Back to Journals » Clinical Epidemiology » Volume 10
Familial risks of second primary cancers and mortality in ovarian cancer patients
Authors Zheng G ![]() , Chattopadhyay S
, Chattopadhyay S ![]() , Försti A
, Försti A ![]() , Sundquist K, Hemminki K
, Sundquist K, Hemminki K ![]()
Received 15 May 2018
Accepted for publication 27 July 2018
Published 11 October 2018 Volume 2018:10 Pages 1457—1466
DOI https://doi.org/10.2147/CLEP.S174173
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Irene Petersen
Guoqiao Zheng,1,2 Subhayan Chattopadhyay,1,2 Asta Försti,1,3 Kristina Sundquist,3–6 Kari Hemminki1,3
1Division of Molecular Genetic Epidemiology, German Cancer Research Center (DKFZ), Heidelberg, Baden-Württemberg, Germany; 2Faculty of Medicine, University of Heidelberg, Heidelberg, Baden-Württemberg, Germany; 3Center for Primary Health Care Research, Lund University, 205 02 Malmö, Skåne County, Sweden; 4Department of Family Medicine and Community Health, 5Department of Population Health Science and Policy, Icahn School of Medicine at Mount Sinai, New York, NY, USA; 6Center for Community-Based Healthcare Research and Education (CoHRE), Department of Functional Pathology, School of Medicine, Shimane University, Izumo, Japan
Background: With improving survival in ovarian cancer, second primary cancers (SPCs) and their etiological foundations are becoming an issue. The ways in which family history may influence the occurrence of SPCs and the related mortality are not well known.
Methods: Based on the Swedish Family-Cancer Database, we identified 11,300 ovarian cancer patients and followed them for diagnoses of SPCs until the end of 2015. Relative risks (RRs) of SPC in patients who had parents or siblings diagnosed with the same cancer (positive family history) were compared to those in patients without a family history (negative family history). Causes of death were compared between patients with and without SPC.
Results: A total of 1,111 (9.8%) ovarian cancer patients developed SPC with a median follow-up of 8 years. The impact of a family history of cancer on the risk of the same cancer as SPC was significant for colon (RRpositive family history [95% CI] vs RRnegative family history [95% CI]: 4.95 [3.03–8.09] vs 2.00 [1.63–2.47]), lung (3.32 [1.88–5.84] vs 1.45 [1.16–1.83]), and breast (2.08 [1.58–2.73] vs 1.01 [0.88–1.15]) cancers. With a family history of any cancer, the RR for non-ovarian SPCs was 1.66 (1.54–1.74), in contrast to 1.38 (1.24–1.54) for SPCs without any family history (P-trend <0.001). Accounting for 42.1% of all deaths, SPC was found to be the main cause of death for patients with SPC.
Conclusion: A family history of a particular cancer contributed to an increased risk of SPC at the same site. Therefore, considering family history at the time of diagnosis of ovarian cancer may alert physicians to a syndromic background, management of which may help the patient and her family members.
Keywords: second primary cancer, familial cancer, cause of death, cumulative incidence
Introduction
The occurrence of multiple primary cancers is increasing with better survival in cancer patients. In cancer registries, such as the Swedish Cancer Registry, multiple primary cancers amounted to 23.1% of the number of first invasive cancers in 2016.1 Most multiple primary cancers are second primary cancers (SPCs). Survival in women with ovarian cancer, following the same pattern, has also been improving, and the 5-year relative survival is about 46% in USA.2 According to a Swedish study, the overall risk of SPCs was increased by about 1.5-fold compared to the risk of first cancer, and the highest excess risks were noted for connective tissue cancer, small intestinal cancers, and leukemia.3 Contributory factors for SPCs may include therapy-related adverse effects, increased surveillance, and the same causes that influenced patients’ first cancers, including family history and environmental factors.4–6 The impact of family history has been demonstrated in Hodgkin lymphoma patients, who had an excess risk of lung, colorectal, and breast cancers when their family members were diagnosed with these cancers.7 The potential importance of family history is emphasized by the fact that about 50% of the cancer patients have a first degree relative diagnosed with some other (discordant) cancer, and for ovarian cancer this proportion is as high as 56%.8 Although SPCs have become an important cause of death in many cancers, data from the US Surveillance, Epidemiology, and End Results program indicate that in ovarian cancer SPCs account for far fewer deaths than ovarian cancer itself.9
In this study, we investigate the impact of family history on the risk of SPC in ovarian cancer patients. According to our hypothesis, if the ovarian cancer patient has a family history of colon cancer she would be at excess risk of a second primary colon cancer compared to the ovarian cancer patient without a family history of colon cancer. Ovarian cancer is a cancer related to BRCA1/2 mutations, which are associated with high-grade serous histology.10,11 Mutations in mismatch repair (MMR) genes manifested in hereditary non-polyposis colorectal cancer (HNPCC) syndrome, predisposing to familial ovarian cancer, preferentially of endometrioid and clear cell histology.12,13 We also explore the effect of the family history after excluding high-risk families possibly due to these gene mutations, using criteria based on age at the time of diagnosis and defined family history. In addition to the familial risk in SPC, we report on causes of death in patients with SPCs, noting their important contribution.
Methods
The Swedish Family-Cancer Database contains information on the national population in families (through the Multigeneration Register) diagnosed with cancer (from the Cancer Registry, started in 1958). The families include the second (offspring) generation born after 1931 and their biological parents (parental generation). Siblings can be identified in the offspring but not in the parental generation. The latest version of the database contains 16.1 million individuals, of whom 2.0 million were diagnosed with cancer by the end of 2015. A total of 46,227 ovarian cancer cases (offspring and parental generation) were found among 4,216,676 women followed. Borderline tumors and in situ tumors have been excluded.
Follow-up for cancer in the offspring generation was started at the beginning of 1958, the birth year, or the immigration year, whichever came latest. The follow-up was terminated when a person was diagnosed with cancer, emigrated, or died, or at the end of 2015, whichever came first. Follow-up for SPC was started at the diagnosis of ovarian cancer and terminated at the diagnosis of SPC, emigration, death, or the end of 2015. Synchronous second malignancies were included as SPC. As the primary treatment for ovarian cancer is surgery which usually includes removal of the uterus, we did not consider the risk of second endometrial cancer, but in the case of overall risk calculation all cancer cases are retained. The absolute risk was calculated as the number of second cancers observed in our cohort divided by the number of person-years at risk, multiplied by 100,000. Cumulative incidence rates of second malignancies were calculated considering death as a competing event. A Poisson regression model was employed to estimate the relative risks (RRs) and corresponding 95% CIs of SPC compared to the incidence of the same cancer as a first primary cancer. Significance levels were also indicated for 1% and 0.1% significance. A family history of cancer was defined through first degree relatives (parents and siblings). The P-value for trend was obtained from a non-parametric test (Median test) by comparing bootstrapped samples of a particular second cancer risk in ovarian cancer patients with a family history of this cancer (RRpositive family history) to those samples in patients without a first-degree relative family history (RRnegative family history). Potential confounders, including age group (5-year bands), parity (0, 1, 2, 3, and ≥4 child births), calendar period (5-year bands), residential area (large cities, South Sweden, North Sweden, or unspecified), and socioeconomic status (blue-collar worker, white-collar worker, farmer, private business, professional, or other/unspecified), were added to the model as covariates.
In sensitivity analyses, we attempted to delete families that may be carriers of known high-risk genes. An increased probability of BRCA1/2 association was defined for families in which at least two members had been diagnosed with breast cancer before the age of 50 years, or at least one member with bilateral breast cancer, counted as two independent cases when diagnosed at <50 years of age, or with a male breast cancer diagnosis;14 high-risk families associated with MMR were defined as families with colorectal or endometrial cancer diagnosed before the age of 50.15 Among all the women followed, 25,188 (0.60%) were from possible high-risk families related to MMR mutations and 14,854 (0.35%) were from possible high-risk families related to BRCA1/2 mutations. In 11,300 ovarian cancer patients, the respective numbers were 108 (0.96%) and 79 (0.70%).
Underlying and contributory causes of death were obtained from the Swedish Cause of Death Register and recorded according to the WHO ICD (ICD-7, 1961; ICD-8, 1969–1986; ICD-9, 1987–1996; ICD-10, 1997–2015). All cancer-related deaths were stratified into ovarian cancer, SPC, and “other cancer”, which includes cancers diagnosed at the time of issue of the death certificate and which are neither the first cancer nor SPC. These death certificate notifications are not used by the Swedish Cancer Registry, in contrast to the other Nordic Cancer Registries.16–18 We found that they often included multiple cancers and cancer of unknown primary (CUP). In our previous analyses, these cancers have been interpreted as metastases.19,20 If the death certificate notification matched the organ site of the reported primary cancer it was classified to that site, but in most cases no such assignment could be made and the classification was to “other cancer”. In some analyses, higher order primary cancers were classified separately. Other non-neoplastic causes of death were reported as “other cause”. Statistical analyses were carried out with SAS version 9.4 (SAS Institute Inc., Cary, NC, USA).
Results
In total, 11,300 ovarian cancers were diagnosed in the 0–83-year-old women in the offspring generation for whom RRs were calculated, and 1,111 patients (9.8%) were diagnosed with SPC (Table 1). Among SPC cases, 751 patients (67.6%) had a family history of cancer in first-degree relatives; for 129 patients, SPC was the same (concordant) cancer that was diagnosed in the family members, and for 622 patients, it was a different (discordant) cancer. The median age at the time of diagnosis of ovarian cancer was 50 years in patients who later developed SPC. The median age at the time of diagnosis of SPC was 62 years. The median time between diagnosis of ovarian cancer and that of SPC was 8 years. In addition, 122 of the patients with an SPC had been diagnosed with a third primary cancer (11.0% of all SPCs).
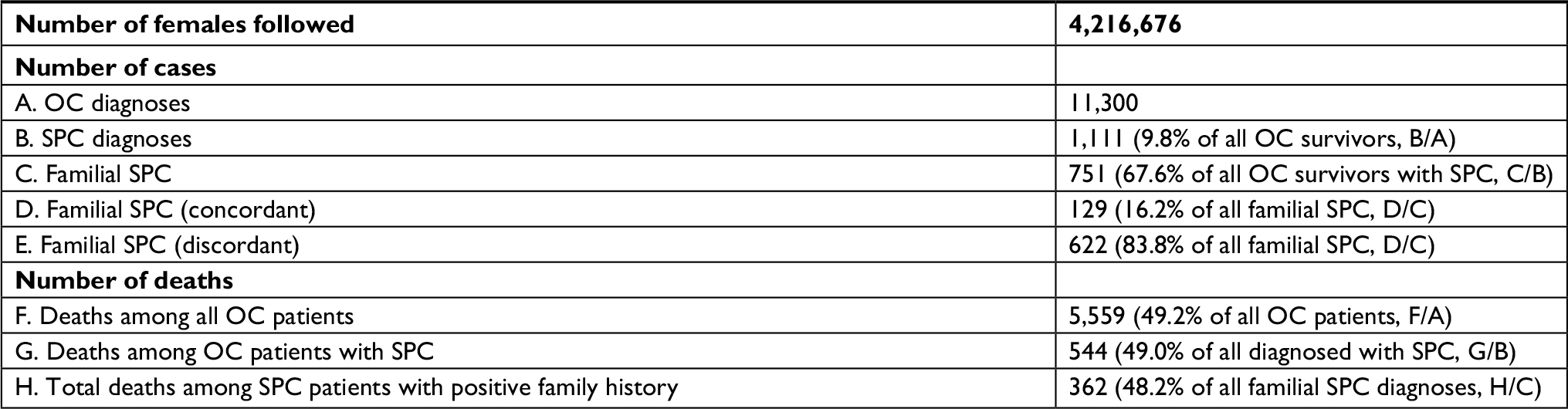 | Table 1 Familial demography of the population followed during the period 1958–2015 Abbreviations: OC, ovarian cancer; SPC, second primary cancer. |
The impact of family history on SPCs without removing high-risk families is assessed in Table 2, where the reference population had neither ovarian cancer nor a family history of SPC of any cancer (in the last row). The risk for SPC with a family history of any cancer was 1.74 (95% CI 1.64–1.87) compared to 1.49 (1.34–1.65) for SPCs lacking such a family history. After removing the second ovarian cancer, the respective RRs were 1.66 (1.54–1.74) and 1.38 (1.24–1.54). For patients lacking a family history, high risks were observed for small intestinal (3.23, 1.73–6.03), connective tissue (3.13, 1.73–5.67), and bladder (2.72, 2.01–3.69) cancers, and for CUP (2.69, 2.06–3.52). The trend test for family history was positive for SPCs with a concordant family history of four cancers, namely colon (RRpositive family history [95% CI] vs RR negative family history [95% CI]: 4.95 (3.03–8.09) vs 2.00 (1.63–2.47)], colorectal (4.17 [2.09–6.00] vs 1.84 [1.53–2.22]), lung (3.32 [1.88–5.84] vs 1.45 [1.16–1.83]), and breast (2.08 [1.58–2.73] vs 1.01 [0.88–1.15]) cancers. In addition, the trend test was of borderline significance for SPC of squamous cell carcinoma of the skin with a family history compared to without a family history (3.25 [1.05–10.02] vs 0.74 [0.48–1.15]). SPCs in patients with a family history of pancreatic and bladder cancers and of CUP displayed high RRs, although the case numbers were few. The most common familial SPCs were breast (52 patients) and colorectal cancers (29 patients). Table 2 does not list cancers with fewer than ten total cases; however, these are included in “All cancers”. Of the 122 third primary cancers, breast (32), colorectal (27), and lung (12) cancers accounted for 58% of the total. After removing high-risk families, the impact of a family history of cancer on the risk of this cancer as an SPC remained significant (Table S1).
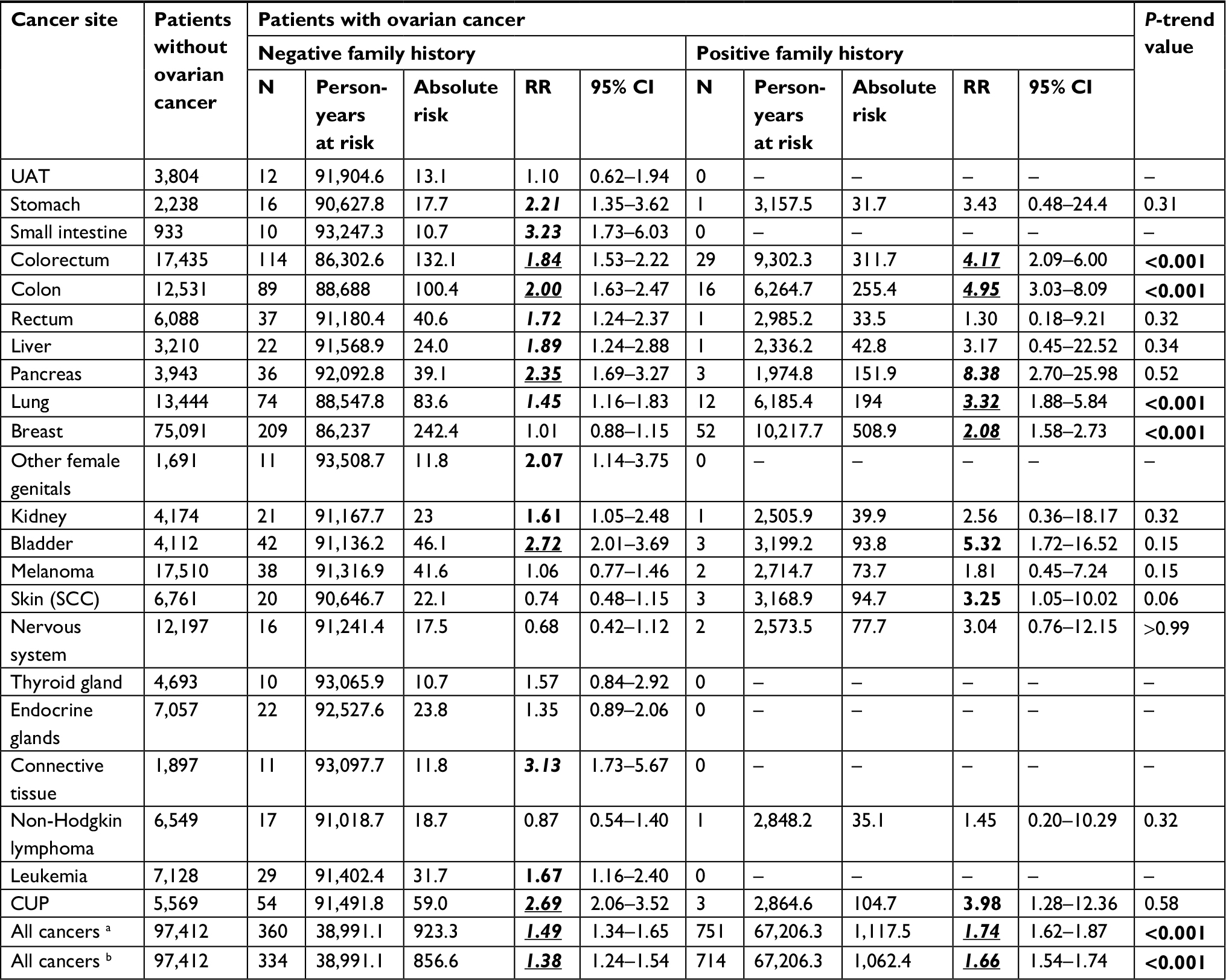 | Table 2 Risk of second primary cancers in ovarian cancer patients with or without a family history of concordant cancer Notes: aAll cancers including second ovarian cancer. bAll cancers excluding second ovarian cancer. Absolute risk = Number/100,000 person-years. Bold, italic and underlined values indicate that the 95% CI, 99% CI and 99.9% CI did not overlap with 1.00 respectively. Abbreviations: CUP, cancer of unknown primary; RR, relative risk; SCC, squamous cell carcinoma; UAT, upper aerodigestive tract. |
The cumulative incidence of second primary breast and colorectal cancer is shown in Figure 1, stratified by age at the time of diagnosis of the SPC. Figure 1 shows plots of SPCs for those with a family history of concordant cancer (solid lines) and without a family history (dashed lines). For SPCs in patients with a family history, the cumulative incidence increased steeply from about the age of 45–60 years and less steeply at higher ages. The cumulative incidence of second primary breast cancer with a family history reached 5.0% at the age of >80 years, somewhat higher than that for second primary colorectal cancers (2.6%). In patients lacking a family history, second primary breast cancer reached a cumulative incidence of 2.4%, compared to that of colorectal cancer of 1.5%.
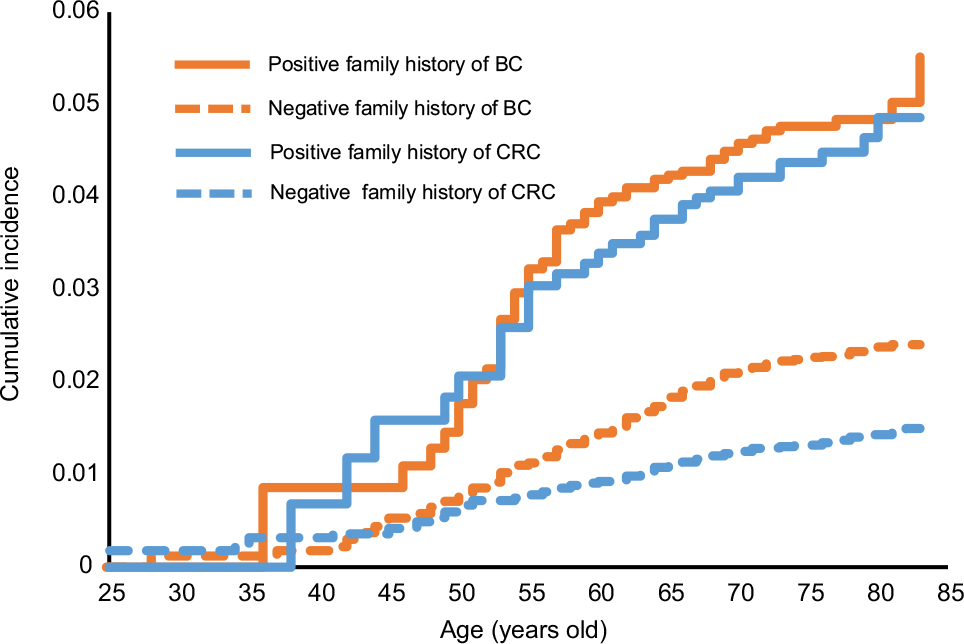 | Figure 1 Cumulative incidence of SPCs according to age at the time of diagnosis of SPC. Abbreviations: BC, breast cancer; CRC, colorectal cancer; SPC, second primary cancer. |
Overall, 5,559 ovarian cancer patients (49.19%) died during the follow-up period, and the proportion was almost identical (48.96%) in those with SPC and those with SPC and a family history (48.20%) (Table 1, bottom part).
Causes of deaths were analyzed for ovarian cancer with or without SPC depending on follow-up time from the diagnosis of ovarian cancer (Table 3). Causes of deaths were defined separately as ovarian cancer, SPC (if present), other cancers, and other (non-neoplastic) causes. For patients with SPC, SPC was the main cause of death in the last two follow-up periods (43.1%, 5–10 years and 50.9%, >10 years) and overall (42.1%). During the follow-up time up to 5 years, ovarian cancer was the main cause of death. Other causes were minor, reaching 15.1% in the last period. For ovarian cancer without SPC, the dominant cause of death was ovarian cancer. In the last follow-up period, other causes emerged, reaching 32.5%. Among 102 other cancers, many death codes indicated multiple and poorly localized cancers or tumors of undefined behavior; CUP was specified for 19 deaths.
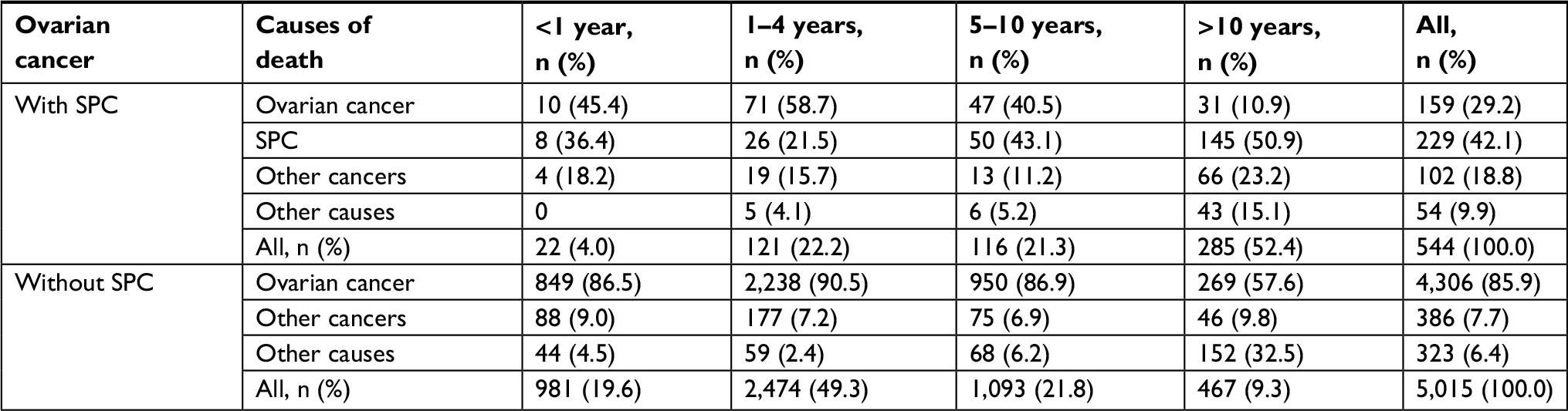 | Table 3 Causes of death in ovarian cancer patients with or without SPC according to follow-up time from ovarian cancer diagnosis Abbreviation: SPC, second primary cancer. |
Of the total 544 deaths in patients with SPC, causes of death were distributed among ovarian cancer (29.2%), SPC (42.1%), other cancers (18.8%), and other causes (9.9%) (Table S2). Among the 102 patients who died from other cancers, 28 deaths were due to higher order primaries (third, fourth, etc, primaries), and the rest were cancers assigned as the cause of death by the death registrar. Among 28 deaths with higher order primaries, eight patients shared second and third primary cancers and cause of death (three were breast cancer and the rest were other individual cancers). Most fatalities were caused by breast (88), colorectal (75), and lung (66) cancers, and by CUP (47). Figure 2 shows the distribution of causes of death for all patients with SPC and for cancers causing at least ten deaths. Pancreatic cancer, leukemia (seven acute myeloid leukemia, three acute monocytic leukemia, and one megakaryocytic type), liver, and lung cancers were most fatal as SPCs and they were responsible for more than 70% of deaths. Melanoma and CUP appeared to be the least fatal types.
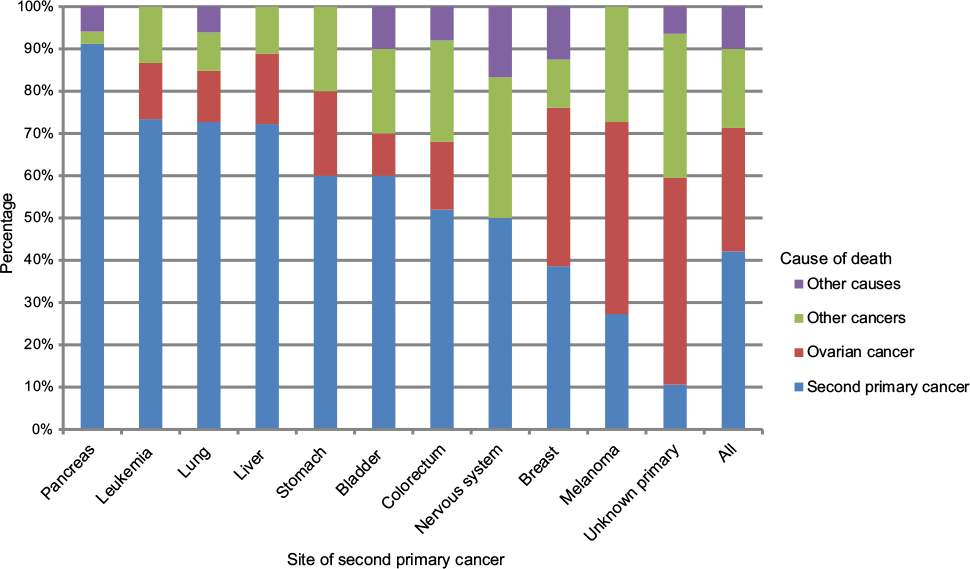 | Figure 2 Distribution of cause of death: second primary cancer, ovarian cancer, other cancers, and other causes among ovarian cancer patients with second primary cancer. Only cancer sites with more than ten cases of death are displayed. |
Discussion
In the present study, 9.8% of the ovarian cancer patients were diagnosed with SPC at a median of 8 years after the initial diagnosis. The proportion of third primaries was approximately similar (11.0% of SPCs). As this was a familial study focusing on the offspring generation, who were only followed to younger ages than their parents, the median diagnostic age of ovarian cancer (50 years) was lower than the median diagnostic age in the whole population including the parents (63 years). Patients’ family history was an important contributor to the risk of SPC and was found in 67.6% of the ovarian cancer patients. A family history of colon cancer was associated with close to a 2.5-fold increased risk and of lung and breast cancers with more than a two-fold risk. The present data show that in patients with SPC, this cancer was the main cause of death, accounting for 42.1% of all deaths. In patients without SPCs, ovarian cancer was the overwhelming cause of death, accounting for 85.9% of all deaths.
The clinical implications of the findings reiterate the notion that multiple primary cancers often emerge in the hereditary cancer setting.4,21 It is thus likely that some of the observed SPCs arose in mutation carriers; for example, breast and pancreatic cancers are found in BRCA1/2 mutation carriers,22 whereas colorectal cancer is the hallmark cancer in HNPCC associated with MMR mutations.23 Population frequencies of deleterious BRCA1/2 or MMR mutations are not known for Sweden but the estimate for MMR mutation carriers is 1/1200 (0.08%).24 Among the US European population, BRCA1/2 mutations are assumed to account for 1.4% of the breast cancer cases, and as the cumulative incidence of breast cancer is 10%, the population frequency of these mutations would be 0.14%;25 the mutation frequency in Sweden is estimated to be 1.8% of all breast cancers, which would translate to a population frequency of 0.18% (Kamila Czene, unpublished). According to a Swedish study, about 11% of the ovarian cancers were caused by germline mutations in the BRCA1/2 and 2% in the MMR genes.26 As we have no genetic data for the Swedish population, we tried to estimate the potential role of these genes in the discordant familial associations by removing high-risk families according to previously defined criteria.14,15 After excluding such patients, the effect of family history on SPC risk remained. This suggests that some other genetic or environmental factors are likely to contribute to the excess number of ovarian cancer patients later developing second cancers.
Even though family history contributed to a larger number of patients with SPCs, the overall proportion of deaths in patients with a family history was in line with overall mortality in ovarian cancer. Although SPCs would be expected to convey an unfavorable prognosis, particularly in cancers with good survival rates, such as breast cancer, survival of ovarian cancer is relatively poor and thus SPCs did not influence mortality.27 However, Figure 2 shows that fatal first primary cancers, such as pancreatic cancer, were also fatal as SPCs, and these influenced survival negatively.
According to Figure 2, CUP was among the least fatal cancers although, as a metastatic cancer, it is known to carry a miserable prognosis, similar to or even worse than pancreatic cancer.28–30 The likely reason for this discrepancy is the death registration practice in Sweden for CUP. The cause of death is listed as the cancer that was thought to kill the patient, ie, if the death registrar reasons that lung metastases killed the patient, the cause of death is assigned to lung cancer.31 Another deviation in mortality was leukemia which, overall, is only moderately fatal as the first primary (5-year survival 62.7%).2 The likely reason for the high mortality of leukemia as an SPC was that ten of eleven cases were acute leukemia.
One of the challenges for studies on SPCs is making an accurate diagnosis to separate the second primary from metastases resulting from the first primary. An ad hoc study on the diagnostic accuracy of second neoplasms in the Swedish Cancer Registry found 98% to be correctly classified; no recorded SPC was found to be a metastasis.32 Another concern for this study is the difficulty in identifying the real cause of death for individuals with multiple primary cancer diagnoses. One study performed in Sweden, based on international standards, indicated 77% agreement between the cause of death from death certificates and the expected cause of death based on case summaries.33 As an additional caveat, clinical, behavioral, and environmental information is not available in the database, such as histology, tumor stage, and treatment for the ovarian cancer, body mass index, behavioral features such as smoking patterns, and oral contraceptive use. However, we adjusted the socioeconomic factors, which removes a certain degree of confounding.
Conclusion
Approximately 10% of the ovarian cancer patients were diagnosed with SPC and two-thirds of the patients had a first degree relative diagnosed with cancer. High familial risks were found for breast and colorectal cancers, which are known to manifest in ovarian cancer-related syndromes. However, a family history of a particular cancer contributed to an increased risk of SPC at the same site, despite the non-inclusion of possible high-risk families. Thus, our data collectively indicate that taking a family history at the time of diagnosis of ovarian cancer may alert physicians to disease management, which may help the patient as well as her family members.
Abbreviations
BC, breast cancer; CRC, colorectal cancer; CUP, cancer of unknown primary; HNPCC, hereditary non-polyposis colorectal cancer; OC, ovarian cancer; RR, relative risk; SCC, squamous cell carcinoma; SPC, second primary cancer; UAT, upper aerodigestive tract.
Ethics approval and informed consent
The study was approved by the ethical committee of Lund University without requirement for informed consent, and the study was conducted in accordance with the tenets of the Declaration of Helsinki. The Swedish government registers every individual residing in Sweden in this database, and data are subject to confidentiality irrespective of individuals’ disease status. Upon accessing the database, a confidentiality agreement was signed by users. Hence, patient confidentiality is assured by approval of the study obtained from the ethics committee.
Data availability
The data that support the findings of this study are available from Lund University but restrictions apply to the availability of these data, which were used under license for the current study and so are not publicly available.
Acknowledgments
This project was supported by grants from German Cancer Aid, the Swedish Research Council (2014-2517, 2014-10134, and 2016-01176), ALF funding from Region Skåne, as well as by the China Scholarship Council (201606100057, for doctoral student GZ). The sponsors had no roles in study design; in the collection, analysis, and interpretation of data; in the writing of the report; or in the decision to submit the article for publication.
Author contributions
KH proposed the research conception and study design. KS, KH, GZ, and SC contributed to the collection and assembly of data. GZ, KH, SC, and AF were responsible for the data analysis and interpretation. KH completed the draft of the manuscript, and all the authors revised it critically for important intellectual content. The final manuscript was reviewed and approved by all authors. All authors are accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
National Board of Health and Welfare [webpage on the Internet]. Statistics on Cancer Incidence 2016. Official Statistics of Sweden; 2017. Available from: http://www.socialstyrelsen.se/statistics/statisticaldatabase/cancer. | ||
Jemal A, Ward EM, Johnson CJ, et al. Annual Report to the Nation on the Status of Cancer, 1975–2014, Featuring Survival. J Natl Cancer Inst. 2017;109(9). | ||
Hemminki K, Aaltonen L, Li X. Subsequent primary malignancies after endometrial carcinoma and ovarian carcinoma. Cancer. 2003;97(10):2432–2439. | ||
Travis LB, Demark Wahnefried W, Allan JM, Wood ME, Ng AK. Aetiology, genetics and prevention of secondary neoplasms in adult cancer survivors. Nat Rev Clin Oncol. 2013;10(5):289–301. | ||
Wood ME, Vogel V, Ng A, Foxhall L, Goodwin P, Travis LB. Second malignant neoplasms: assessment and strategies for risk reduction. J Clin Oncol. 2012;30(30):3734–3745. | ||
Schaapveld M, Aleman BM, van Eggermond AM, et al. Second Cancer Risk Up to 40 Years after Treatment for Hodgkin’s Lymphoma. N Engl J Med. 2015;373(26):2499–2511. | ||
Sud A, Thomsen H, Sundquist K, Houlston RS, Hemminki K. Risk of Second Cancer in Hodgkin Lymphoma Survivors and Influence of Family History. J Clin Oncol. 2017;35(14):1584–1590. | ||
Frank C, Sundquist J, Yu H, Hemminki A, Hemminki K. Concordant and discordant familial cancer: Familial risks proportions and population impact. Int J Cancer. 2017;140(7):1510–1516. | ||
Donin N, Filson C, Drakaki A, et al. Risk of second primary malignancies among cancer survivors in the United States, 1992 through 2008. Cancer. 2016;122(19):3075–3086. | ||
Lakhani SR, Manek S, Penault-Llorca F, et al. Pathology of ovarian cancers in BRCA1 and BRCA2 carriers. Clin Cancer Res. 2004;10(7):2473–2481. | ||
Pal T, Permuth-Wey J, Betts JA, et al. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer. 2005;104(12):2807–2816. | ||
Geary J, Sasieni P, Houlston R, et al. Gene-related cancer spectrum in families with hereditary non-polyposis colorectal cancer (HNPCC). Fam Cancer. 2008;7(2):163–172. | ||
Watson P, Bützow R, Lynch HT, et al. The clinical features of ovarian cancer in hereditary nonpolyposis colorectal cancer. Gynecol Oncol. 2001;82(2):223–228. | ||
Lorenzo Bermejo J, Hemminki K. A population-based assessment of the clustering of breast cancer in families eligible for testing of BRCA1 and BRCA2 mutations. Ann Oncol. 2005;16(2):322–329. | ||
Bermejo JL, Eng C, Hemminki K. Cancer characteristics in Swedish families fulfilling criteria for hereditary nonpolyposis colorectal cancer. Gastroenterology. 2005;129(6):1889–1899. | ||
Brooke HL, Talbäck M, Hörnblad J, et al. The Swedish cause of death register. Eur J Epidemiol. 2017;32(9):765–773. | ||
Ji J, Sundquist K, Sundquist J, Hemminki K. Comparability of cancer identification among Death Registry, Cancer Registry and Hospital Discharge Registry. Int J Cancer. 2012;131(9):2085–2093. | ||
Pukkala E, Engholm G, Højsgaard Schmidt LK, et al. Nordic Cancer Registries - an overview of their procedures and data comparability. Acta Oncol. 2018;57(4):440–455. | ||
Riihimäki M, Hemminki A, Fallah M, et al. Metastatic sites and survival in lung cancer. Lung Cancer. 2014;86(1):78–84. | ||
Riihimäki M, Hemminki A, Sundquist J, Hemminki K. Patterns of metastasis in colon and rectal cancer. Sci Rep. 2016;6:29765. | ||
Vogt A, Schmid S, Heinimann K, et al. Multiple primary tumours: challenges and approaches, a review. ESMO Open. 2017;2(2):e000172. | ||
Moran A, O’Hara C, Khan S, et al. Risk of cancer other than breast or ovarian in individuals with BRCA1 and BRCA2 mutations. Fam Cancer. 2012;11(2):235–242. | ||
Bonadona V, Bonaïti B, Olschwang S, et al; French Cancer Genetics Network Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA. 2011;305(22):2304-10. | ||
Lagerstedt-Robinson K, Rohlin A, Aravidis C, et al. Mismatch repair gene mutation spectrum in the Swedish Lynch syndrome population. Oncol Rep. 2016;36(5):2823–2835. | ||
Palomaki GE. Is it time for BRCA1/2 mutation screening in the general adult population?: impact of population characteristics. Genet Med. 2015;17(1):24–26. | ||
Malander S, Rambech E, Kristoffersson U, et al. The contribution of the hereditary nonpolyposis colorectal cancer syndrome to the development of ovarian cancer. Gynecol Oncol. 2006;101(2):238–243. | ||
Riihimäki M, Thomsen H, Brandt A, Sundquist J, Hemminki K. Death causes in breast cancer patients. Ann Oncol. 2012;23(3):604–610. | ||
Hemminki K, Bevier M, Hemminki A, Sundquist J. Survival in cancer of unknown primary site: population-based analysis by site and histology. Ann Oncol. 2012;23(7):1854–1863. | ||
Riihimäki M, Hemminki A, Sundquist K, Hemminki K. Time trends in survival from cancer of unknown primary: small steps forward. Eur J Cancer. 2013;49(10):2403–2410. | ||
Riihimäki M, Hemminki A, Sundquist K, Hemminki K. Causes of death in patients with extranodal cancer of unknown primary: searching for the primary site. BMC Cancer. 2014;14:439. | ||
Hemminki K, Bevier M, Sundquist J, Hemminki A. Cancer of unknown primary (CUP): does cause of death and family history implicate hidden phenotypically changed primaries? Ann Oncol. 2012;23(10):2720–2724. | ||
Frödin JE, Ericsson J, Barlow L. Multiple primary malignant tumors in a national cancer registry--reliability of reporting. Acta Oncol. 1997;36(5):465–469. | ||
Johansson LA, Björkenstam C, Westerling R. Unexplained differences between hospital and mortality data indicated mistakes in death certification: an investigation of 1,094 deaths in Sweden during 1995. J Clin Epidemiol. 2009;62(11):1202–1209. |
Supplementary materials
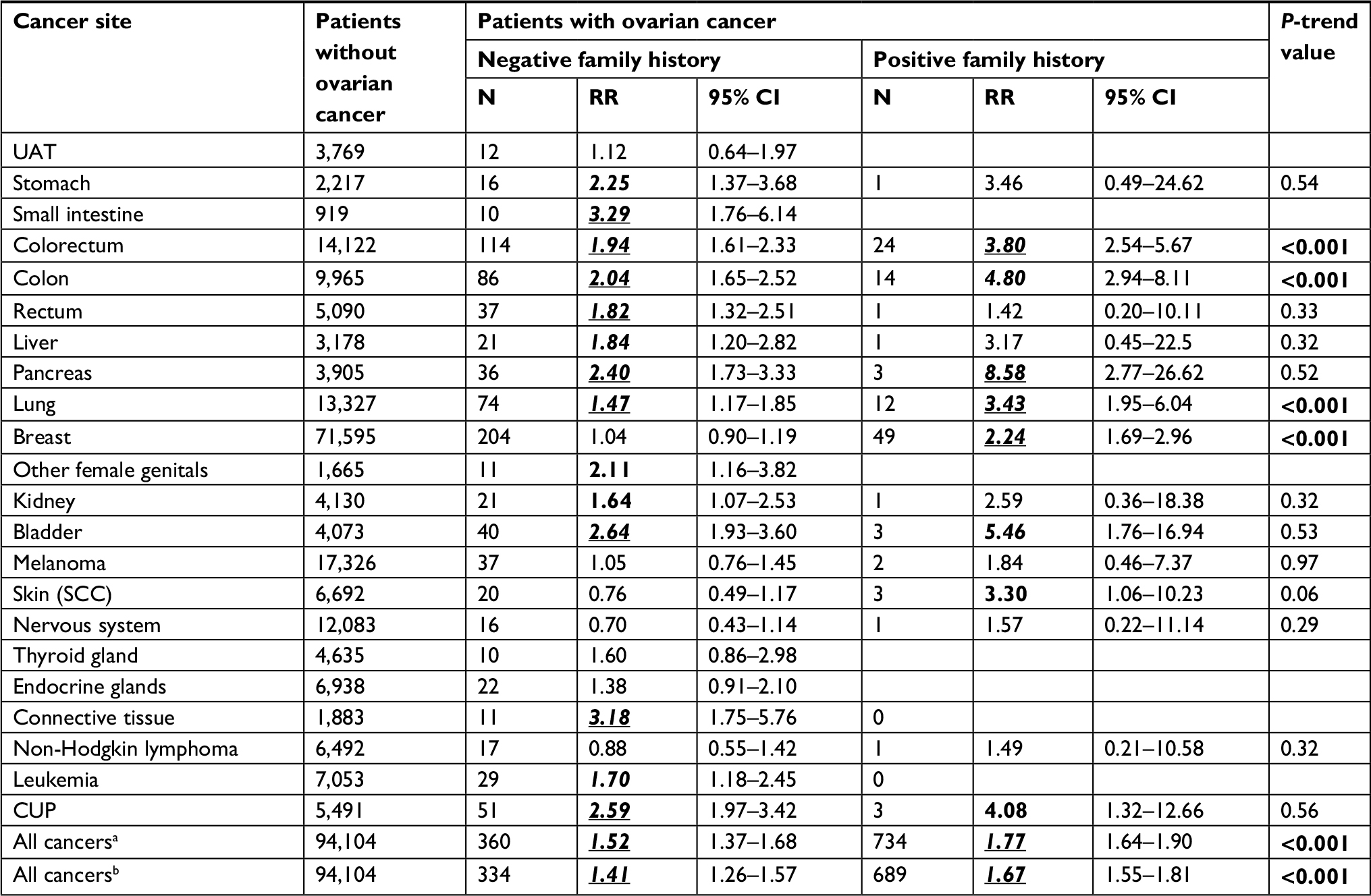 | Table S1 Risk of second primary cancers in ovarian cancer patients with or without a family history of concordant cancer, after removing high-risk families Notes: aAll cancers including second ovarian cancer. bAll cancers excluding second ovarian cancer. Bold, italic and underlined values indicate that the 95% CI, 99% CI and 99.9% CI did not overlap with 1.00 respectively. Abbreviations: CUP, cancer of unknown primary; RR, relative risk; SCC, squamous cell carcinoma; UAT, upper aerodigestive tract. |
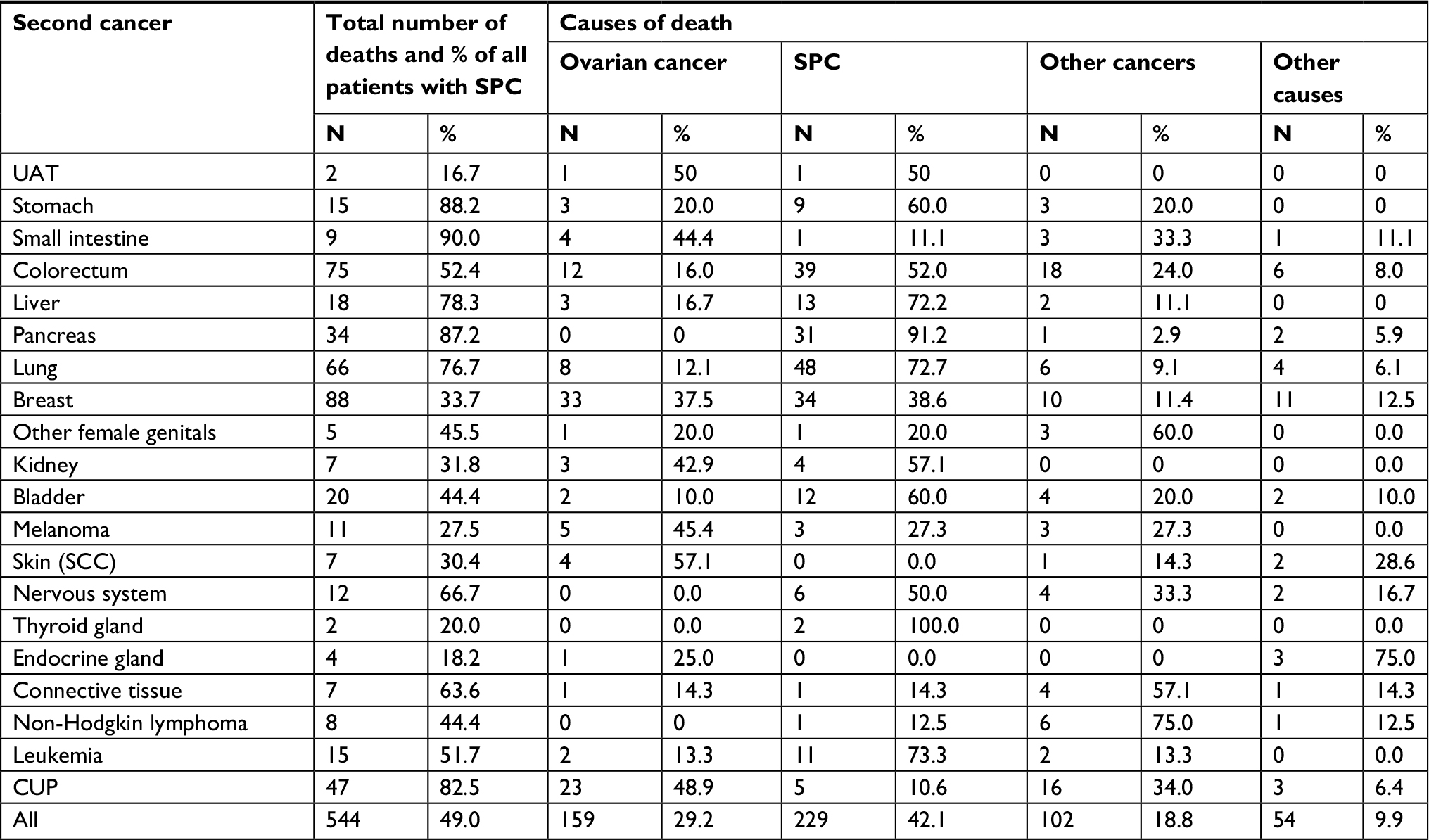 | Table S2 Causes of death in ovarian cancer patients diagnosed with SPC Abbreviations: CUP, cancer of unknown primary; SCC, squamous cell carcinoma; SPC, second primary cancer; UAT, upper aerodigestive tract. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.