeutic effects in patients with PAH, especially in patients with congenital heart defects and severe PAH.13–15 Furthermore, dichloroacetate (DCA), a metabolic modulator, has been reported to be able to prevent and reverse both chronic hypoxia- and monocrotaline-induced PAH by inhibiting pulmonary vasoconstriction and PASMC proliferation.16,17 Recently, DCA has been shown to improve the hemodynamics of patients with idiopathic PAH in clinical trials.18
Based on these findings, fasudil dichloroacetate (FDCA), a novel synthetic water-soluble oral drug synthesized from these two drugs, may have dual and synergistic effects by inhibiting the activation of Rho-kinase and regulating mitochondrial function. Our previous pharmacokinetic parameters demonstrated that FDCA enhanced the plasma concentrations of fasudil, and slightly prolonged the half-life.19 Compared to the traditional fasudil hydrochloride, salifying with dichloroacetic acid may slightly influence the binding of fasudil with the ROCK protein.19 However, the effects of FDCA on pulmonary vascular remodeling and vasoconstriction in PAH are still not well understood. Therefore, we investigated the effects of FDCA on pulmonary hemodynamics, and PA and RV remodeling in a SU5416 plus hypoxia (SuHx)-induced rat model of PAH. Additionally, the potential underlying cellular and molecular mechanisms were investigated in vitro.
Materials and Methods
Animals
All studies were carried out in line with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals (NIH publication no. 85–23, revised 1996) based on a protocol approved by the Institutional Animal Care and Use Committee of Nanjing Medical University (NJMU/IACUC-2005021).
Male Sprague-Dawley rats weighing about 250 g (Bikai Laboratory Animal Company, Shanghai, China) were randomly assigned to six experimental groups (n = 6–8 per group): control, SuHx, SuHx+FDCA (15 mg/kg), SuHx+FDCA (45 mg/kg), SuHx+FDCA (135 mg/kg), and SuHx+bosentan (100 mg/kg) as a positive control group.20 SU5416 (APExBIO, Houston, TX, USA) was dissolved in a solvent containing 0.5% (wt/vol) carboxymethylcellulose sodium, 0.9% (wt/vol) sodium chloride, 0.4% (vol/vol) polysorbate, and 0.9% (vol/vol) benzyl alcohol in deionized water. The SuHx-treated rats were subcutaneously injected with a single dose of SU5416 (20 mg/kg) and then exposed to 10% O2 hypoxia from the first day after SU5416 injection for 3 weeks using a hypoxic chamber (Aipu Instrument Equipment Co., Ltd, Hangzhou, China). The rats in the control group were maintained in room air after injection of solvent only. From the first day of the 3-week hypoxia period, the rats in the various experimental groups received saline (the vehicle control), FDCA (State Key Laboratory of Natural Medicine, China Pharmaceutical University), or bosentan (Widely Chemical Technology Co., Ltd, Hubei, China).
Hemodynamics Analysis and Right Heart Hypertrophy Assessment
Three weeks later, the rats were anesthetized by intraperitoneal injection of 2% sodium pentobarbital (Sigma-Aldrich, St. Louis, MO, USA). The anesthetized rats were fixed on the animal operating table, and a polyethylene catheter was placed in the RV via the right common jugular vein. Right heart catheterization was performed for RV systolic pressure (RVSP) measurement using a Power Lab data acquisition system (ADI Instruments, Australia). After the rats were euthanized, the heart and lung tissues were immediately collected for subsequent biochemical experiments. The RV free wall and left ventricle and septum (LV+S) were separated and weighed separately, and the RV/(LV+S) weight ratio was calculated to assess right heart hypertrophy.
Histological Analysis
The isolated RV and lung tissues were soaked in 4% paraformaldehyde for 24 h and embedded in paraffin. PA wall thickness (PAWT) and cardiomyocyte hypertrophy (calculated based on the cardiomyocyte cross-sectional area (CSA)) were determined by hematoxylin and eosin (H&E) staining. Twenty PAs (diameter 30–100 µm) were randomly selected at high magnification (×400) to analyze PAWT (calculated as medial wall thickness index (%) = (external diameter–internal diameter)/external diameter×100).
To assess the fibrosis of the RV and lung tissues, the tissues were subjected to Masson’s trichrome staining and analyzed based on the percentage of collagen fiber deposition.
Immunohistochemical staining of lung sections with anti-alpha smooth muscle actin (α-SMA, 1:200, Proteintech Group, Rosemont, IL, USA) antibody was performed to identify the PASMC and to assess the degree of PA muscularization, based on the proportion of α-SMA-positive sites of circumference in PA medial wall: non-muscularized (none): ≤25%, partly muscularized (partly): 25–75%, and fully muscularized (fully): ≥75%.
Culture of Human PASMCs
Human PASMCs (ScienCell, Carlsbad, CA, USA) were maintained in smooth muscle cell medium (SMCM, ScienCell, Carlsbad, CA, USA) supplemented with 2% fetal bovine serum, 1% growth supplement, and 1% penicillin/streptomycin solution at 37°C in 5% CO2. The cells were then cultured with equimolar doses of fasudil, DCA, fasudil+DCA, or FDCA under 1% O2 hypoxia using an anoxic cell incubator (Huaxi Electronic Technology Co., Ltd, Changsha, China) prior to the following experimental assessments.
Cell Viability and Proliferation Assays
PASMCs were seeded into 96-well plates with 10,000 cells per well. After stimulation with different concentrations of FDCA for 24 h, 10 μL Cell Counting Kit-8 solution (CCK-8, APExBIO, Houston, TX, USA) was added to each well for 4 h at 37°C, and the optical density (OD) was measured at a wavelength of 450 nm using a microplate reader (Thermo Scientific, CA, USA) to assess PASMC viability. To assess PASMC proliferation, we used a 5-ethynyl-2′deoxyuridine (EdU) incorporation assay kit (RiboBio, Guangzhou, China) and calculated the proportion of EdU-positive cells in each group.
Cell Migration Assay
PASMC migration was assessed using Transwell chambers (8 µm membrane pore size, Corning Costar, Cambridge, MA, USA). 50,000 cells were seeded in the upper chamber in 100 µL serum-free SMCM, and fasudil, DCA, fasudil+DCA, or FDCA were added to the lower chamber in 600 µL 0.2%-serum SMCM under normoxia or hypoxia (1% O2) for 24 h. The cells were then fixed in 4% paraformaldehyde for 30 min and stained with 5% crystal violet (Beyotime Biotech, Nantong, China) for 30 min. The number of cells that had migrated to the lower chamber was calculated based on at least five randomly chosen fields per membrane.
Cell Contraction Assay
A cell contraction assay kit (Cell Biolabs, San Diego, CA, USA) was used to evaluate the PASMC contractile capability. PASMCs (2×106 cells/mL) were suspended in basic medium, mixed with collagen gel working solution, placed into a 24-well plate, and left in an incubator at 37°C for 1 h. After the gel was polymerized, 1 mL basic medium with fasudil, DCA, fasudil+DCA, or FDCA was added to each collagen gel lattice under normoxia or hypoxia (1% O2) at 37°C. After 48 h of cell culture, the collagen gel was gently released from the side. Finally, the collagen gel size change in response to hypoxia (relative to the initial collagen gel lattice size) were imaged and quantified using Image-J software (NIH, Bethesda, MD, USA).
Measurement of Mitochondrial Function and [Ca2+]i
A mitochondrial membrane potential (Δψm) assay kit with JC-1 (Beyotime Biotech, Nantong, China) and a mitochondrial permeability transition pore (MPTP) assay kit (Beyotime Biotech, Nantong, China) were used to evaluate the mitochondria function. In the Δψm assay, red fluorescence indicates polarized mitochondria and green fluorescence indicates depolarized mitochondria. In the MTPT assay kit, stronger green fluorescence indicates reduced MPTP opening. [Ca2+]i was assessed using Fluo-4 AM (Beyotime Biotech, Nantong, China), a fluorescent dye that can penetrate the cell membrane and specifically bind to Ca2+. PASMCs were cultured in 24-well plates at a density of 50,000 cells/well with fasudil, DCA, fasudil+DCA, or FDCA for 24 h. The assay working solutions, configured according to the manufacturer’s instructions, were added to 24-well plates and incubated for 30 min at 37°C in the dark. Finally, the plates were observed using a fluorescence microscope, and random fields were imaged.
Western Blot Analysis
Cell total proteins were obtained using radioimmunoprecipitation assay (RIPA) lysis buffer (Thermo Fisher Scientific, Rockford, IL, USA) supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF, Beyotime Biotech, Nantong, China) and 1% protease inhibitor cocktail (Roche, Basel, Switzerland). A bicinchoninic acid (BCA) kit (Beyotime Biotech, Nantong, China) was used to measure the protein concentrations. Protein samples were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride membranes (Millipore, Bedford, MA, USA) in a Trans-Blot system (Bio-Rad, Hercules, CA, USA). Next, the membranes were blocked with 5% non-fat milk powder in Tris-buffered saline with Tween 20 (TBST) for 1 h at room temperature. They were then probed at 4°C overnight with primary antibody: anti-Ca2+/calmodulin-dependent protein kinase II (CaMK II), anti-p-CaMK II (1:1000, Santa Cruz Biotechnology, Dallas, TX, USA), anti-Ca2+/calmodulin-dependent MLC kinase (MLCK), anti-p-MLCK (1:1000, Affinity Biosciences, Cincinnati, OH, USA), anti-myosin phosphatase-targeting subunit 1 (MYPT1), anti-p-MYPT1 (1:1000, Cell Signaling Technology, Danvers, MA, USA), anti-Rho-associated coiled-coil-containing protein kinase (ROCK), and anti-β-actin (1:1000, Proteintech Group, Rosemont, IL, USA). Thereafter, the blots were incubated with horseradish peroxidase-conjugated AffiniPure goat anti-mouse or anti-rabbit IgG (H+L, 1:5000, Proteintech Group, Rosemont, IL, USA). Finally, the protein signals were visualized using a Western Bright ECL reagent kit (Thermo Fisher Scientific, Rockford, IL, USA) and the band intensities were measured using Image Lab 4.1 software (Bio-Rad, Hercules, CA, USA).
Statistical Analysis
Differences in means were determined by one-way analysis of variance (ANOVA) with the least significant difference (LSD) post hoc test. Data analysis was performed using SPSS v22 software (IBM Corp., Armonk, NY, USA), and statistical significance was defined as P<0.05. All bar graphs show the mean ± standard deviation (SD).
Results
FDCA Alleviated Increased RVSP in the SuHx-Induced Rat Model of PAH
As shown in Figure 1A, SuHx group rats developed severe PAH with significantly increased RVSP (51.4±3.86 mmHg) compared to the controls (30.1±2.99 mmHg). Intragastric administration of FDCA (15, 45, and 135 mg/kg) once daily for 3 weeks from the first day after the SU5416 injection significantly decreased SuHx-induced elevation of RVSP in a dose-dependent manner (45.0±1.15, 44.4±3.88, and 38.6±3.79 mmHg, respectively. P<0.05). Regarding the positive control, bosentan (100 mg/kg) significantly reduced the SuHx-induced increase of RVSP (42.2±6.16 mmHg), indicating the reliability of our experimental system (P<0.01).
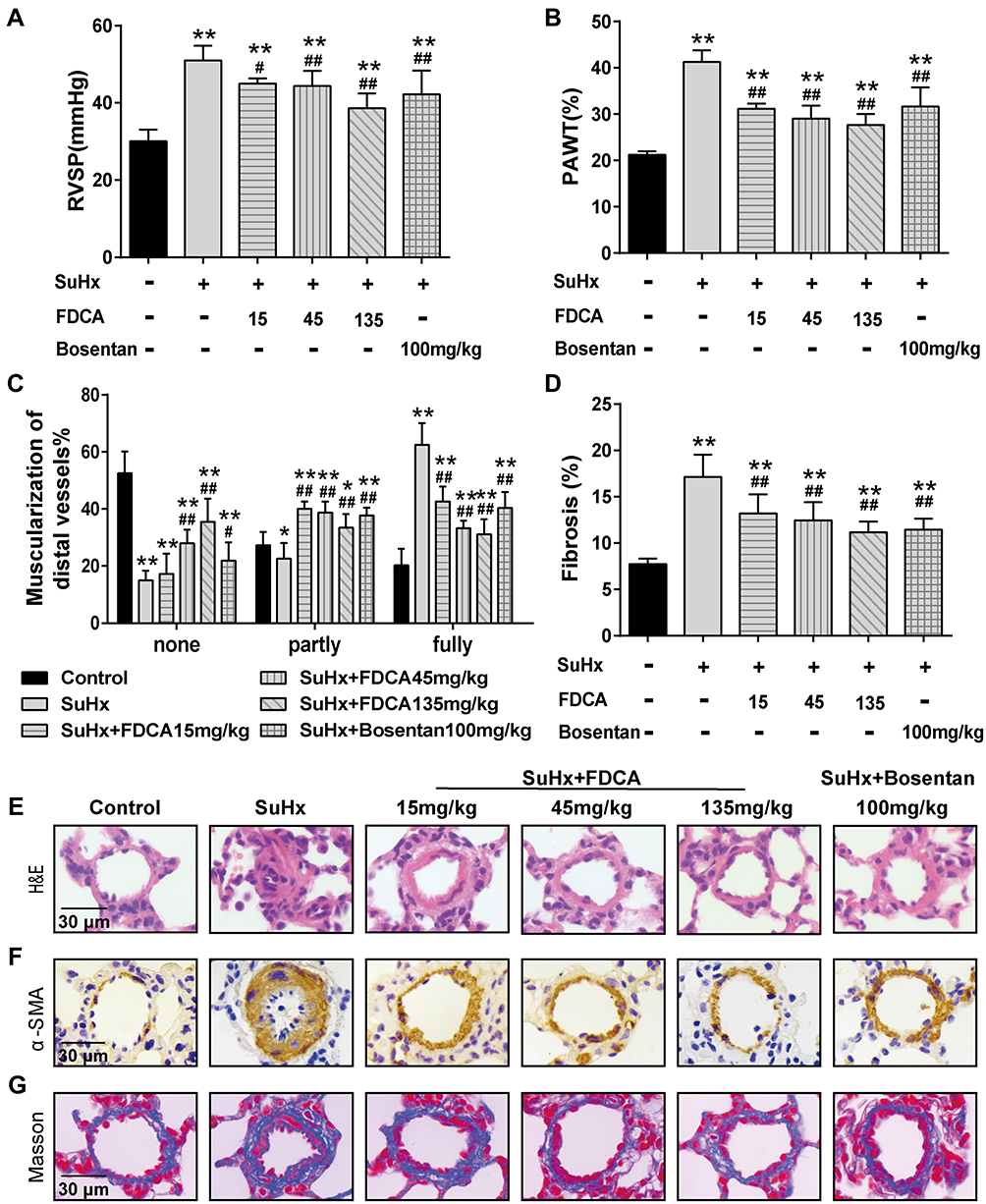 |
Figure 1 FDCA attenuated hemodynamics and pulmonary vascular remodeling in the SU5416 plus hypoxia (SuHx)-induced rat model of pulmonary arterial hypertension (PAH). (A) Hemodynamics were assessed based on right ventricular systolic pressure (RVSP). (B) Degree of pulmonary vascular remodeling was evaluated based on the pulmonary arterial wall thickness (PAWT). (C) Muscularization of distal pulmonary arteries was defined based on the proportion of alpha smooth muscle actin (α-SMA)-positive sites of circumference in PA medial wall: non-muscularized (none): ≤25%, partly muscularized (partly): 25–75%, and fully muscularized (fully): ≥75%. (D) Degree of pulmonary vascular adventitial fibrosis was evaluated based on the percentage of the collagen-positive area. (E) Representative images of hematoxylin-eosin (H&E) staining. (F) Representative images of immunohistochemical staining with α-SMA antibody (stained brown). (G) Representative images of Masson’s trichrome staining (collagen is stained blue) of lung sections. Data are expressed as mean ± SD (n=6-8). *P<0.05, **P<0.01 vs normoxia control group; #P<0.05, ##P<0.01 vs SuHx group. |
FDCA Inhibited Pulmonary Vascular Remodeling in the SuHx-Induced Rat Model of PAH
Pulmonary vascular remodeling in PAH is characterized by vascular wall thickening, an increased proportion of muscularized vessels, and excessive collagen deposition. As shown in Figure 1B and E, compared to the SuHx group, FDCA (15, 45, and 135 mg/kg) dose-dependently decreased the PAWT by 24.3%, 30.0%, and 33.4%, respectively (P<0.05). The proportion of α-SMA-positive sites of circumference in PA medial wall was used to evaluate the degree of muscularization of PAs of diameter 30–100 μm. The majority PAs of control group in this range were non-muscularized, while there was a significant increase in fully muscularized PAs in the SuHx group. FDCA (15, 45, and 135 mg/kg) treatment resulted in a dose-dependent reduction in fully muscularized PAs and a dose-dependent increase in non-muscularized PAs (P<0.01, Figure 1C and F). Moreover, Masson’s trichrome staining indicated that FDCA (15, 45, and 135 mg/kg) dose-dependently decreased SuHx-induced collagen deposition in the distal PAs by 21.6%, 27.5%, and 34.5%, respectively (P<0.01, Figure 1D and G).
FDCA Reversed RV Remodeling in the SuHx-Induced Rat Model of PAH
As shown in Figure 2A and D, the RV/(LV+S) values indicated that SuHx challenge resulted in significant right heart hypertrophy (0.42±0.048) compared to the controls (0.23±0.027). FDCA (15, 45, and 135 mg/kg) dose-dependently reduced the RV/(LV+S) values to 0.35±0.044, 0.31±0.026, and 0.30±0.022, respectively (P<0.01). The cardiomyocyte CSA was assessed to further evaluate the effect of FDCA on RV hypertrophy. As shown in Figure 2B and E, the CSA data showed that FDCA (15, 45, and 135 mg/kg) dose-dependently inhibited SuHx-induced cardiomyocyte hypertrophy (372.9±64.63, 347.1±45.74, and 237.4±16.10 μm2, respectively) compared to the SuHx group (608.3±66.62 μm2. P<0.01), with 135 mg/kg FDCA completely reversing the CSA to the level in the control group (222.5±40.68 μm2. P>0.05). Similar results were found regarding RV fibrosis. As shown in Figure 2C and F, daily administration of FDCA (15, 45, and 135 mg/kg) dose-dependently decreased the RV fibrosis in SuHx-treated rats by 35.7%, 44.6% and 60.7%, respectively (P<0.01), and 135 mg/kg FDCA even reversed collagen deposition to the level in the control group (P>0.05).
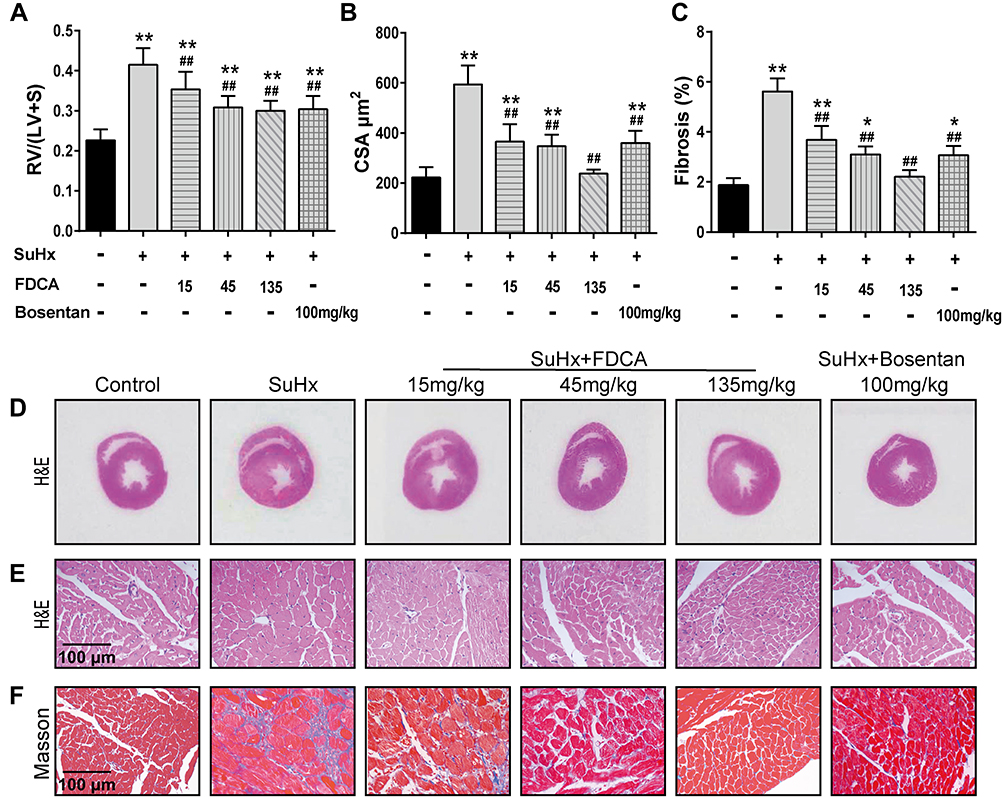 |
Figure 2 FDCA ameliorated right ventricular (RV) hypertrophy and fibrosis in SuHx-induced rat model of PAH. (A) RV hypertrophy was assessed based on the ratio of the RV weight to the left ventricle and interventricular septum (LV+S) weight (RV/(LV+S)). (B) RV remodeling was also evaluated based on RV cardiomyocyte cross-sectional area (CSA). (C) Degree of fibrosis in RV tissue was calculated based on the percentage of the collagen-positive area. (D) Representative images of H&E staining of full heart cross-sections. (E) Representative images of H&E staining of RV cross-sections. (F) Representative images of Masson’s trichrome staining of RV cross-sections (collagen is stained blue). Data are expressed as mean ± SD (n=6-8). *P<0.05, **P<0.01 vs normoxia control group; ##P<0.01 vs SuHx group. |
FDCA Inhibited Hypoxia-Induced Human PASMC Proliferation in vitro
Enhanced proliferation of PASMC is a key mechanism underlying pulmonary vascular remodeling in PAH. The CCK-8 results revealed that FDCA at ≤50 μM had no effect on cell viability under normoxia (P>0.05), while 50 μM FDCA attenuated the hypoxia-induced enhancement of PASMC viability (P<0.05, Figure 3A and B). Therefore, 50 μM FDCA and equimolar doses of fasudil, DCA, or fasudil+DCA were used in the subsequent in vitro experiments. As shown in Figure 3C and D, the EdU incorporation assay showed that hypoxia induced a significant increase of EdU-positive PASMCs compared to controls (15.0%±1.19% vs 6.4%±0.25%). FDCA (7.4%±0.40%) or fasudil+DCA (7.2%±0.05%) significantly reduced the hypoxia-induced increase in the proportion of EdU-positive PASMCs, displaying better efficiency than equimolar doses of fasudil (12.2%±0.31%) or DCA (11.5%±0.83%) alone regarding inhibiting PASMC proliferation (P<0.01).
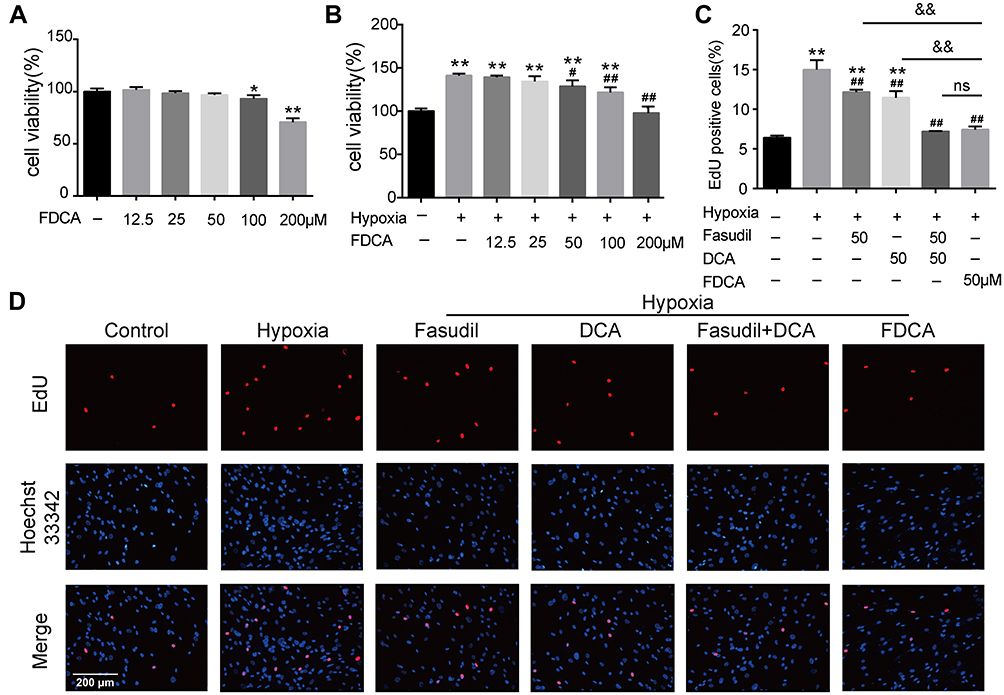 |
Figure 3 FDCA inhibited hypoxia-induced proliferation of human pulmonary arterial smooth muscle cells (PASMCs). (A, B) Cell viability based on Cell Counting Kit-8 assay. (C) Quantification of 5-ethynyl-2ʹdeoxyuridine (EdU)-positive cells. (D) Representative images of EdU incorporation assay. Proliferated cells were labeled with EdU (red) and cell nuclei labeled with Hoechst 33342 (blue). Data are expressed as mean ± SD (n=3). *P<0.05, **P<0.01 vs normoxia control group; #P<0.05, ##P<0.01 vs hypoxia group; &&P<0.01 vs hypoxia+FDCA group, ns (not significant). |
FDCA Suppressed Hypoxia-Induced Human PASMC Migration and Contraction in vitro
Hypoxia-induced abnormal PASMC migration and contraction are also prominent pathophysiological processes of PAH. As shown in Figure 4A and C, exposure to hypoxia significantly increased the number of cells that migrated to the lower side of the membrane in the Transwell chambers compared with controls, which was partially inhibited by fasudil or DCA, and fully reversed by FDCA and fasudil+DCA. The contractile capability of PASMC under hypoxia, as assessed by the percentage of constriction relative to the initial collagen gel lattice size, was increased compared to controls. FDCA or fasudil+DCA suppressed PASMC contraction more potently than either fasudil or DCA alone (P<0.05, Figure 4B and D)
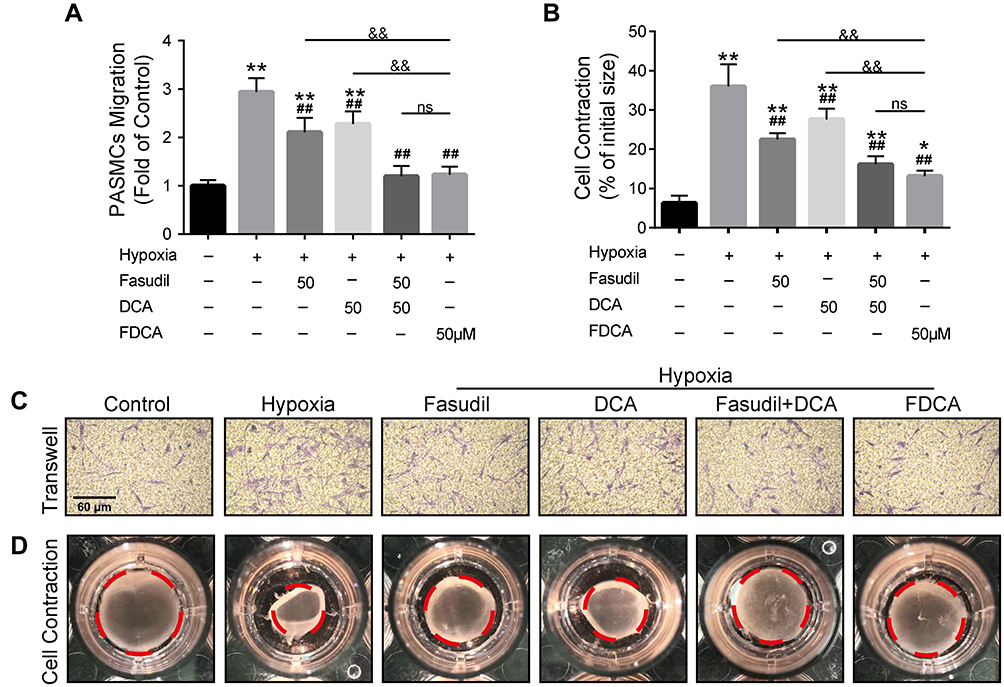 |
Figure 4 FDCA inhibited hypoxia-induced migration and contraction of human PASMCs. (A) Quantification of PASMC migration. (B) Quantification of collagen gel shrinkage relative to the initial collagen gel lattice size. (C) Representative images of PASMCs that migrated through the Transwell migration chamber membrane. (D) Representative images of cell contraction assay. The red dotted circles represent the gel area. Data are expressed as mean ± SD (n=3). *P<0.05, **P<0.01 vs normoxia control group; ##P<0.01 vs hypoxia group; &&P<0.01 vs hypoxia+FDCA group, ns (not significant). |
FDCA Alleviated Hypoxia-Induced Mitochondrial Dysfunction in PASMCs
Hypoxia-induced mitochondrial dysfunction is considered to be one of the causes of PASMC dysfunction in PAH. As shown in Figure 5A and C, hypoxia significantly increased red JC-1 fluorescence and decreased green JC-1 fluorescence compared to controls, indicating increased Δψm in the PASMCs. FDCA or fasudil+DCA significantly decreased the hypoxia-induced rise in Δψm with better efficiency than an equimolar dose of DCA (P<0.05), while fasudil had no effect on Δψm. Similar results were obtained in the MPTP assay. As shown in Figure 5B and D, the MTPT assay fluorescence intensity in PASMCs was significantly increased under hypoxia, indicating decreased MPTP opening, which was partially reversed by FDCA, fasudil+DCA, and DCA (P<0.05). However, fasudil alone did not restore the MPTP opening degree.
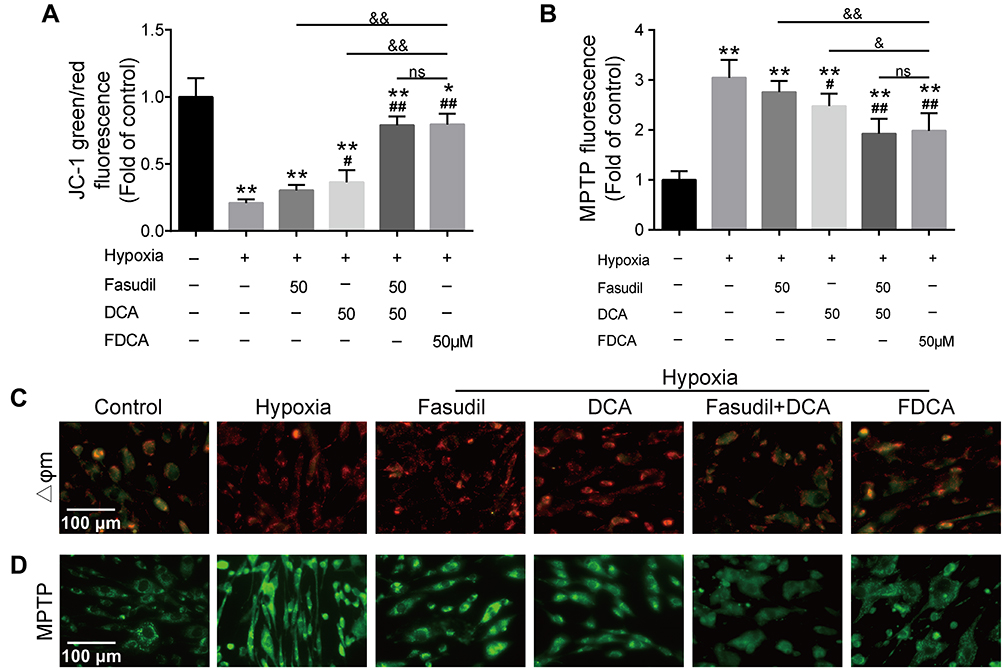 |
Figure 5 FDCA alleviated hypoxia-induced mitochondrial dysfunction in human PASMCs. (A) Quantitative analysis of JC-1 green/red fluorescence intensity. (B) Quantitative analysis of mitochondrial permeability transition pore (MPTP) assay fluorescence intensity. (C) Representative images of mitochondrial membrane potential (Δψm). Red fluorescence indicates normal mitochondria and green fluorescence indicates depolarized mitochondria. (D) Representative images of MPTP opening. Stronger green fluorescence intensity indicates reduced MPTP opening. Data are expressed as mean ± SD (n=3-4). *P<0.05, **P<0.01 vs normoxia control group; #P<0.05, ##P<0.01 vs hypoxia group; &P<0.05, &&P<0.01 vs hypoxia+FDCA group, ns (not significant). |
FDCA Inhibited Hypoxia-Induced Activation of the Ca2+/CaMK Signaling Pathway
Ca2+-related signals play vital roles in PASMC proliferation, migration, and contraction. We evaluated [Ca2+]i in PASMCs using Fluo-4 AM, a green fluorescent Ca2+ indicator. As shown in Figure 6A and B, although fasudil or DCA suppressed the hypoxia-induced [Ca2+]i increase, FDCA or fasudil+DCA exhibited more potent inhibitory effects on [Ca2+]i than fasudil or DCA alone. Thereafter, two main CaMKs (CaMK II and MLCK) were assessed. As shown in Figure 6C–E, hypoxia exposure for 30 min enhanced CaMK II and MLCK phosphorylation in PASMCs, which was fully blocked by pretreatment with FDCA or fasudil+DCA, but only partially blocked by fasudil or DCA alone (P<0.01).
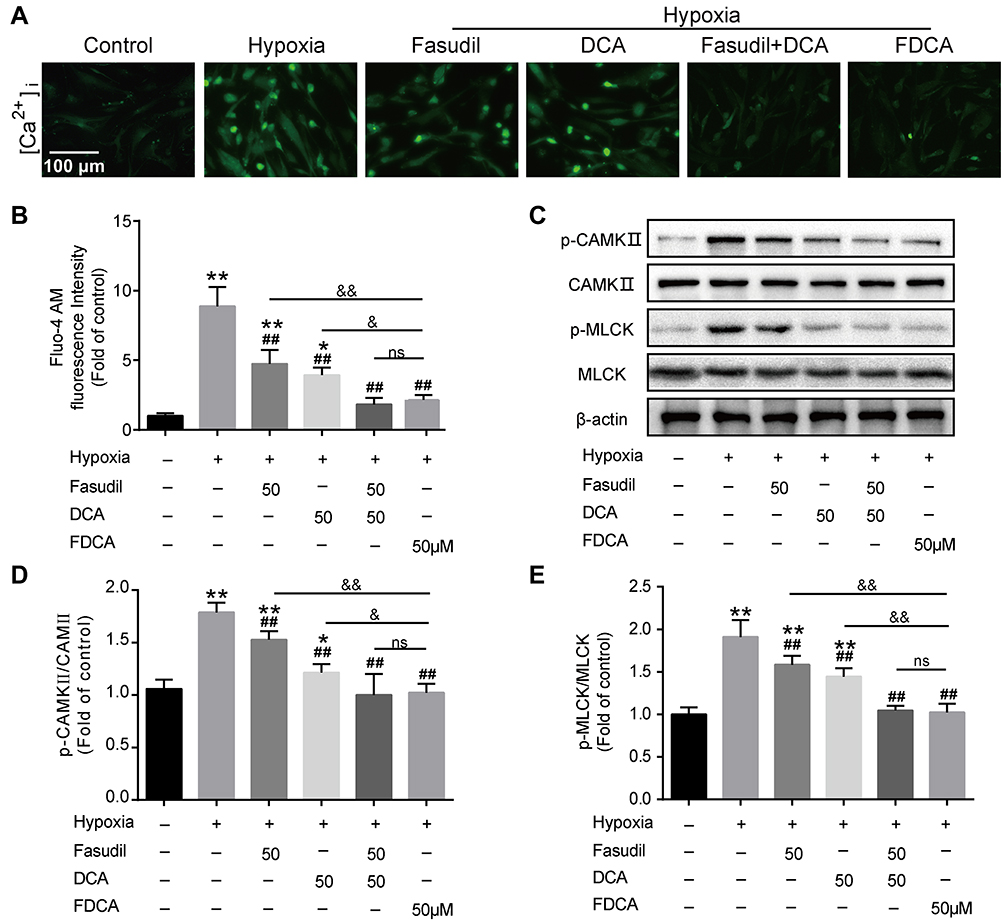 |
Figure 6 FDCA inhibited hypoxia-induced activation of the intracellular Ca2+ signaling pathway. (A) Representative images of [Ca2+]i. (B) Quantitative analysis of Fluo-4 AM fluorescence intensity. (C) Representative Western blot bands. (D) Quantitative analysis of CaMK II/p-CaMK II. (E) Quantitative analysis of p-MLCK/MLCK. Data are expressed as mean ± SD (n=3-5). *P<0.05, **P<0.01 vs normoxia control group; ##P<0.01 vs hypoxia group; &P<0.05, &&P<0.01 vs hypoxia+FDCA group, ns (not significant). |
FDCA Inhibited Hypoxia-Induced Activation of the Rho-Kinase Pathway
ROCK, also known as Rho-associated kinase, is one of the key contributors to PASMC dysfunction in PAH. As shown in Figure 7A, hypoxia for 24 h significantly upregulated ROCK compared to controls, which was partially reversed by fasudil or DCA, and fully reversed by FDCA or fasudil+DCA. To determine the activity of ROCK, the phosphorylation of MYPT1, a substrate of ROCK, was assessed. As shown in Figure 7B, pretreated with FDCA or fasudil+DCA fully reversed hypoxia-induced upregulation of p-MYPT1, while fasudil or DCA alone only partially reversed it (P<0.05).
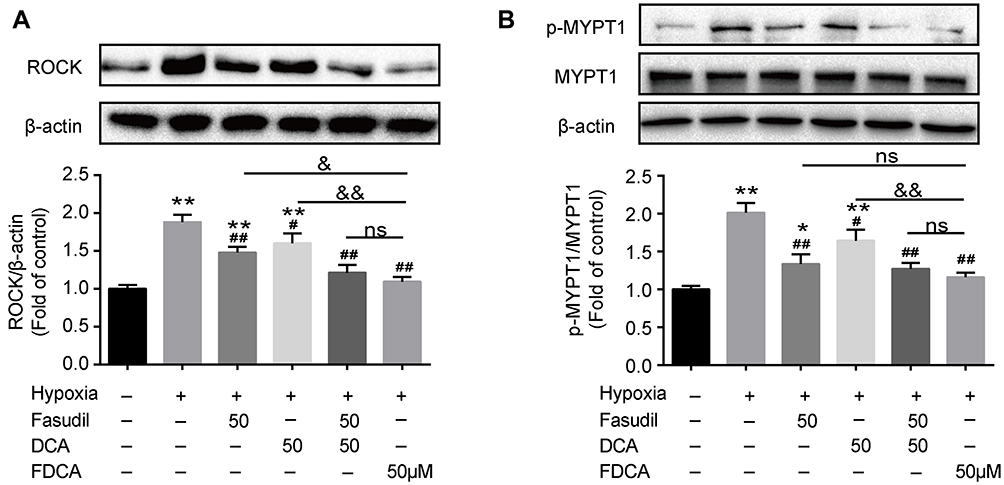 |
Figure 7 FDCA inhibited hypoxia-induced activation of the Rho-kinase pathway. (A) Representative Western blot bands and quantitative analysis of ROCK protein levels. (B) Representative Western blot bands and quantitative analysis of MYPT1/p-MYPT1. Data are expressed as mean ± SD (n=3). *P<0.05, **P<0.01 vs normoxia control group; #P<0.05, ##P<0.01 vs hypoxia group; &P<0.05, &&P<0.01 vs hypoxia+FDCA group, ns (not significant). |
Discussion
In this study, FDCA effectively alleviated the RVSP increase, pulmonary vascular remodeling, and RV remodeling in the SuHx-induced rat model of PAH. In vitro, FDCA suppressed hypoxia-induced PASMC proliferation, migration, and contraction. Moreover, cellular and biochemical assays indicated that FDCA potently maintained mitochondrial homeostasis, inhibited intracellular Ca2+ overload and dysregulation of Ca2+/CaMK signaling, as well as Rho-kinase signaling, in human PASMCs under hypoxia.
The SU5416 plus hypoxia-induced rat model of PAH, a widely used animal model simulating the pathological characteristics of human PAH, is frequently used in preclinical research to explore the efficacy and underlying mechanisms of new pharmacotherapies for PAH.21,22 In this study, we observed significant increases in RVSP, PAWT, PA muscularization, perivascular fibrosis, as well as RV hypertrophy and fibrosis in the SuHx-induced rat model. As a positive control, bosentan inhibited SuHx-induced PAH, with significant reductions in RVSP, accompanied by significant reductions in pulmonary vascular remodeling and RV hypertrophy, confirming the validity and reliability of our experimental system. In our previous research, FDCA exhibited a therapeutic effect on monocrotaline-induced PAH in rats, demonstrating a more potent effect than equimolar doses of fasudil or DCA,19 but the mechanism remained unknown. In this study, by using the SuHx-induced PAH rat model, we observed that FDCA also alleviated the PAH-related hemodynamic and pathological changes mentioned above, confirming the reliable effect of FDCA on pulmonary vascular and RV remodeling in vivo.
PASMC dysfunction plays a pivotal role in pulmonary vascular remodeling and vasoconstriction, which are two predominant pathophysiological features of PAH.23 During the development of PAH, chronic hypoxia resulted in dysregulation of energy metabolism, continuous proliferation, and excessive migration of PASMCs, which eventually leads to pulmonary vascular remodeling.4 Moreover, PASMC contraction directly controls the size of the vascular lumen and increases of pulmonary vascular resistance.5 In our study, we found that FDCA or fasudil+DCA effectively suppressed hypoxia-induced PASMC proliferation, migration, and contraction with better efficiency than equimolar doses of fasudil or DCA alone. These results indicated that FDCA could inhibit PASMC-mediated pulmonary vascular remodeling and vasoconstriction at the same time. Therefore, combined usage of fasudil and DCA may have synergistic therapeutic effects on hypoxia-induced PASMC dysfunction in vitro.
A large number of studies indicate that chronic hypoxia disturbs intracellular Ca2+ signaling dynamics, including the elevation of basal [Ca2+]i level and the enhancement of Rho-kinase-mediated Ca2+ sensitization, which subsequently leads to PASMC dysfunction.9,24–28 Intracellular Ca2+ overload can persistently activate downstream Ca2+/CaMK II signaling and thereby stimulate cell proliferation, inhibit apoptosis, and promote cell movement, which eventually leads to pulmonary vascular remodeling. At the same time, an elevated concentration of Ca2+/calmodulin activates MLCK and promotes the PASMC contraction.29–31 In this study, we confirmed the significant hypoxia-induced increase in [Ca2+]i, accompanied by CaMK phosphorylation in PASMCs. It has been reported that hypoxia can directly activate intracellular Ca2+ release from intracellular Ca2+ stores and then trigger the opening of voltage-dependent Ca2+ channels to further increase [Ca2+]i.32 Mitochondria, one of the main components of the intracellular Ca2+ pools, can not only directly store and release Ca2+, but also modulate hypoxia-induced Ca2+ release from the sarcoplasmic reticulum (another component of the intracellular Ca2+ pools).33 As a mitochondrial regulator, DCA can depolarize the Δψm and restore the MPTP opening degree by inhibiting mitochondrial pyruvate dehydrogenase kinase (PDK) and thereby bringing about a series of electrophysiological responses.34 These responses eventually inhibit Ca2+ outflow from the mitochondria and Ca2+ influx from the voltage-gated Ca2+ channels, and promote the recovery of intracellular Ca2+ by intracellular calcium pools.16,18,35 In this study, Δψm and MPTP opening assays confirmed that DCA can inhibit hypoxia-induced Ca2+/CaMK signaling by maintaining mitochondrial homeostasis. Notably, FDCA, synthesized from DCA and the selective Rho-kinase inhibitor fasudil, exhibited better effects than DCA alone regarding protecting mitochondrial function, lowering [Ca2+]i, and inhibiting CaMK phosphorylation. This indicated a potential link between mitochondrial function and Rho-kinase regarding regulating Ca2+/CaMK signaling.
The Rho-associated kinase ROCK, an important signaling molecule in the regulation of cell morphology, adhesion, and motility, can inhibit MLC phosphorylase (MLCP) by increasing p-MYPT1, thereby increasing MLC phosphorylation and smooth muscle cell contraction, which is also known as ROCK-mediated Ca2+ sensitization of contractions.36,37 Moreover, the Rho-kinase inhibitor fasudil directly blocks the hypoxia-induced increases in [Ca2+]i in distal PASMCs by directly inhibiting Ca2+ release from the sarcoplasmic reticulum, and Ca2+ entry via store- and voltage-operated Ca2+ channels.28,38 In this study, fasudil blocked hypoxia-induced ROCK signaling in PASMCs and thereby partially decreased [Ca2+]i and CaMK phosphorylation. Additionally, FDCA suppressed Ca2+/CaMK signaling to a greater degree than fasudil, further confirming an interaction between mitochondria and Rho-kinase.
In this study, FDCA was more efficient at suppressing ROCK signaling than fasudil. Notably, DCA alone also showed inhibitory effects on hypoxia-induced activation of ROCK, indicating that mitochondrial function can regulate the activation of Rho-kinase signaling. Mitochondria may be involved in regulating ROCK expression and distribution during Drosophila dorsal closure, but the mechanism remains unclear.39 Although our data support the hypothesis that there is an interaction between mitochondrial function and activation of Rho-kinase signaling in hypoxia-treated PASMCs, the exact relationship remains unclear. Moreover, fasudil alone did not restore the mitochondrial function in our study, which conflicts with a recent study showing that fasudil can ameliorate mitochondrial stress via stabilization of the mitochondrial membrane potential in hippocampal neurons.40 Therefore, the interaction between mitochondria and ROCK in PASMCs may be tissue- and cell-specific. Collectively, these results demonstrated that alleviating DCA-induced alleviation of mitochondrial dysfunction may facilitate the inhibitory effects of fasudil on ROCK signaling in pulmonary arterial smooth muscle under hypoxia.
There is a potential interaction loop among mitochondrial function, the Ca2+/CaMK signaling pathway, and the Rho/ROCK pathway. Hypoxia-induced mitochondrial dysfunction directly or indirectly increases [Ca2+]i, and activates Rho-kinase. Additionally, the direct hypoxia-induced activation of ROCK not only augments Ca2+ sensitization to enhance cell contraction, but also directly promotes Ca2+ influx and further increases [Ca2+]i (Figure 8). In our study, we provided new evidence on the interactions between mitochondria and the Rho-kinase pathway in hypoxia-induced PASMC dysfunction. However, the precise relationship between mitochondria and Rho-kinase is not yet completely clear. The specific feedback effects of intracellular Ca2+ overload on mitochondrial function and Rho-kinase activity in the abovementioned interaction loop remain to be elucidated. Exactly, mitochondrial function, the Ca2+/CaMK signaling, and the Rho-kinase pathway do not act independently, but may converge. Therefore, the protective effect of FDCA may be attributable to the synergistic roles between maintaining mitochondrial function and ROCK inhibition, which further ensure Ca2+ homeostasis during hypoxia.
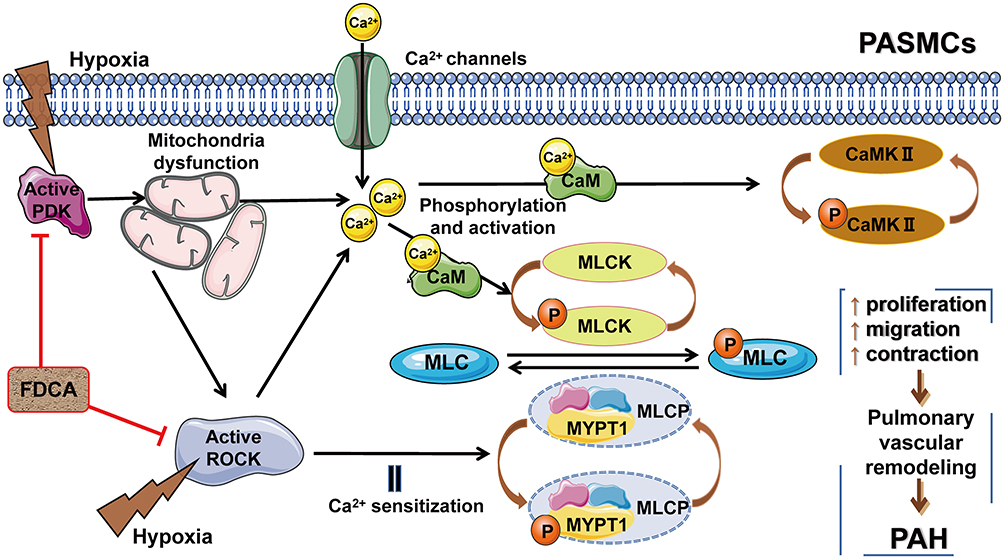 |
Figure 8 Potential interaction loop among mitochondrial function, the Ca2+/CaMK signaling pathway, and the ROCK pathway. FDCA inhibited hypoxia-induced PASMC proliferation, migration, and contraction by restoring mitochondrial function, reducing [Ca2+]i, and inhibiting the Ca2+/CaMK activity and the ROCK pathway activity. Abbreviations: PDK, pyruvate dehydrogenase kinase; Ca2+/CaMKs, calcium/calmodulin-dependent kinases; MLCK, Ca2+/calmodulin-dependent myosin light chain kinase; MYPT1, myosin phosphatase-targeting subunit 1; MLCP, myosin light chain phosphorylase. |
Conclusion
Taken together, our results demonstrate that FDCA alleviated SuHx-induced PAH by ameliorating PASMC dysfunction under hypoxia. FDCA represents a promising drug for the treatment of PAH.
Ethics Approval
The study was approved by the Institutional Animal Care and Use Committee of Nanjing Medical University (NJMU/IACUC-2005021).
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grants 81273571, 81870054), and the Key Project of National Science & Technology for Infectious Diseases of China (grants 2015ZX10003001, 2018ZX10722301).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1):1801913. doi:10.1183/13993003.01913-2018
2. Humbert M, Guignabert C, Bonnet S, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J. 2019;53(1):1801887. doi:10.1183/13993003.01887-2018
3. Galiè N, Channick RN, Frantz RP, et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J. 2019;53(1):1801889. doi:10.1183/13993003.01889-2018
4. Pullamsetti SS, Savai R, Seeger W, Goncharova EA. Translational advances in the field of pulmonary hypertension. from cancer biology to new pulmonary arterial hypertension therapeutics. targeting cell growth and proliferation signaling hubs. Am J Respir Crit Care Med. 2017;195(4):425–437. doi:10.1164/rccm.201606-1226PP
5. Brozovich FV, Nicholson CJ, Degen CV, Gao YZ, Aggarwal M, Morgan KG. Mechanisms of Vascular Smooth Muscle Contraction and the Basis for Pharmacologic Treatment of Smooth Muscle Disorders. Pharmacol Rev. 2016;68(2):476–532. doi:10.1124/pr.115.010652
6. Hong Z, Chen KH, DasGupta A, et al. MicroRNA-138 and MicroRNA-25 Down-regulate Mitochondrial Calcium Uniporter, Causing the pulmonary arterial hypertension cancer phenotype. Am J Respir Crit Care Med. 2017;195(4):515–529. doi:10.1164/rccm.201604-0814OC
7. Yuan JX, Aldinger AM, Juhaszova M, et al. Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation. 1998;98(14):1400–1406. doi:10.1161/01.CIR.98.14.1400
8. Ghisdal P, Vandenberg G, Morel N. Rho-dependent kinase is involved in agonist-activated calcium entry in rat arteries. J Physiol. 2003;551(Pt 3):855–867. doi:10.1113/jphysiol.2003.047050
9. Liu Z, Khalil RA. Evolving mechanisms of vascular smooth muscle contraction highlight key targets in vascular disease. Biochem Pharmacol. 2018;153:91–122. doi:10.1016/j.bcp.2018.02.01
10. Klinger JR, Elliott CG, Levine DJ, et al. Therapy for pulmonary arterial hypertension in adults: update of the CHEST Guideline and expert panel report. Chest. 2019;155(3):565–586. doi:10.1016/j.chest.2018.11.030
11. Mouchaers KT, Schalij I, de Boer MA, et al. Fasudil reduces monocrotaline-induced pulmonary arterial hypertension: comparison with bosentan and sildenafil. Eur Respir J. 2010;36(4):800–807. doi:10.1183/09031936.00130209
12. Keshavarz A, Alobaida A, McMurtry IF, Nozik-Grayck E, Stenmark KR, Ahsan F. CAR, a homing peptide, prolongs pulmonary preferential vasodilation by increasing pulmonary retention and reducing systemic absorption of liposomal fasudil. Mol Pharm. 2019;16(8):3414–3429. doi:10.1021/acs.molpharmaceut.9b00208
13. Ruan H, Zhang Y, Liu R, Yang X. The acute effects of 30 mg vs 60 mg of intravenous Fasudil on patients with congenital heart defects and severe pulmonary arterial hypertension. Congenit Heart Dis. 2019;14(4):645–650. doi:10.1111/chd.12764
14. Jiang X, Wang YF, Zhao QH, et al. Acute hemodynamic response of infused fasudil in patients with pulmonary arterial hypertension: a randomized, controlled, crossover study. Int J Cardiol. 2014;177(1):61–65. doi:10.1016/j.ijcard.2014.09.101
15. Fukumoto Y, Yamada N, Matsubara H, et al. Double-blind, placebo-controlled clinical trial with a rho-kinase inhibitor in pulmonary arterial hypertension. Circ J. 2013;77(10):2619–2625. doi:10.1253/circj.CJ-13-0443
16. McMurtry MS, Bonnet S, Wu X, et al. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res. 2004;95(8):830–840. doi:10.1161/01.RES.0000145360.16770.9f
17. Li T, Li S, Feng Y, et al. Combination of dichloroacetate and atorvastatin regulates excessive proliferation and oxidative stress in pulmonary arterial hypertension development via p38 signaling. Oxid Med Cell Longev. 2020;2020:6973636. doi:10.1155/2020/6873636
18. Michelakis ED, Gurtu V, Webster L, et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci Transl Med. 2017;9(413):eaao4583. doi:10.1126/scitranslmed.aao4583
19. Qi L, Lv T, Cheng Y, et al. Fasudil dichloroacetate (FDCA), an orally available agent with potent therapeutic efficiency on monocrotaline-induced pulmonary arterial hypertension rats. Bioorg Med Chem Lett. 2019;29(14):1812–1818. doi:10.1016/j.bmcl.2019.05.006
20. Chaumais MC, Djessas MRA, Thuillet R, et al. Additive protective effects of sacubitril/valsartan and bosentan on vascular remodeling in experimental pulmonary hypertension. Cardiovasc Res. 2020. doi:10.1093/cvr/cvaa200
21. Abe K, Toba M, Alzoubi A, et al. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation. 2010;121(25):2747–2754. doi:10.1161/CIRCULATIONAHA.109.927681
22. de Raaf MA, Schalij I, Gomez-Arroyo J, et al. SuHx rat model: partly reversible pulmonary hypertension and progressive intima obstruction. Eur Respir J. 2014;44(1):160–168. doi:10.1183/09031936.00204813
23. Stenmark KR, Frid MG, Graham BB, Tuder RM. Dynamic and diverse changes in the functional properties of vascular smooth muscle cells in pulmonary hypertension. Cardiovasc Res. 2018;114(4):551–564. doi:10.1093/cvr/cvy004
24. Herbert LM, Resta TC, Jernigan NL. RhoA increases ASIC1a plasma membrane localization and calcium influx in pulmonary arterial smooth muscle cells following chronic hypoxia. Am J Physiol Cell Physiol. 2018;314(2):C166–c176. doi:10.1152/ajpcell.00159.2017
25. Aaronson PI, Robertson TP, Knock GA, et al. Hypoxic pulmonary vasoconstriction: mechanisms and controversies. J Physiol. 2006;570(Pt 1):53–58. doi:10.1113/jphysiol.2005.098855
26. Kuhr FK, Smith KA, Song MY, Levitan I, Yuan JX. New mechanisms of pulmonary arterial hypertension: role of Ca2⁺ signaling. Am J Physiol Heart Circ Physiol. 2012;302(8):H1546–H1562. doi:10.1152/ajpheart.00944.2011
27. Leggett K, Maylor J, Undem C, et al. Hypoxia-induced migration in pulmonary arterial smooth muscle cells requires calcium-dependent upregulation of aquaporin 1. Am J Physiol Lung Cell Mol Physiol. 2012;303(4):L343–L353. doi:10.1152/ajplung.00130.2012
28. Wang J, Weigand L, Foxson J, Shimoda LA, Sylvester JT. Ca2+ signaling in hypoxic pulmonary vasoconstriction: effects of myosin light chain and Rho kinase antagonists. Am J Physiol Lung Cell Mol Physiol. 2007;293(3):L674–L685. doi:10.1152/ajplung.00141.2007
29. Marganski WA, Gangopadhyay SS, Je HD, Gallant C, Morgan KG. Targeting of a novel Ca+2/calmodulin-dependent protein kinase II is essential for extracellular signal-regulated kinase-mediated signaling in differentiated smooth muscle cells. Circ Res. 2005;97(6):541–549. doi:10.1161/01.RES.0000182630.29093.0d
30. Saddouk FZ, Ginnan R, Singer HA. Ca(2+)/calmodulin-dependent protein kinase II in vascular smooth muscle. Adv Pharmacol. 2017;78:171–202. doi:10.1016/bs.apha.2016.08.003
31. Ikebe M, Hartshorne DJ. Phosphorylation of smooth muscle myosin at two distinct sites by myosin light chain kinase. J Biol Chem. 1985;260(18):10027–10031. doi:10.1016/S0021-9258(17)39206-2
32. Gelband CH, Gelband H. Ca2+ release from intracellular stores is an initial step in hypoxic pulmonary vasoconstriction of rat pulmonary artery resistance vessels. Circulation. 1997;96(10):3647–3654. doi:10.1161/01.CIR.96.10.3647
33. Kang TM, Park MK, Uhm DY. Characterization of hypoxia-induced [Ca2+]i rise in rabbit pulmonary arterial smooth muscle cells. Life Sci. 2002;70(19):2321–2333. doi:10.1016/S0024-3205(02)01497-2
34. Bonnet S, Archer SL, Allalunis-Turner J, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11(1):37–51. doi:10.1016/j.ccr.2006.10.020
35. Gupte SA, Wolin MS. Mitochondrial calcium transport: a potentially prominent, therapeutically targetable contributor to pulmonary arterial hypertension progression. Am J Respir Crit Care Med. 2017;195(4):420–421. doi:10.1164/rccm.201609-1896ED
36. Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83(4):1325–1358. doi:10.1152/physrev.00023.2003
37. Sakurada S, Takuwa N, Sugimoto N, et al. Ca2+-dependent activation of Rho and Rho kinase in membrane depolarization-induced and receptor stimulation-induced vascular smooth muscle contraction. Circ Res. 2003;93(6):548–556. doi:10.1161/01.RES.0000090998.08629.60
38. Krauszman A, Mak TW, Szaszi K, Kuebler WM. Role of phosphatase and tensin homolog in hypoxic pulmonary vasoconstriction. Cardiovasc Res. 2017;113(8):869–878. doi:10.1093/cvr/cvx076
39. Muliyil S, Narasimha M. Mitochondrial ROS regulates cytoskeletal and mitochondrial remodeling to tune cell and tissue dynamics in a model for wound healing. Dev Cell. 2014;28(3):239–252. doi:10.1016/j.devcel.2013.12.019
40. Gao Y, Yan Y, Fang Q, et al. The Rho kinase inhibitor fasudil attenuates Aβ(1-42)-induced apoptosis via the ASK1/JNK signal pathway in primary cultures of hippocampal neurons. Metab Brain Dis. 2019;34(6):1787–1801. doi:10.1007/s11011-019-00487-0
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.