Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
Familial Partial Lipodystrophy (FPLD): Recent Insights
Authors Bagias C ![]() , Xiarchou A, Bargiota A, Tigas S
, Xiarchou A, Bargiota A, Tigas S
Received 17 January 2020
Accepted for publication 31 March 2020
Published 6 May 2020 Volume 2020:13 Pages 1531—1544
DOI https://doi.org/10.2147/DMSO.S206053
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Christos Bagias,1 Angeliki Xiarchou,1 Alexandra Bargiota,2 Stelios Tigas1
1Department of Endocrinology, University of Ioannina, Ioannina, Greece; 2Department of Endocrinology, University of Thessaly, Larissa, Greece
Correspondence: Stelios Tigas
Department of Endocrinology, University of Ioannina, Ioannina 45110, Greece
Tel +30 2651007800
Email [email protected]
Abstract: Lipodystrophies are a heterogeneous group of congenital or acquired disorders, characterized by partial or generalized loss of adipose tissue. Familial partial lipodystrophy (FPLD) presents with genetic and phenotypic variability with insulin resistance, hypertriglyceridemia and hepatic steatosis being the cardinal metabolic features. The severity of the metabolic derangements is in proportion with the degree of lipoatrophy. The underpinning pathogenetic mechanism is the limited capacity of adipose tissue to store lipids leading to lipotoxicity, low-grade inflammation, altered adipokine secretion and ectopic fat tissue accumulation. Advances in molecular genetics have led to the discovery of new genes and improved our knowledge of the regulation of adipose tissue biology. Diagnosis relies predominantly on clinical findings, such as abnormal fat tissue topography and signs of insulin resistance and is confirmed by genetic analysis. In addition to anthropometry and conventional imaging, new techniques such as color-coded imaging of fat depots allow more accurate assessment of the regional fat distribution and differentiation of lipodystrophic syndromes from common metabolic syndrome phenotype. The treatment of patients with lipodystrophy has proven to be challenging. The use of a human leptin analogue, metreleptin, has recently been approved in the management of FPLD with evidence suggesting improved metabolic profile, satiety, reproductive function and self-perception. Preliminary data on the use of glucagon-like peptide 1 receptor agonists (GLP1 Ras) and sodium-glucose co-transporter 2 (SGLT2) inhibitors in cases of FPLD have shown promising results with reduction in total insulin requirements and improvement in glycemic control. Finally, investigational trials for new therapeutic agents in the management of FPLD are underway.
Keywords: lipodystrophy, partial lipodystrophy, familial lipodystrophy, leptin
Introduction
Lipodystrophies are a heterogeneous group of disorders characterized by selective partial or generalized loss of adipose tissue. They can be either congenital or acquired in origin, and according to the distribution of adipose tissue loss, lipodystrophies are categorized to localized, partial, or generalized. Lipodystrophy is an ultra-rare syndrome with an estimated prevalence of 1.3–4.7 cases per million,1 although this might be an underestimate, as many cases remain unrecognized. Congenital lipodystrophies, based on the pattern of adipose tissue loss, can be categorized into either familial partial lipodystrophy or congenital generalized lipodystrophy (CGL). Familial partial lipodystrophy (FPLD) is a disease with considerable genetic and phenotypic variability that was first described in the 1970s2,3 but attracted greater attention in the last 20 years due to the expanded knowledge on genetics, adipose tissue biology and discovery of the leptin gene. Depending on the extent of fat loss, described as lipoatrophy by many authors, affected individuals may present with primarily cosmetic problems whereas others may develop severe metabolic complications such as insulin resistance and diabetes, hypertriglyceridemia, non-alcoholic steatohepatitis (NASH) and premature atherosclerosis. Treating these patients has proven to be challenging, but new modalities such as recombinant human leptin therapy have shown some promising results. The purpose of this review is to summarize the latest insights and findings for FPLD, focusing on new knowledge on the pathogenesis, molecular genetics and therapeutic options.
Pathophysiology of Metabolic Disorders Associated with FPLD
So far, six different types of FPLD (FPLD1 to FPLD6) and several other unclassified forms of partial lipodystrophy in the context of rare genetic syndromes with distinct phenotypic characteristics and genetic causes have been described (Table 1).4,5
 |
Table 1 Classification of Partial Lipodystrophies, Prevalence and OMIM (Online Mendelian Inheritance in Man) Phenotype Number |
FPLDs are generally dominantly inherited and are characterized by loss of subcutaneous adipose tissue (usually during late childhood or puberty) from the upper and lower extremities as well as from the truncal region.6 Common clinical features of patients with lipodystrophies are shown in Table 2. Limited capacity of adipose tissue to store lipids and failure of buffering post-prandial lipid refluxes have a fundamental role in the pathogenesis of the metabolic disorders associated with lipodystrophies. It has been proposed that, when the genetically determined capacity of adipose tissue to expand is surpassed, lipotoxicity, macrophage infiltration, mitochondrial dysfunction and oxidative stress develop within dysfunctional lipodystrophic adipocytes leading to altered adipokine secretion.7–10 As a result, ectopic fat tissue accumulates in the liver, muscle, pancreas and the vasculature, resulting in insulin resistance and abnormal lipid and glucose metabolism.7 The metabolic derangement is generally in proportion with the degree of lipoatrophy.
 |
Table 2 Clinical Features of Familial Partial Lipodystrophies |
Low levels of leptin and adiponectin may in part explain some of the metabolic and reproductive changes observed in those patients with more extensive fat loss.11 Indeed, low adiponectin levels promote insulin resistance and are closely related to the development of diabetes and cardiovascular disease whereas low leptin levels result in hyperphagia and reduced energy expenditure.12 Lipodystrophy-associated metabolic derangements have recently been associated with increased levels of mitochondrial DNA damage and transcriptional activation of genes involved with antioxidant response and DNA repair.13 Features of polycystic ovarian syndrome and subfertility are common in women with lipodystrophies. Although leptin receptors are present in the ovaries and the prostate gland, current evidence suggests that the primary leptin actions on human reproductive function are central and that its effects on peripheral reproductive tissues are most likely indirect.14,15
Interestingly, data suggest the existence of tissue-selective insulin resistance8 with inhibition of insulin signaling in liver and muscle leading to increased gluconeogenesis, increased lipogenesis and reduced peripheral glucose uptake. On the other hand, insulin seems to preserve its mitogenic effect on the ovarian theca cells and its anabolic action causing hyperandrogenemia and acromegaloid features, respectively.16 This pathway selective insulin resistance leads to the development of the cardinal metabolic disorders found in lipodystrophic syndromes, namely diabetes mellitus, dyslipidemia, hepatic steatosis and reproductive dysfunction (reduced fertility, PCOS). Furthermore, recent evidence from genome-wide association studies (GWAS) in a general population suggests that genetic variants related to gluteofemoral versus abdominal fat storage are associated with increased risk of cardiovascular disease and type 2 diabetes.17 These findings support the hypothesis that similar genetically determined mechanisms may play a role in the pathogenesis of the common metabolic syndrome and of the much rarer lipodystrophy syndromes.5
Molecular Pathogenesis, Genetics and Association with Clinical Phenotype
In recent years. research has shed light on the molecular pathogenesis of familial lipodystrophies. Despite progress, in many cases no known mutations are found in patients presenting with clinical features of lipodystrophy, suggesting that there are still more genes to be identified. In general, the responsible genes can be categorized into those involved in: i) adipogenesis ii) lipid droplet formation and lipolysis iii) cell membrane integrity and iv) DNA repair (Table 3; Figure 1). FPLD type 1 (Köbberling type lipodystrophy) is thought to have a polygenic etiology and will be described separately.18
 |
Table 3 Gene Mutations Involved in Familial Partial Lipodystrophy (FPLD) Pathogenesis, Mode of Inheritance and Corresponding Phenotypes |
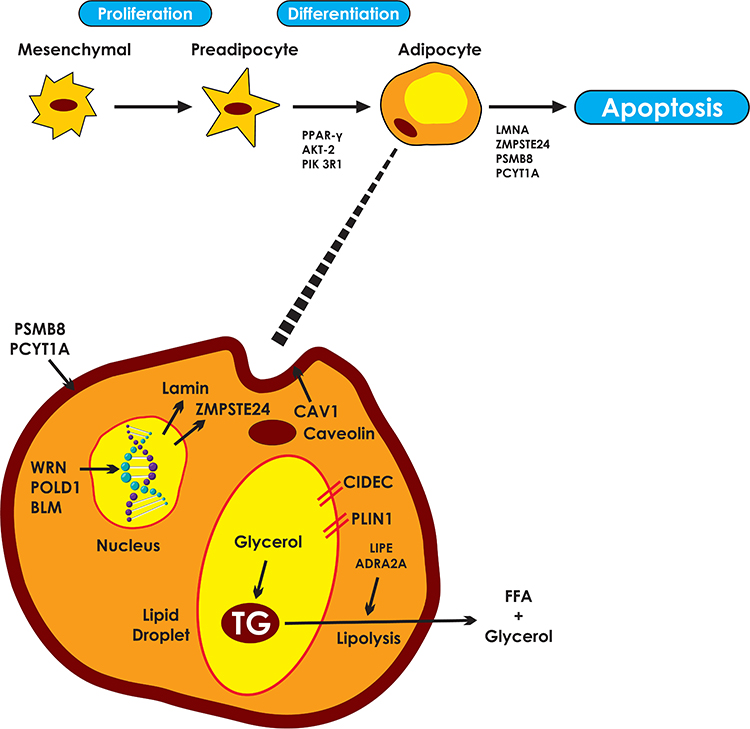 |
Figure 1 Pathways involved in the molecular pathogenesis of familial partial lipodystrophies. Responsible genes may affect adipocyte differentiation, cell membrane integrity, DNA repair, lipid droplet formation and lipolysis. PPARγ, the “master regulator” of adipogenesis coordinates the transcription of proteins central to the adipocyte function. AKT2 and PIK3R1 are involved in insulin signaling pathways, mediating adipocyte differentiation. LMNA gene encodes lamins A/C which are essential components of the nuclear envelope. ZMPSTE24 is responsible for the proteolysis of prelamin A to mature/active lamin A. PSM8 and PCYT1A are responsible for the composition and integrity of cell membranes. Mutations lead to intracellular oxidative stress, inflammation and apoptosis. WRN, POLD1 and BLM participate in DNA repair and replication, ensuring genomic stability. Caveolin 1, the product of CAV1, participates in the formation of caveolae. Caveolin vesicles are created from endocytosis of caveolae which carry cholesterol and sphingolipids from the cell surface. Caveolin vesicles are merged with the lipid droplet, mediating the transcytosis of fatty acids. CIDEC is responsible for the formation of unilocular lipid droplets and PLIN1 for the structure of the lipid droplets. Finally, LIPE and ADRA2A regulate triglyceride lipolysis to free fatty acids and glycerol. |
Adipogenesis – Adipocyte Differentiation-Related Genes
In this category belong the two most common genetic causes for FPLD, that is the pathogenic variants in the peroxisome proliferator-activated receptor γ (PPARγ) and LMNA genes as well as some rarer gene mutations (Table 3).
PPARγ gene variants are associated with variable lipodystrophy phenotypes.19 Dominant-negative mutations in the PPARγ are related to FPLD type 3.20 Patients typically present in late childhood with loss of subcutaneous fat in the extremities and gluteofemoral region and “reactive” fat deposition in the viscera. Face and neck regions are usually spared. Metabolic complications secondary to insulin resistance present in adulthood and are more prominent in women. Early cardiovascular disease has also been reported.21
PPARγ, the master regulator of adipogenesis and adipose tissue maintenance,22 is expressed in many tissues but predominantly found in white and brown adipose tissue.23 It promotes lipogenesis without creating larger adipocytes; more and smaller are created instead.24 Furthermore, its action is to promote subcutaneous to visceral fat differentiation.25
LMNA gene encodes intermediate filament proteins called lamins. Lamins A and C are the major proteins expressed by the gene and provide structural stability to the nuclear envelope and the cytoskeleton. Lamins are expressed in all cell types and mutated variants lead to premature apoptosis of the cells.26 Autosomal dominant mutations of LMNA gene are associated with FPLD type 2 (Dunnigan type), the most common form of FPLD,27 whereas autosomal recessive mutations are linked to mandibuloacral dysplasia (MAD) type A.28 The onset of lipodystrophy in FPLD2 is gradual; lipodystrophic phenotypic characteristics may not be present until puberty, a period when fat depots expand due to sex hormone abundance.29 However, recent data from Patni et al suggest that body fat distribution may change at earlier stages. Using objective measurements of adiposity (dual-energy X-ray absorptiometry, DXA) in a cohort of 46 patients with FPLD2, the authors showed that distal lipoatrophy is present earlier than thelarche.30 Muscular “pseudohypertrophy” is commonly observed especially in females, and accumulation of fat on the face, neck and supraclavicular areas is also noted, giving patients a “Cushingoid appearance”.6 Insulin resistance is present in >80% of the cases leading to hepatic steatosis, hypertriglyceridemia, acanthosis nigricans and early atherosclerotic disease. Patients with LMNA-related lipodystrophy may also manifest muscular dystrophy,31 dilated cardiomyopathy32 and proteinuric nephropathy.33
Mandibuloacral dysplasia (MAD) is an extremely rare autosomal recessive disorder with progeroid features and lipodystrophy. These patients develop two distinct patterns of lipoatrophy; in type A (MADA), which is due to LMNA gene mutations, there is loss of fat from the extremities but normal or excessive fat deposition in the neck and trunk whereas in type B (MADB), which is caused by mutations of the zinc metalloproteinase (ZMPSTE24) gene, loss of subcutaneous fat is more generalized.34 ZMPSTE24 gene encodes a Zinc metalloproteinase which is responsible for the proteolysis of prelamin A to mature lamin A. Deficiency in ZMPSTE24 causes toxic prelamin accumulation35 and is linked to a spectrum of phenotypes, varying from restrictive dermopathy (total loss of enzyme activity) to MADB and generalized lipodystrophy.36 Both types of MAD share common characteristics such as mandibular and clavicular hypoplasia, acroosteolysis and delayed dentition.37 Progeroid features such as pointed nose, microstomy, high-pitched voice, skin atrophy, nail dysplasia and alopecia characterize both types but are more prominent in MADB.38 Metabolic complications are mild in both types, whereas patients with MADB are at risk of myopathy,39 calcified skin nodules40 and focal segmental glomerulosclerosis.41
AKT2 (protein kinase B) is involved in the insulin signaling pathway, mediating adipocyte differentiation. Missense mutation in AKT2 gene has been reported in only one family (four subjects) who presented with severe insulin resistance and partial distal lipodystrophy.42
PIK3R1 encodes the 85KD regulator subunit of phosphatidylinositol 3 kinase, an enzyme playing a key role in insulin signaling. Missense mutation of PIK3R1 gene is related to SHORT syndrome43,44 which manifests with short stature, hyperextensibility of the joints, ocular depression, Rieger anomaly, and teething delay. Patients present with partial lipoatrophy involving the face, arms and upper trunk, with lower extremities being spared. Severe insulin resistance is present, and the majority of cases develop diabetes mellitus after adolescence.45 Recently, a mutation in PRKCE gene encoding protein kinase c epsilon, a protein involved in apoptosis and insulin signaling, was identified in a patient with clinical characteristics of SHORT syndrome but negative PIK3R1 mutation.46
Genes Involved in Lipid Droplet Formation and Lipolysis
This category of FPLD genetic causes includes the pathogenic variants in CIDEC, PLIN1, LIPE, ADRA2A and CAV1.
CIDEC encoded protein promotes the formation of unilocular lipid droplets by transferring lipids from the smaller to larger droplets.47 Up to date, one case of partial lipodystrophy has been recognized as secondary to nonsense CIDEC mutation. The patient presented with lack of subcutaneous fat from the lower limbs and the femorogluteal region but preserved visceral and neck fat. Metabolically, insulin-resistant diabetes, hepatic steatosis, hypertriglyceridemia and hypertension were present. Fat biopsy revealed white adipocytes with multilocular lipid droplets.48
PLIN1 is a structural protein of adipocyte lipid droplets, which also regulates lipolysis by controlling intracellular lipases.49 Autosomal dominant frameshift mutations have been associated with FPLD type 4, a syndrome manifesting with lower limb lipoatrophy, facial acromegaloid features and muscular hypertrophy. All affected cases (eight families) share common metabolic characteristics, such as insulin resistance, hyperandrogenaemia, hypertension, hypertriglyceridemia and hepatic steatosis.50 Recent work though by Laver et al51 suggested that null variants of PLIN1 are not related to overt lipodystrophy and should not be considered pathogenic. This observation suggests that only PLIN1 variants with specific genetic mechanisms are associated with lipodystrophic syndromes.
LIPE encodes hormone-sensitive lipase, an enzyme highly expressed in adipose tissue responsible for hydrolyzing esters to free fatty acids. Mutations lead to dysmorphic and dysfunctional white adipocytes in FPLD type 6. Phenotypic manifestations include distal lipoatrophy with increased visceral fat depots and progressive myopathy. Insulin resistance with dyslipidemia and hepatic steatosis is present, and vitiligo has also been described.52,53
ADRA2A is a non-adrenergic receptor regulating lipolysis in the adipocytes. Heterozygous mutations of the relevant gene have been described in one African-American pedigree. Affected individuals present with distal lipoatrophy and increased deposition of fat in the face and intraabdominal region. Increased muscularity is also present.54
CAV1 gene encodes caveolin-1, a protein that serves in the formation of caveolae in plasma membranes. By binding fatty acids on the plasma membrane of the adipocyte and translocating them into lipid droplets, caveolin-1 appears to play a role in lipid droplet formation and adipocyte differentiation.55 Homozygous and heterozygous CAV1 variants cause CGL type356 and neonatal onset GL, respectively.57 However, Cao et al also described three cases of different heterozygous CAV1 mutations causing atypical partial lipodystrophy.58 Patients had subcutaneous fat loss affecting the face and upper part of the body, hypertriglyceridemia and congenital cataracts with or without neurological findings.
Cell Membrane Integrity-Related Genes
PCYT1A is involved in the phosphatidylcholine synthesis, a major component of all cell membranes. Phenotypic heterogeneity, depending on the affected protein domain, exists and patients can present with distal lipoatrophy, short stature, visual impairment due to cone-rod dystrophy and skeletal anomalies due to spondylometaphyseal dysplasia.59,60
Proteasome subunit beta type 8 (PSMB8) encodes a protein responsible for cell homeostasis and its deficiency leads to hypersecretion of interferons, cellular stress, inflammation and is linked to the Chronic Atypical Neutrophilic Dermatosis (CANDLE) syndrome (also known as JMP or Nakajo–Nishimura syndrome).61 Up to date, more than 60 cases of this autosomal recessive syndrome have been described. Episodes of recurrent fever, with formation of cutaneous, erythematous, annular plaques present during the first months of life. Patients have typical facial characteristics with violaceous periorbital and periocular oedema. Lipodystrophy begins in early childhood and follows a progressive pattern, primarily affecting the face, upper trunk and upper limbs. Other clinical features include arthralgia without arthritis, finger clubbing, joint contractures and cardiomyopathy. Lipodystrophy follows the development of autoimmune panniculitis and is attributed to the toxic effect of interferons on adipose tissue.62
Genes Involved in DNA Repair
WRN encodes a helicase that plays an important role in repairing and maintaining the DNA structure.63 Null mutations of WRN gene, inherited in an autosomal recessive way, are responsible for Werner syndrome. Patients typically develop normally until puberty when growth ceases resulting in short stature. Progeroid characteristics and distal lipoatrophy with truncal obesity develop in the late 20s. Patients develop premature cataracts, hypogonadism, osteoporosis, atherosclerosis and are at higher risk of malignancies. “Bird – like” facial appearance and Achilles tendon ulcerations are pathognomonic of the syndrome.64
POLD1 encodes the catalytic subunit of the DNA polymerase delta, an enzyme responsible for DNA repair and stability. Mutations are related to MDPL syndrome, manifesting with mandibular hypoplasia, sensorineural deafness, progeroid features and acral lipoatrophy. Visceral fat depots are markedly increased. Other common characteristics include reduced muscle bulk in the limbs, hypogonadism, undescended testicles in males and poor breast development in females.65
BLM gene is responsible for the RecQ helicase production, a protein participating in the unwinding of the DNA helix. Autosomal recessive mutations in the BLM gene are related to the Bloom syndrome.66 Patients present with pre and postpartum growth restriction, short stature, microcephaly and distal lipoatrophy. Photosensitivity and telangiectasia are also described. The syndrome is characterized by high mortality due to immune system deficiency leading to recurrent infections and increased cancer prevalence. Male sterility and female infertility due to early menopause are other features of the syndrome.67
FPLD Type 1 (Köbberling-Type Lipodystrophy)
The syndrome described by Köbberling typically presents with distal lipoatrophy and increased facial, neck and visceral adiposity.68 The hallmark diagnostic feature is the formation of a ledge between affected (lipodystrophic) and non-affected areas. Metabolic complications present in early adulthood and include insulin resistance, hypertriglyceridemia and NASH.69 FPLD1 is commonly diagnosed on a clinical basis, mostly in female patients with limb lipoatrophy, central obesity and severe insulin resistance after exclusion of FPLD3 (PPARG mutations) which manifests a similar phenotype. It has recently been suggested that this phenotype has a strong polygenic contribution, illustrating the contribution of common alleles to severe forms of insulin resistance.18
In conclusion, lipodystrophies despite being classified as monogenic disorders, exhibit great genetic, allelic and phenotypic variability (Supplementary Table 1).70 Accumulating evidence suggests that patients’ phenotype is dictated not only by a given gene mutation but is also susceptible to genetic and environmental modifiers. Gender, race, coding and non-coding single nucleotide variants (SNVs) across the genome can alter expression of genes, leading to variable clinical features. Epigenetic (DNA methylation, histone modification) and environmental factors, such as nutrition, pregnancy and ageing, may also modulate gene expression.19
Diagnosis
Clinical features of lipoatrophy with insulin resistance, dyslipidemia and hepatic steatosis should always pose lipodystrophy as part of the differential diagnosis. Information regarding the onset and severity of symptoms and family penetration is crucial in establishing the lipodystrophy subtype.4,71 Thorough clinical examination may reveal abnormal topography of adipose tissue deposition, increased or prominent musculature, phlebomegaly and signs of insulin resistance such as acanthosis nigricans.
Anthropometry such as skinfold thickness and waist to hip ratio are also used to support the diagnosis. Objective measurements of adiposity (DXA and whole-body magnetic resonance imaging-MRI) provide information on the regional distribution of fat. Recently, Meral et al72 described a simple method to diagnose lipodystrophic syndromes called “Fat Shadows”. Images derived from DXA scans are reconstructed in order to present adipose tissue as “shadow” (Figure 2). Radiological signs, such as the “Dunnigan sign” (hypertrophy of mons pubis fat surrounded by lipoatrophy), can be easily identified by this method allowing for early diagnosis of lipodystrophy subtypes.
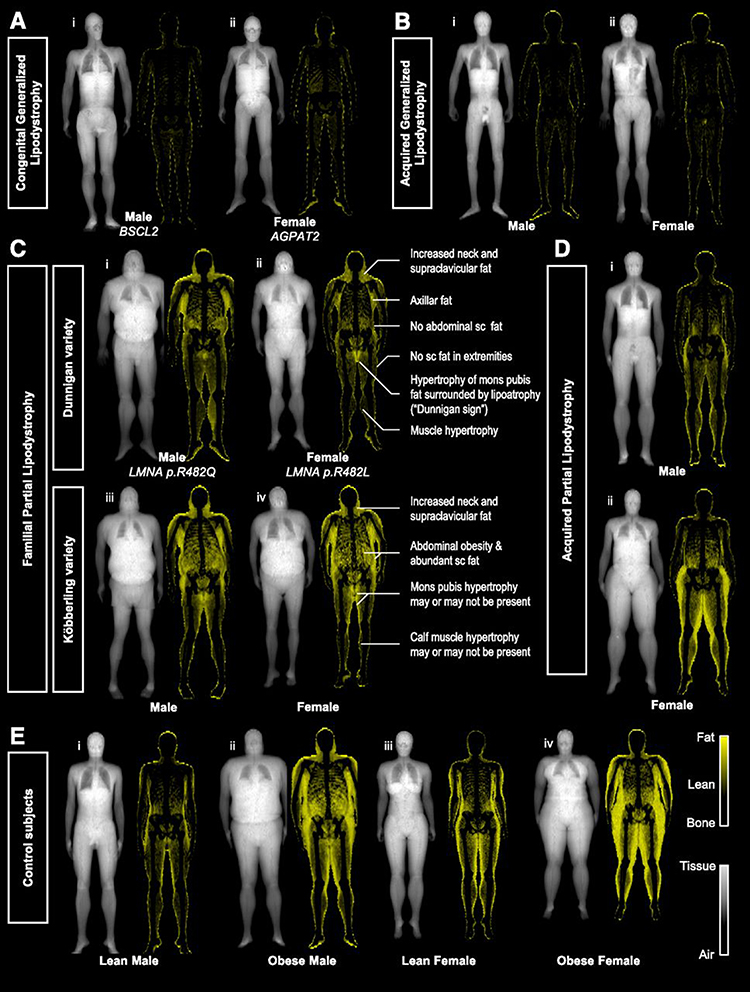 |
Figure 2 “Fat shadows”: reconstructed DXA scan images using color-coded representation to highlight adipose tissue. Patients with (A) congenital type 2 (i) and type 1 (ii) and (B) acquired generalized lipodystrophy have minimal residual fat depots. Patients with (C) FPLD of the Dunnigan (i and ii), and Köbberling variety (iii and iv), present with lipoatrophy of subcutaneous fat and hypertrophy of adipose tissue in the neck and upper trunk. Identification of the “Dunnigan sign” is diagnostic of FLPD2. Patients with acquired partial lipodistrophy (D) have loss of fat from the upper extremities and truncal region with lower half depots being either normal (i) or hypertrophied (ii). (E) Lean (i and iii) and obese (ii and iv) control subjects present gender-specific fat distribution. American Diabetes Association. "Fat Shadows" From DXA for the Qualitative Assessment of Lipodystrophy: When a Picture Is Worth a Thousand Numbers, Diabetes Care, 2018. Copyright and all rights reserved. Material from this publication has been used with the permission of American Diabetes Association.72 |
Diagnosis of FPLD cannot be confirmed or excluded based on serum leptin levels, as these are widely variable. Furthermore, due to lack of specific reference data, quantification of adipocytokines (such as leptin or adiponectin levels in serum) is not currently recommended as a diagnostic tool. Genetic analysis including single or a panel of candidate gene sequencing or whole-exome/whole-genome sequencing is required to establish the diagnosis and to guide further screening and counseling although a negative result cannot exclude lipodystrophic syndromes.71
Treatment
The management of patients with FPLD is challenging, and requires a multidisciplinary approach focusing on both prevention and therapy of metabolic complications as well as cosmetic interventions.71 Lifestyle changes with diet modification and exercise are recommended. Patients should be advised to follow a balanced diet containing approximately 50–60% of carbohydrates, 20–30% fat, and 20% protein.4 Individualized dietary advice should be provided in patients with diabetes or severe hypertriglyceridemia. Overfeeding to achieve “catch up” growth or improved cosmetic appearances is strongly contraindicated as it may increase the risk of metabolic complications. On the other hand, food restriction, which might be needed to manage hyperphagia and obesity, is quite challenging during puberty and adolescence and caloric intake should match the developmental needs. Patients with FPLD should also be encouraged to engage in physical activity; however, those predisposed or already suffering from structural heart disease should avoid strenuous exercise.
In terms of hyperglycemia management, insulin sensitizers are a useful treatment option in patients with FPLD. Metformin is the first-line therapy although some patients may require additional agents including insulin. Paradoxically, metformin was recently shown to increase insulin resistance in a patient with SHORT syndrome, but further studies are required to confirm these findings.73
Thiazolidinediones (TZDs), which are direct agonists of PPARγ receptors leading to improved insulin sensitivity and conversion of visceral to subcutaneous fat, have been used in FPLD with mixed results. Some studies have shown an improvement in metabolic markers,74 such as hyperglycemia, dyslipidemia and hyperandrogenism, whilst others have demonstrated increased overall adiposity due to fat accumulation in non-lipoatrophic areas.75 Different mutations in the ligand-binding domain of PPARγ may affect the binding capacity to synthetic ligands, thus affecting the responsiveness of different subtypes of lipodystrophy to TZD.76 Cardiomyopathy, when present, restricts the use of these medications.
Insulin treatment might also be needed and in cases of severe insulin resistance, concentrated forms of insulin (U500) can be used. High doses and multiply daily injections, of even the concentrated forms, are required with not always satisfactory results on glycemic control.
The newer classes of hypoglycemic agents have also been used in patients with FPLD. The use of a combined intravenous glucose tolerance-euglycemic clamp in cases of FPLD revealed not only increased insulin resistance but also impaired first-phase insulin secretion.77 In addition, Valerio et al78 showed that the increased visceral adiposity characterizing FPLD is directly related to low dipeptidyl peptidase 4 (DPP4) levels. The above observations led researchers to investigate the role of glucagon-like peptide 1 receptor agonists (GLP1 Ras) in the management of overt diabetes in FPLD cases with promising results such as improvement of glycemic control and reduced insulin requirements.77,79
The sodium-glucose co-transporter 2 (SGLT2) inhibitors have an insulin-independent effect on glucose control and have been proven to be beneficial in cardiovascular and renal disease in type 2 diabetes mellitus. Evidence from cases of congenital generalized lipodystrophy has showed that use of SGLT2 inhibitors is associated with improvement in all metabolic markers and reduced incidence of cardiomyopathy.80 Preliminary data in FPLD reveal an improvement in the metabolic profile and a reduction in total insulin requirements.81,82 Further studies are however needed to confirm the clinical benefits of the newer diabetes treatments (GLP1 Ras, SGLT2 inhibitors) in the management of FPLD.
Management of hypertriglyceridemia can be challenging and requires the combination of diet and pharmacologic treatment with fibrates and long-chain omega-3 fatty acids. Caution is advised when combinations of fibrates with statins are used in patients with coexisting myopathy. Volanesorsen, an inhibitor of hepatic Apolipoprotein C-III, has been found to significantly reduce triglycerides in cases of familial chylomicronaemia83 and is currently under study in patients with FPLD and resistant hypertriglyceridemia in a randomized control trial (BROADEN study, Ionis Pharmaceuticals Inc., ClinicalTrials.gov Identifier: NCT02527343). Plasmapheresis might be needed in refractory cases to avoid pancreatitis.
Roux-en-Y surgery has been considered in cases of FPLD and refractory metabolic disease. The procedure has been successfully tried in cases of FPLD 1 and 2, associated with central adiposity, with promising results such as sustained weight loss, improvement of metabolic parameters and even withdrawal of insulin treatment.84,85 The dramatic improvement in glycemic control has been attributed to reduced lipotoxicity as well as increased postprandial levels of GLP-1.86
Efficacy of cosmetic treatment has been predominantly studied in cases of acquired partial lipodystrophy. Procedures such as liposuction, gluteoplasty, breast implants, facial reconstruction and autologous fat transplantation have been tried with variable results in an effort to improve appearances and self-esteem in affected patients.87,88
Metreleptin, a recombinant analogue of human leptin, is the only specific therapy available for the management of human lipodystrophies.89,90 The drug was approved in the US, in 2014 for patients with congenital or acquired generalized lipodystrophy and is also approved in Japan for the treatment of diabetes and/or hypertriglyceridemia in patients with congenital or acquired lipodystrophy (generalized or partial). In 2018, metreleptin was approved in the European Union, for adults and children aged ≥2 years with generalized lipodystrophy as well as for adults and children aged ≥12 years with partial lipodystrophy (familial or acquired) for whom standard treatments have not achieved adequate metabolic control (https://www.ema.europa.eu/en/medicines/human/EPAR/myalepta).
Leptin’s main role is to reflect energy reserves and regulate appetite by signaling the hypothalamus. In this regard, it was assumed that the beneficial effects of leptin replacement in patients with lipodystrophy arise from reduced caloric intake. However, Brown et al suggested that independent of food intake, metreleptin has an additional favorable metabolic effect due to an improvement of insulin sensitivity.91 Recently though, Püschel et al92 after studying four cases of FPLD treated with metreleptin for over 3 years, demonstrated improved satiety, reduced hunger and meal frequency in agreement with the results of an older study with a shorter duration of leptin treatment.93 Metreleptin has been shown to improve atherogenic profile, HbA1c, serum triglycerides and hepatic steatosis in patients with acquired and congenital lipodystrophy (generalized or partial).89,94,95 Long-term effectiveness and safety of metreleptin has been demonstrated in patients with FPLD due to both PPARG and LMNA pathogenic variants.96,97 Interestingly, results suggest that those patients with more severe baseline metabolic characteristics benefit most from metreleptin treatment.98,99
The pleiotropic effects of leptin replacement therapy on the gonadal axis have been described in many studies. Patients with lipodystrophy demonstrate increased gonadotropic (luteinising hormone; LH) secretion following metreleptin treatment.100 Female patients present with increased fertility and menstrual cycle normalization100,101 whereas testosterone levels increase in males.102 Furthermore, metreleptin use improves urinary protein excretion in CGL but has no significant effect on proteinuria in patients with FPLD.103 It should however be noted that experience is limited as proteinuria occurs less frequently in partial lipodystrophy and that the degree of proteinuria was less severe in patients with FPLD compared to CGL in the study by Lee at al.33,103 Patients with FPLD suffer social stigma with devastating effects on their psychology and self-esteem. In one of the largest cohorts studied up to date, patients treated with metreleptin for over a year experienced improvement in their appearance which had a positive impact on their self-perception.104 Metreleptin is given subcutaneously, by once-daily injections and when body weight is >40kg, the initial dose in females is 5mg and in males 2.5 mg. The dose is adjusted according to the clinical response.
Treatment with metreleptin is generally well tolerated but poor adherence therapy is noted in up to 30% of the cases.104 Injection site reactions due to lipoatrophy and the daily reconstitution from the powder form have been described as the main contributing factors. Hypoglycemia secondary to reduced insulin requirements, after treatment initiation, has also been described. Paradoxical worsening of hyperphagia and adverse metabolism in metreleptin treated patients have been attributed to the development of neutralizing antibodies. At the same time, whereas the majority of metreleptin treated patients develop antibodies, only a small number will develop neutralizing activity.105 Lymphoma development has been described in very few patients with acquired lipodystrophy who received metreleptin therapy, although this may be attributed to pre-existing immunodeficiency rather than to metreleptin use.71,89,106
Currently, investigational trials for new therapeutic agents are underway.89 Results regarding the efficacy of lipid-lowering agents such as gemcabene (dialkyl ether dicarboxylic acid) and evinacumab (monoclonal antibody to ANGPTL3) as well as anorexigenic hormones modulators (setmelanotide – melanocortin receptor agonist) in the management of FPLD are highly anticipated.
Conclusion
FPLD is a group of genetic disorders affecting predominantly adipogenesis, lipid droplet structure and function leading to altered adipose tissue topography. The altered storing capacity and endocrine function of affected adipocytes result into insulin resistance and ectopic fat accumulation with severe metabolic complications. Raised awareness amongst clinicians will lead to early diagnosis of the syndrome and management of comorbidities. In this regard, recently acquired knowledge on the genetics and the effectiveness of new treatment modalities will dictate new diagnostic and therapeutic strategies for these rare metabolic diseases.
Abbreviations
CGL, congenital generalized lipodystrophy; DPP4, dipeptidyl peptidase 4; DXA, dual-energy absorptiometry; FPLD, familial partial lipodystrophy; GLP! Ras, glucagon-like peptide 1 receptor agonists; GWAS, genome-wide association studies; HbA1c, hemoglobin A1c; LH, luteinizing hormone; MAD, mandibuloacral dysplasia; MRI, magnetic resonance imaging; NASH, non-alcoholic steatohepatitis; PCOS, polycystic ovarian syndrome; PCYT1A, phosphate cytidyltransferase 1A; PIK3R1, phosphatidylinositol 3 kinase regulatory subunit; PPARγ, peroxisome proliferator-activated receptor γ; PSMB8, proteasome subunit beta type 8; SGLT2, sodium-glucose co-transporter 2; SNVs, single nucleotide variants; TZDs, thiazolidinediones; ZMPSTE24, zinc metalloproteinase STE24.
Acknowledgments
We would like to thank Prof. Ioannis Georgiou and Dr Charilaos Kostoulas, from the Laboratory of Medical Genetics in Clinical Practice (Medical School, University of Ioannina, Greece), for their assistance reviewing the evidence on the genetic variants of FPLD.
Disclosure
Stelios Tigas and Alexandra Bargiota have received travel grants from Aegerion Pharmaceuticals.
References
1. Chiquette E, Oral EA, Garg A, Araújo-Vilar D, Dhankhar P. Estimating the prevalence of generalized and partial lipodystrophy: findings and challenges. Diabetes Metab Syndr Obes. 2017;10:375. doi:10.2147/DMSO.S130810
2. Köbberling J, Willms B, Kattermann R, Creutzfeldt W. Lipodystrophy of the extremities. A dominantly inherited syndrome associated with lipatrophic diabetes. Humangenetik. 1975;29(2):111–120. doi:10.1007/bf00430347
3. Dunnigan MG, Cochrane MA, Kelly A, Scott JW. Familial lipoatrophic diabetes with dominant transmission: a new syndrome. QJM. 1974;43(1):33–48.
4. Araújo-Vilar D, Santini F. Diagnosis and treatment of lipodystrophy: a step-by-step approach. J Endocrinol Invest. 2019;42(1):61–73. doi:10.1007/s40618-018-0887-z
5. Melvin A, Stears A, Savage DB. Recent developments in lipodystrophy. Curr Opin Lipidol. 2019;30(4):284–290. doi:10.1097/MOL.0000000000000613
6. Hussain I, Garg A. Lipodystrophy syndromes. Endocrinol Metab Clin. 2016;45(4):783–797. doi:10.1016/j.ecl.2016.06.012
7. Vigouroux C, Caron-Debarle M, Le Dour C, Magré J, Capeau J. Molecular mechanisms of human lipodystrophies: from adipocyte lipid droplet to oxidative stress and lipotoxicity. Int J Biochem Cell Biol. 2011;43(6):862–876. doi:10.1016/j.biocel.2011.03.002
8. Huang-Doran I, Sleigh A, Rochford JJ, O’Rahilly S, Savage DB. Lipodystrophy: metabolic insights from a rare disorder. J Endocrinol. 2010;207(3):245–255. doi:10.1677/JOE-10-0272
9. Virtue S, Vidal-Puig A. It’s not how fat you are, it’s what you do with it that counts. PLoS Biol. 2008;6(9):e237. doi:10.1371/journal.pbio.0060237
10. Wong SPY, Huda M, English P, et al. Adipokines and the insulin resistance syndrome in familial partial lipodystrophy caused by a mutation in lamin A/C. Diabetologia. 2005;48(12):2641–2649. doi:10.1007/s00125-005-0038-x
11. Haque WA, Shimomura I, Matsuzawa Y, Garg A. Serum adiponectin and leptin levels in patients with lipodystrophies. J Clin Endocrinol Metab. 2002;87(5):2395–2398. doi:10.1210/jcem.87.5.8624
12. Blüher M, Mantzoros CS. From leptin to other adipokines in health and disease: facts and expectations at the beginning of the 21st century. Metabolism. 2015;64(1):131–145. doi:10.1016/j.metabol.2014.10.016
13. Sarmento ASC, Lima JG, de Souza Timoteo AR, et al. Changes in redox and endoplasmic reticulum homeostasis are related to congenital generalized lipodystrophy type 2. Biochim Biophys Acta Mol Cell Biol Lipids. 2020;158610.
14. Mathew H, Castracane VD, Mantzoros C. Adipose tissue and reproductive health. Metabolism. 2018;86:18–32. doi:10.1016/j.metabol.2017.11.006
15. Kawwass JF, Summer R, Kallen CB. Direct effects of leptin and adiponectin on peripheral reproductive tissues: a critical review. Mol Hum Reprod. 2015;21(8):617–632. doi:10.1093/molehr/gav025
16. Dib K, Whitehead JP, Humphreys PJ, et al. Impaired activation of phosphoinositide 3-kinase by insulin in fibroblasts from patients with severe insulin resistance and pseudoacromegaly. A disorder characterized by selective postreceptor insulin resistance. J Clin Invest. 1998;101(5):1111–1120. doi:10.1172/JCI119884
17. Lotta LA, Wittemans LBL, Zuber V, et al. Association of genetic variants related to gluteofemoral vs abdominal fat distribution with type 2 diabetes, coronary disease, and cardiovascular risk factors. JAMA. 2018;320(24):2553–2563. doi:10.1001/jama.2018.19329
18. Lotta LA, Gulati P, Day FR, et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet. 2017;49(1):17. doi:10.1038/ng.3714
19. Broekema MF, Savage DB, Monajemi H, Kalkhoven E. Gene-gene and gene-environment interactions in lipodystrophy: lessons learned from natural PPARγ mutants. Biochim Biophys Acta Mol Cell Biol Lipids. 2019.
20. Campeau PM, Astapova O, Martins R, et al. Clinical and molecular characterization of a severe form of partial lipodystrophy expanding the phenotype of PPARγ deficiency. J Lipid Res. 2012;53(9):1968–1978. doi:10.1194/jlr.P025437
21. Hegele RA, Cao H, Frankowski C, Mathews ST, Leff T. PPARG F388L, a transactivation-deficient mutant, in familial partial lipodystrophy. Diabetes. 2002;51(12):3586–3590. doi:10.2337/diabetes.51.12.3586
22. Strissel KJ, Stancheva Z, Miyoshi H, et al. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes. 2007;56(12):2910–2918. doi:10.2337/db07-0767
23. Sears IB, MacGinnitie MA, Kovacs LG, Graves RA. Differentiation-dependent expression of the brown adipocyte uncoupling protein gene: regulation by peroxisome proliferator-activated receptor gamma. Mol Cell Biol. 1996;16(7):3410–3419. doi:10.1128/MCB.16.7.3410
24. Okuno A, Tamemoto H, Tobe K, et al. Troglitazone increases the number of small adipocytes without the change of white adipose tissue mass in obese Zucker rats. J Clin Invest. 1998;101(6):1354–1361. doi:10.1172/JCI1235
25. Kelly IE, Han TS, Walsh K, Lean ME. Effects of a thiazolidinedione compound on body fat and fat distribution of patients with type 2 diabetes. Diabetes Care. 1999;22(2):288–293. doi:10.2337/diacare.22.2.288
26. Agarwal AK, Barnes RI, Garg A. Genetic basis of congenital generalized lipodystrophy. Int J Obes. 2004;28(2):336. doi:10.1038/sj.ijo.0802487
27. Shackleton S, Lloyd DJ, Jackson SNJ, et al. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet. 2000;24(2):153. doi:10.1038/72807
28. Garg A. Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab. 2011;96(11):3313–3325. doi:10.1210/jc.2011-1159
29. Garg A, Peshock RM, Fleckenstein JL. Adipose tissue distribution pattern in patients with familial partial lipodystrophy (Dunnigan variety). J Clin Endocrinol Metab. 1999;84(1):170–174.
30. Patni N, Li X, Adams-Huet B, Vasandani C, Gomez-Diaz RA, Garg A. Regional body fat changes and metabolic complications in children with Dunnigan lipodystrophy-causing LMNA variants. J Clin Endocrinol Metab. 2018;104(4):1099–1108. doi:10.1210/jc.2018-01922
31. Vantyghem MC, Pigny P, Maurage CA, et al. Patients with familial partial lipodystrophy of the Dunnigan type due to a LMNA R482W mutation show muscular and cardiac abnormalities. J Clin Endocrinol Metab. 2004;89(11):5337–5346. doi:10.1210/jc.2003-031658
32. Pasotti M, Klersy C, Pilotto A, et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol. 2008;52(15):1250–1260. doi:10.1016/j.jacc.2008.06.044
33. Fountas A, Giotaki Z, Dounousi E, et al. Familial partial lipodystrophy and proteinuric renal disease due to a missense c. 1045C> T LMNA mutation. Endocrinol Diabetes Metab Case Rep. 2017;2017(1). doi:10.1530/EDM-17-0049
34. Simha V, Agarwal AK, Oral EA, Fryns J-P, Garg A. Genetic and phenotypic heterogeneity in patients with mandibuloacral dysplasia-associated lipodystrophy. J Clin Endocrinol Metab. 2003;88(6):2821–2824. doi:10.1210/jc.2002-021575
35. Spear ED, Hsu E-T, Nie L, Carpenter EP, Hrycyna CA, Michaelis S. ZMPSTE24 missense mutations that cause progeroid diseases decrease prelamin A cleavage activity and/or protein stability. Dis Model Mech. 2018;11(7):dmm033670. doi:10.1242/dmm.033670
36. Cassini TA, Robertson AK, Bican AG, et al. Phenotypic heterogeneity of ZMPSTE24 deficiency. Am J Med Genet A. 2018;176(5):1175–1179. doi:10.1002/ajmg.a.38493
37. Agarwal AK, Fryns J-P, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. 2003;12(16):1995–2001. doi:10.1093/hmg/ddg213
38. Gonzalo S, Kreienkamp R, Askjaer P. Hutchinson-Gilford progeria syndrome: a premature aging disease caused by LMNA gene mutations. Ageing Res Rev. 2017;33:18–29. doi:10.1016/j.arr.2016.06.007
39. Bonne G, Yaou RB, Navarro C, et al. Type B mandibuloacral dysplasia with congenital myopathy due to homozygous ZMPSTE24 missense mutation. Eur J Hum Genet. 2011;19(6):647–654. doi:10.1038/ejhg.2010.256
40. Kwan JM, Satter EK. A 53-year-old woman with multifocal subcutaneous nodules. J Am Acad Dermatol. 2015;72(5):924–926. doi:10.1016/j.jaad.2012.06.032
41. Agarwal AK, Zhou XJ, Hall RK, et al. Focal segmental glomerulosclerosis in patients with mandibuloacral dysplasia owing to ZMPSTE24 deficiency. J Investigat Med. 2006;54(4):208–213. doi:10.2310/6650.2006.05068
42. George S, Rochford JJ, Wolfrum C, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304(5675):1325–1328. doi:10.1126/science.1096706
43. Klatka M, Rysz I, Kozyra K, Polak A, Kołłątaj W. SHORT syndrome in a two-year-old girl–case report. Ital J Pediatr. 2017;43(1):44. doi:10.1186/s13052-017-0362-z
44. Thauvin-Robinet C, Auclair M, Duplomb L, et al. PIK3R1 mutations cause syndromic insulin resistance with lipoatrophy. Am J Hum Genet. 2013;93(1):141–149. doi:10.1016/j.ajhg.2013.05.019
45. Dyment DA, Smith AC, Alcantara D, et al. Mutations in PIK3R1 cause SHORT syndrome. Am J Hum Genet. 2013;93(1):158–166. doi:10.1016/j.ajhg.2013.06.005
46. Alcantara D, Elmslie F, Tetreault M, et al. SHORT syndrome due to a novel de novo mutation in PRKCE (Protein Kinase Cɛ) impairing TORC2-dependent AKT activation. Hum Mol Genet. 2017;26(19):3713–3721. doi:10.1093/hmg/ddx256
47. Chen FJ, Yin Y, Chua BT, Li P. CIDE family proteins control lipid homeostasis and the development of metabolic diseases. Traffic. 2020;21(1):94–105.
48. Rubio‐Cabezas O, Puri V, Murano I, et al. Partial lipodystrophy and insulin resistant diabetes in a patient with a homozygous nonsense mutation in CIDEC. EMBO Mol Med. 2009;1(5):280–287. doi:10.1002/emmm.200900037
49. Ajjaji D, Ben M’barek K, Mimmack ML, et al. Dual binding motifs underpin the hierarchical association of perilipins1–3 with lipid droplets. Mol Biol Cell. 2019;30(5):703–716. doi:10.1091/mbc.E18-08-0534
50. Jéru I, Vantyghem M-C, Bismuth E, et al. Diagnostic challenge in PLIN1-Associated familial partial lipodystrophy. J Clin Endocrinol Metab. 2019;104(12):6025–6032. doi:10.1210/jc.2019-00849
51. Laver TW, Patel KA, Colclough K, et al. PLIN1 haploinsufficiency is not associated with lipodystrophy. J Clin Endocrinol Metab. 2018;103(9):3225–3230. doi:10.1210/jc.2017-02662
52. Zolotov S, Xing C, Mahamid R, Shalata A, Sheikh‐Ahmad M, Garg A. Homozygous LIPE mutation in siblings with multiple symmetric lipomatosis, partial lipodystrophy, and myopathy. Am J Med Genet A. 2017;173(1):190–194. doi:10.1002/ajmg.a.37880
53. Albert JS, Yerges-Armstrong LM, Horenstein RB, et al. Null mutation in hormone-sensitive lipase gene and risk of type 2 diabetes. N Engl J Med. 2014;370(24):2307–2315. doi:10.1056/NEJMoa1315496
54. Garg A, Sankella S, Xing C, Agarwal AK. Whole-exome sequencing identifies ADRA2A mutation in atypical familial partial lipodystrophy. JCI Insight. 2016;1(9). doi:10.1172/jci.insight.86870
55. Garg A, Agarwal AK. Caveolin-1: A New Locus for Human Lipodystrophy. Oxford University Press; 2008.
56. Kim CA, Delépine M, Boutet E, et al. Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J Clin Endocrinol Metab. 2008;93(4):1129–1134. doi:10.1210/jc.2007-1328
57. Garg A, Kircher M, Del Campo M, Amato RS, Agarwal AK, University of Washington Center for Mendelian G. Whole exome sequencing identifies de novo heterozygous CAV1 mutations associated with a novel neonatal onset lipodystrophy syndrome. Am J Med Genet A. 2015;167(8):1796–1806. doi:10.1002/ajmg.a.37115
58. Cao H, Alston L, Ruschman J, Hegele RA. Heterozygous CAV1 frameshift mutations (MIM 601047) in patients with atypical partial lipodystrophy and hypertriglyceridemia. Lipids Health Dis. 2008;7(1):3. doi:10.1186/1476-511X-7-3
59. Yamamoto GL, Baratela WAR, Almeida TF, et al. Mutations in PCYT1A cause spondylometaphyseal dysplasia with cone-rod dystrophy. Am J Hum Genet. 2014;94(1):113–119. doi:10.1016/j.ajhg.2013.11.022
60. Hoover-Fong J, Sobreira N, Jurgens J, et al. Mutations in PCYT1A, encoding a key regulator of phosphatidylcholine metabolism, cause spondylometaphyseal dysplasia with cone-rod dystrophy. Am J Hum Genet. 2014;94(1):105–112. doi:10.1016/j.ajhg.2013.11.018
61. Liu Y, Ramot Y, Torrelo A, et al. Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 2012;64(3):895–907. doi:10.1002/art.33368
62. Torrelo A. CANDLE syndrome as a paradigm of proteasome-related autoinflammation. Front Immunol. 2017;8:927. doi:10.3389/fimmu.2017.00927
63. Yu C-E, Oshima J, Fu Y-H, et al. Positional cloning of the Werner’s syndrome gene. Science. 1996;272(5259):258–262. doi:10.1126/science.272.5259.258
64. Oshima J, Sidorova JM, Monnat JRJ. Werner syndrome: clinical features, pathogenesis and potential therapeutic interventions. Ageing Res Rev. 2017;33:105–114. doi:10.1016/j.arr.2016.03.002
65. Weedon MN, Ellard S, Prindle MJ, et al. An in-frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy. Nat Genet. 2013;45(8):947. doi:10.1038/ng.2670
66. Cunniff C, Bassetti JA, Ellis NA. Bloom’s syndrome: clinical spectrum, molecular pathogenesis, and cancer predisposition. Mol Syndromol. 2017;8(1):4–23. doi:10.1159/000452082
67. Sanz MM, German J, Cunniff C. Bloom’s Syndrome Mar 22 [Updated 2016 Apr 7]. In: Pagon RA, Adam MP, Ardinger HH, et al, editors. GeneReviews® [Internet]. Seattle, WA, USA:University of Washington; 2006:1993–2017.
68. Köbberling J, Dunnigan MG. Familial partial lipodystrophy: two types of an X linked dominant syndrome, lethal in the hemizygous state. J Med Genet. 1986;23(2):120–127. doi:10.1136/jmg.23.2.120
69. Herbst KL, Tannock LR, Deeb SS, Purnell JQ, Brunzell JD, Chait A. Köbberling type of familial partial lipodystrophy: an underrecognized syndrome. Diabetes Care. 2003;26(6):1819–1824. doi:10.2337/diacare.26.6.1819
70. Stenson PD, Ball EV, Mort M, et al. Human gene mutation database (HGMD®): 2003 update. Hum Mutat. 2003;21(6):577–581. doi:10.1002/humu.10212
71. Brown RJ, Araujo-Vilar D, Cheung PT, et al. The diagnosis and management of lipodystrophy syndromes: a multi-society practice guideline. J Clin Endocrinol Metab. 2016;101(12):4500–4511. doi:10.1210/jc.2016-2466
72. Meral R, Ryan BJ, Malandrino N, et al. “Fat Shadows” from DXA for the qualitative assessment of lipodystrophy: when a picture is worth a thousand numbers. Diabetes Care. 2018;41(10):2255–2258. doi:10.2337/dc18-0978
73. Lewandowski KC, Dąbrowska K, Brzozowska M, Kawalec J, Lewiński A. Metformin paradoxically worsens insulin resistance in SHORT syndrome. Diabetol Metab Syndr. 2019;11(1):81. doi:10.1186/s13098-019-0477-z
74. Arioglu E, Duncan-Morin J, Sebring N, et al. Efficacy and safety of troglitazone in the treatment of lipodystrophy syndromes. Ann Intern Med. 2000;133(4):263–274. doi:10.7326/0003-4819-133-4-200008150-00009
75. Simha V, Rao S, Garg A. Prolonged thiazolidinedione therapy does not reverse fat loss in patients with familial partial lipodystrophy, Dunnigan variety. Diabetes Obes Metab. 2008;10(12):1275–1276. doi:10.1111/j.1463-1326.2008.00978.x
76. Agostini M, Schoenmakers E, Beig J, et al. A pharmacogenetic approach to the treatment of patients with PPARG mutations. Diabetes. 2018;67(6):1086–1092. doi:10.2337/db17-1236
77. Banning F, Rottenkolber M, Freibothe I, Seissler J, Lechner A. Insulin secretory defect in familial partial lipodystrophy type 2 and successful long‐term treatment with a glucagon‐like peptide 1 receptor agonist. Diabet Med. 2017;34(12):1792–1794. doi:10.1111/dme.13527
78. Valerio CM, de Almeida JS, Moreira RO, et al. Dipeptidyl peptidase-4 levels are increased and partially related to body fat distribution in patients with familial partial lipodystrophy type 2. Diabetol Metab Syndr. 2017;9(1):26. doi:10.1186/s13098-017-0226-0
79. Oliveira J, Lau E, Carvalho D, Freitas P. Glucagon-like peptide-1 analogues-an efficient therapeutic option for the severe insulin resistance of lipodystrophic syndromes: two case reports. J Med Case Rep. 2017;11(1):12. doi:10.1186/s13256-016-1175-1
80. Ulahannan TJ, D’Emden M. Sustained, successful treatment of diabetes in lipodystrophy by dapagliflozin. 2018.
81. Hamaguchi T, Hirota Y, Takeuchi T, et al. Treatment of a case of severe insulin resistance as a result of a PIK 3R1 mutation with a sodium–glucose cotransporter 2 inhibitor. J Diabetes Investig. 2018;9(5):1224–1227. doi:10.1111/jdi.12825
82. Kawana Y, Imai J, Sawada S, Yamada T, Katagiri H. Sodium–glucose cotransporter 2 inhibitor improves complications of lipodystrophy: a case report. Ann Intern Med. 2017;166(6):450–451. doi:10.7326/L16-0372
83. Witztum JL, Gaudet D, Freedman SD, et al. Volanesorsen and triglyceride levels in familial chylomicronemia syndrome. N Engl J Med. 2019;381(6):531–542. doi:10.1056/NEJMoa1715944
84. Ciudin A, Baena‐Fustegueras JA, Fort JM, Encabo G, Mesa J, Lecube A. Successful treatment for the Dunnigan‐type familial partial lipodystrophy with Roux‐en‐Y gastric bypass. Clin Endocrinol. 2011;75(3):403. doi:10.1111/j.1365-2265.2011.04057.x
85. Utzschneider KM, Trence DL. Effectiveness of gastric bypass surgery in a patient with familial partial lipodystrophy. Diabetes Care. 2006;29(6):1380–1382. doi:10.2337/dc06-0130
86. Melvin A, Adams C, Flanagan C, et al. Roux-en-Y gastric bypass surgery in the management of familial partial lipodystrophy type 1. J Clin Endocrinol Metab. 2017;102(10):3616–3620. doi:10.1210/jc.2017-01235
87. Dollfus C, Blanche S, Trocme N, Funck‐Brentano I, Bonnet F, Levan P. Correction of facial lipoatrophy using autologous fat transplants in HIV‐infected adolescents. HIV Med. 2009;10(5):263–268. doi:10.1111/j.1468-1293.2008.00682.x
88. Neto B, Andrade G, Lima R, Barros M, Junior J. Surgical lipodystrophy correction associated with the use of antiretroviral therapy: an analysis of procedures performed and impact on the patients. Brasileira De Cirurgia Plástica. 2001;30(2):250–257.
89. Akinci B, Meral R, Oral EA. Update on therapeutic options in lipodystrophy. Curr Diab Rep. 2018;18(12):139. doi:10.1007/s11892-018-1100-7
90. Friedman JM, Mantzoros CS. 20 years of leptin: from the discovery of the leptin gene to leptin in our therapeutic armamentarium. Metabolism. 2015;64(1):1–4. doi:10.1016/j.metabol.2014.10.023
91. Brown RJ, Oral EA, Cochran E, et al. Long-term effectiveness and safety of metreleptin in the treatment of patients with generalized lipodystrophy. Endocrine. 2018;60(3):479–489. doi:10.1007/s12020-018-1589-1
92. Püschel J, Miehle K, Müller K, et al. Beneficial effects of leptin substitution on impaired eating behavior in lipodystrophy are sustained beyond 150 weeks of treatment. Cytokine. 2019;113:400–404. doi:10.1016/j.cyto.2018.10.012
93. McDuffie JR, Riggs PA, Calis KA, et al. Effects of exogenous leptin on satiety and satiation in patients with lipodystrophy and leptin insufficiency. J Clin Endocrinol Metab. 2004;89(9):4258–4263. doi:10.1210/jc.2003-031868
94. Kinzer AB, Shamburek RD, Lightbourne M, Muniyappa R, Brown RJ. Advanced lipoprotein analysis shows atherogenic lipid profile that improves after metreleptin in patients with lipodystrophy. J Endocr Soc. 2019;3(8):1503–1517. doi:10.1210/js.2019-00103
95. Chong AY, Lupsa BC, Cochran EK, Gorden P. Efficacy of leptin therapy in the different forms of human lipodystrophy. Diabetologia. 2010;53(1):27. doi:10.1007/s00125-009-1502-9
96. Oral EA, Gorden P, Cochran E, et al. Long-term effectiveness and safety of metreleptin in the treatment of patients with partial lipodystrophy. Endocrine. 2019;64(3):500–511
97. Sekizkardes H, Cochran E, Malandrino N, Garg A, Brown RJ. Efficacy of metreleptin treatment in familial partial lipodystrophy due to PPARG vs LMNA pathogenic variants. J Clin Endocrinol Metab. 2019;104(8):3068–3076. doi:10.1210/jc.2018-02787
98. Ajluni N, Dar M, Xu J, Neidert AH, Oral EA. Efficacy and safety of metreleptin in patients with partial lipodystrophy: lessons from an expanded access program. J Diabetes Metab. 2016;7(3).
99. Simha V, Subramanyam L, Szczepaniak L, et al. Comparison of efficacy and safety of leptin replacement therapy in moderately and severely hypoleptinemic patients with familial partial lipodystrophy of the Dunnigan variety. J Clin Endocrinol Metab. 2012;97(3):785–792. doi:10.1210/jc.2011-2229
100. Oral EA, Ruiz E, Andewelt A, et al. Effect of leptin replacement on pituitary hormone regulation in patients with severe lipodystrophy. J Clin Endocrinol Metab. 2002;87(7):3110–3117. doi:10.1210/jcem.87.7.8591
101. Ebihara K, Kusakabe T, Hirata M, et al. Efficacy and safety of leptin-replacement therapy and possible mechanisms of leptin actions in patients with generalized lipodystrophy. J Clin Endocrinol Metab. 2006;92(2):532–541. doi:10.1210/jc.2006-1546
102. Musso C, Cochran E, Javor E, Young J, DePaoli AM, Gorden P. The long-term effect of recombinant methionyl human leptin therapy on hyperandrogenism and menstrual function in female and pituitary function in male and female hypoleptinemic lipodystrophic patients. Metabolism. 2005;54(2):255–263. doi:10.1016/j.metabol.2004.08.021
103. Lee HL, Waldman MA, Auh S, et al. Effects of metreleptin on proteinuria in patients with lipodystrophy. J Clin Endocrinol Metab. 2019;104(9):4169–4177. doi:10.1210/jc.2019-00200
104. Vatier C, Kalbasi D, Vantyghem M-C, et al. Adherence with metreleptin therapy and health self-perception in patients with lipodystrophic syndromes. Orphanet J Rare Dis. 2019;14(1):177. doi:10.1186/s13023-019-1141-2
105. Chan JL, Koda J, Heilig JS, et al. Immunogenicity associated with metreleptin treatment in patients with obesity or lipodystrophy. Clin Endocrinol (Oxf). 2016;85(1):137–149. doi:10.1111/cen.12980
106. Aslam A, Savage DB, Coulson IH. Acquired generalized lipodystrophy associated with peripheral T cell lymphoma with cutaneous infiltration. Int J Dermatol. 2015;54(7):827–829. doi:10.1111/ijd.12185
107. Semple RK, Chatterjee VKK, O’Rahilly S. PPARγ and human metabolic disease. J Clin Invest. 2006;116(3):581–589. doi:10.1172/JCI28003
108. Barroso I, Gurnell M, Crowley VEF, et al. Dominant negative mutations in human PPARγ associated with severe insulin resistance, diabetes mellitus and hypertension. Nature. 1999;402(6764):880. doi:10.1038/47254
109. Cao H, Hegele RA. Nuclear lamin A/C R482Q mutation in Canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet. 2000;9(1):109–112. doi:10.1093/hmg/9.1.109
110. Eriksson M, Brown WT, Gordon LB, et al. Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. Nature. 2003;423(6937):293. doi:10.1038/nature01629
111. De Sandre-giovannoli A, Bernard R, Cau P, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300(5628):2055. doi:10.1126/science.1084125
112. Novelli G, Muchir A, Sangiuolo F, et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am J Hum Genet. 2002;71(2):426–431. doi:10.1086/341908
113. Shastry S, Simha V, Godbole K, et al. A novel syndrome of mandibular hypoplasia, deafness, and progeroid features associated with lipodystrophy, undescended testes, and male hypogonadism. J Clin Endocrinol Metab. 2010;95(10):E192–E197. doi:10.1210/jc.2010-0419
114. Grahn THM, Zhang Y, Lee M-J, et al. FSP27 and PLIN1 interaction promotes the formation of large lipid droplets in human adipocytes. Biochem Biophys Res Commun. 2013;432(2):296–301. doi:10.1016/j.bbrc.2013.01.113
115. Kozusko K, Tsang VHM, Bottomley W, et al. Clinical and molecular characterization of a novel PLIN1 frameshift mutation identified in patients with familial partial lipodystrophy. Diabetes. 2015;64(1):299–310. doi:10.2337/db14-0104
116. Gandotra S, Le Dour C, Bottomley W, et al. Perilipin deficiency and autosomal dominant partial lipodystrophy. N Engl J Med. 2011;364(8):740–748. doi:10.1056/NEJMoa1007487
117. Farhan SMK, Robinson JF, McIntyre AD, et al. A novel LIPE nonsense mutation found using exome sequencing in siblings with late-onset familial partial lipodystrophy. Can J Cardiol. 2014;30(12):1649–1654. doi:10.1016/j.cjca.2014.09.007
118. Cavalcante MPV, Brunelli JB, Miranda CC, et al. CANDLE syndrome: chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature—a rare case with a novel mutation. Eur J Pediatr. 2016;175(5):735–740. doi:10.1007/s00431-015-2668-4
119. Agarwal AK, Xing C, DeMartino GN, et al. PSMB8 encoding the β5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am J Hum Genet. 2010;87(6):866–872. doi:10.1016/j.ajhg.2010.10.031
120. Donadille B, D’Anella P, Auclair M, et al. Partial lipodystrophy with severe insulin resistance and adult progeria Werner syndrome. Orphanet J Rare Dis. 2013;8(1):106. doi:10.1186/1750-1172-8-106
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.